

Abstract
Paramyxoviruses are a diverse family which utilizes a fusion (F) protein to enter cells via fusion of the viral lipid bilayer with a target cell membrane. Although certain regions of F are known to play critical roles in membrane fusion, the function of much of the protein remains unclear. Sequence alignment of a set of paramyxovirus F proteins and analysis utilizing Block Maker identified a region of conserved amino acid sequence in a large domain between the heptad repeats of F1, designated CBF1. We employed site-directed mutagenesis to analyze the function of completely conserved residues of CBF1 in both the simian virus 5 (SV5) and Hendra virus F proteins. The majority of CBF1 point mutants were deficient in homotrimer formation, proteolytic processing, and transport to the cell surface. For some SV5 F mutants, proteolytic cleavage and surface expression could be restored by expression at 30°C, and varying levels of fusion promotion were observed at this temperature. In addition, the mutant SV5 F V402A displayed a hyperfusogenic phenotype at both 30°C and 37°C, indicating this mutation allows for efficient fusion with only an extremely small amount of cleaved, active protein. The recently published prefusogenic structure of PIV5/SV5 F [Yin, H.S., et al. (2006) Nature 439, 38–44] indicates that residues within and flanking CBF1 interact with the fusion peptide domain. Together, these data suggest that CBF1-fusion peptide interactions are critical for the initial folding of paramyxovirus F proteins from across this important viral family, and can also modulate subsequent membrane fusion promotion.
The family Paramyxoviridae comprises many diverse members, including well-known human pathogens such as measles, mumps and respiratory syncytial virus (RSV), as well as animal pathogens like parainfluenza virus 5 (PIV5/SV5), and the newly emerged, zoonotic Hendra and Nipah viruses. Hendra virus first emerged in 1994 and caused an outbreak of severe respiratory illness near Brisbane, Australia. This resulted in the deaths of fourteen horses and two out of three humans infected, succumbing either to respiratory illness or to viral meningoencephalitis (1, 2). Nipah virus was responsible for an outbreak of viral encephalitis in Malaysia in 1998, which resulted in the deaths of 105 people and the preventative destruction of over one million swine (3). Hendra and Nipah virus are classified as NIAID priority pathogens and DHHS Select Agents, and no antiviral therapies currently exist for these fatal viruses. Hendra and Nipah are grouped into the Henipavirus genus within the family, due in part to the fact that while they possess 88% homology to each other, they share less than 30% homology with the rest of the family (4, 5).
While some enveloped viruses are known to promote virus-cell or cell-cell membrane fusion via a pH-dependent mechanism within the endosomal pathway (6), the majority of paramyxoviruses enter cells at the plasma membrane at a neutral pH (7). Recent studies on the paramyxovirus human metapneumovirus (HMPV) indicate that the fusion protein of this virus displays enhanced cell-cell fusion promotion at a slightly acidic pH (8). To enter a cell, paramyxoviruses utilize two surface glycoproteins: a fusion (F) protein, which promotes membrane fusion, and an attachment protein designated H, HN or G, which allows for initial binding between the virus and the target cell.
The F protein is a type I integral membrane protein and a class I viral fusion protein, which exists as an inactive F0 precursor protein when it is initially folded. Within the endoplasmic reticulum, F0 undergoes disulfide bond formation, N-linked glycosylation and oligomerization into a homotrimer. At a later stage, F0 is cleaved into two disulfide-linked subunits: F1 and F2 (Figure 1). Furin, a member of the proprotein convertase family, is known to be the protease responsible for the cleavage of many paramyxovirus F proteins, including those of measles virus (9), human parainfluenza virus 3 (HPIV3) (10), SV5 (11) and RSV (12). This cleavage occurs within the trans-Golgi network (TGN) (9). Cathepsin L, an endosomal/lysosomal protease, was recently identified as the protease responsible for cleavage of both Hendra and Nipah virus F proteins (13, 14). Data indicate that Hendra and Nipah F are transported from the TGN to the cell surface and subsequently internalized, cleaved and then recycled to the cell surface (15, 16). All of these folding and processing events, along with a final targeting to the cell surface, must occur in order for the F protein to be fusogenically active.
Figure 1.

Schematic of a paramyxovirus F protein and identification of CBF1. FP = fusion peptide; HRA/B = heptad repeat A/B; TM = transmembrane domain; residues in black = completely conserved amino acids; residues in dark grey = conserved across most members; residues in light grey = similar amino acids; residues underlined = mutations made to alanine within SV5 F and HeV F.
In comparing paramyxovirus F proteins to other class I fusion proteins such as HIV env/gp160 and influenza virus hemagglutinin (HA), similarities and distinctions can be made. For example, the C-terminal F1 subunit of paramyxoviruses corresponds to the HIV gp41 subunit and to influenza virus HA2. This subunit contains a hydrophobic N-terminal fusion peptide domain, which is responsible for insertion into the target cell membrane during fusion (17, 18), a C-terminal transmembrane anchor and two heptad repeats, HRA and HRB, which are known to fold onto each other into a six-helix bundle to drive the merger of viral and target cell membranes (19, 20). However, a major difference between the paramyxovirus F1 subunit and HIV gp41 and influenza virus HA2 is the length of peptide sequence between the two heptad repeat regions. In HIV gp41 and influenza virus HA2, there are only 22 and 8 amino acids, respectively, whereas in F1 there are ~ 250 amino acids in this intervening region (21). It is extremely likely that there are important functions for this large domain within the paramyxovirus F protein, either in F protein folding, processing or fusion.
Although there are certain regions within paramyxovirus fusion proteins with well-defined roles, the function of the majority of the F protein is unknown. To identify regions of conserved amino acid sequence within these proteins, sequence alignment of the F proteins from six paramyxovirus family members, representing four genera, was performed and analyzed for blocks of conservation. One conserved block was identified in the fusion peptide/heptad repeat A region, known to play critical roles in F-mediated membrane fusion and to be affected by mutagenesis (22–24). Two other blocks of conservation were identified: one in the F2 subunit (Gardner and Dutch, manuscript submitted) and the other in a large intervening region between the heptad repeats of F1, designated as the Conserved Block in F1/CBF1 (Figure 1).
We hypothesized that CBF1 was conserved across a diverse viral family due to a critical role in folding or fusion. To address this, point mutations were introduced using site-directed mutagenesis into the completely conserved residues within CBF1 for the fusion proteins of the disparate SV5/PIV5 and Hendra virus. The amino acids which define CBF1 include residues 374–406 in SV5 F and 380–412 in the Hendra virus F protein (HeV F). With one exception, point mutations to alanine of completely conserved residues within CBF1 resulted in F proteins which did not properly fold and oligomerize, and thus were not significantly proteolytically processed or expressed on the cell surface. A number of the SV5 F mutants could be induced to fold properly when expressed at a reduced temperature, and variations in fusogenic activity were observed, suggesting that interactions in this region can modulate fusion if a sufficient level of folded, activated protein is available on the cell surface. Four of the ten conserved cysteines within paramyxovirus F proteins are located within or just outside of CBF1 and participate in intramolecular disulfide bonds (25). Mutation of any of these cysteines resulted in malfolded F proteins. These data indicate a critical role for CBF1 in initial folding of paramyxovirus fusion proteins. A recently published structure of the prefusogenic form of the parainfluenza 5 (PIV5/SV5) F protein reveals that CBF1 from one monomer flanks the hydrophobic fusion peptide domain from another monomer (26). Our results therefore indicate that interactions between this conserved region and the fusion peptide are critical for folding of diverse F proteins.
EXPERIMENTAL PROCEDURES
Cell lines and viruses
CV-1, HeLa T4, BSR (provided by Karl-Klauss Conzelmann, Pettenkofer Institut) and BHK 21F cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). CV-1 cells were used to propagate the recombinant vaccinia virus vTF7-3, which expresses T7 polymerase (27).
Plasmids
Robert Lamb (Howard Hughes Medical Institute, Northwestern University, Evanston, IL) kindly provided the pGEM2X-SV5 F and pCAGGS-SV5 F and HN plasmids. A plasmid containing the Hendra virus F gene was kindly provided by Lin-Fa Wang (Australian Animal Health Laboratory). The F gene was cloned into pGEM4Z and gene sequence integrity confirmed as previously described (28). Site-directed mutagenesis was performed on pGEM2X-SV5 F and pGEM4Z-HeV F using the QuikChange site-directed mutagenesis system (Stratagene, La Jolla, CA), and mutant F genes were sequenced to verify that no secondary mutations had occurred. Specific F protein mutants were subcloned into the pCAGGS expression plasmid as previously described (28). Wt HeV F and G (also provided by Lin-Fa Wang) genes were ligated into pCAGGS as described previously (29).
Antibodies
SV5 F antipeptide antibodies to amino acids 82 to 96, within the SV5 F2 subunit, were provided by Robert Lamb. Antipeptide antibodies (Genemed Custom Peptide Antibody Service, San Francisco, CA) were generated to amino acids 526 to 539 within the cytoplasmic tail of the Hendra virus F protein. The primary monoclonal antibody F1a, provided by Richard Randall (University of St Andrews, Fife, UK) (30) and the secondary antibody fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse immunoglobulin G (Jackson Immunoresearch Laboratories, West Grove, PA) were used for flow cytometry.
Expression of wt F and F protein mutants, metabolic labeling and immunoprecipitation
Subconfluent monolayers of HeLa T4 or CV-1 cells were first infected with vTF7-3 recombinant vaccinia virus [1×107 pfu/cell in DMEM + 1% bovine serum albumin (BSA)] for 60 min to allow for expression of the T7 polymerase (27). Cells were washed with phosphate-buffered saline (PBS) and transfected with 2 μg pGEM2X-SV5 F or pGEM4Z-HeV F wt or mutant constructs, using Lipofectamine Plus reagents (Invitrogen Life Technologies, Carlsbad, CA) per the manufacturer’s protocol (HeLa) or cationic liposomes (CV-1) prepared as described previously (31) in Opti-MEM (Gibco Invitrogen, Carlsbad, CA) according to established laboratory protocols. Following a 3 h incubation at 37°C, cells were washed with PBS, and either starved in cysteine- and methionine-deficient DMEM for 30 min and metabolically labeled for 30 min to 3 h with Tran[35S] (100 μCi/ml; MP Biomedicals, Inc., Irvine, CA) in cysteine- and methionine-deficient DMEM, or labeled overnight with Tran[35S] (50 μCi/ml). Cells were then washed with PBS and either lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (32) and 25 mM iodoacetamide or incubated in cold (non-radioactive) DMEM for 2 to 3 h at 37°C prior to lysis. Immunoprecipitations were performed as described previously (33) using SV5 F or HeV F antipeptide antibodies and protein A-conjugated sepharose beads (Amersham/GE Healthcare Bio-Sciences, Piscataway, NJ) to pull down antibody-bound proteins. Samples were separated on a 15% polyacrylamide gel in the presence of the reducing agent dithiothreitol (DTT, Bio-Rad, Hercules, CA) to allow visualization of both the F1 and F2 subunits and analyzed by storage phosphor autoradiography using a Storm or Typhoon imaging system (Amersham/GE Healthcare Bio-Sciences, Piscataway, NJ).
Flow cytometry to detect surface populations
SV5 F wt or mutant proteins were expressed using the recombinant vTF7-3/pGEM expression system in HeLa T4 cells using cationic liposomes as described above. After the 3 h incubation at 37°C, cells were shifted to 33°C overnight. Cells were prepared for flow cytometry as described previously (22) with the primary monoclonal antibody F1a and the secondary antibody fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse immunoglobulin G. Fluorescence intensity of 10,000 cells was measured by flow cytometry (Flow Cytometry Core Facility, University of Kentucky and FACSCalibur; Becton Dickinson, Mountain View, CA).
Biotinylation of cell surface proteins
Following transient transfection of BSR cells with wt or mutant HeV F proteins using pGEM vector expression, cells were metabolically labeled overnight and chased for 2 h. Cells were placed on ice and washed three times with ice-cold PBS deficient in calcium and magnesium chloride (PBS−) at pH 8. Cells were then incubated in 1 ml of the cell-impermeable biotin analog EZ-Link sulfo-N-hydroxysuccinimide-biotin (sulfo-NHS-biotin, 1 mg/ml; Pierce, Rockford, IL) in PBS− (pH 8), first rocking gently at 4°C for 10 min and then resting at 4°C for 25 min. Cells were then washed six times with ice-cold PBS− (pH 8) and lysed in RIPA buffer and immunoprecipitated as described for metabolic labeling experiments. One hundred microliters of 10% SDS were added to the protein A-sepharose beads, and the samples boiled for 10 min and beads pelleted. The supernatant was removed and saved. Fifty microliters of 10% SDS were added to the protein A-sepharose beads, and samples boiled for another 10 min and beads pelleted. The supernatant was removed and added to the previous supernatant. Thirty microliters (one-fifth) of the combined supernatant was saved for analysis of the total F protein in the lysed cells. To the remaining 120 μl, 400 μl of biotinylation dilution buffer (20 mM Tris [pH 8], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.2% bovine serum albumin) and 30 μl of immobilized streptavidin beads (Pierce) were added, and the samples were rocked at 4°C for 1 h. Samples were washed with RIPA buffer as described previously. Both total and surface portions were analyzed on 15% polyacrylamide gels under reducing conditions and visualized by using the Storm or Typhoon imaging system.
Chemical cross-linking
SV5 F and HeV F wt and mutant proteins were expressed in HeLa T4 cells from pGEM constructs using vTF7-3 infection and transfection with Lipofectamine Plus reagents. Following overnight incubation, cells were starved and labeled for 30 min with Tran[35S] (100 μCi/ml) and then chased in cold DMEM for 1 h. Cells were removed from culture dishes using PBS− + 50mM EDTA. Chemical cross-linking using the reagent 3,3′-dithiobi(sulfosuccinimidyl propionate) (DTSSP, 1 mM; Pierce Biotechnology Inc., Rockford, IL) was performed in the presence of 0.2% NP-40 as described previously (34). Samples were lysed and immunoprecipitated as described above, resolved under nonreducing conditions on a 3.5% acrylamide gel and visualized with the Storm system.
Syncytia assay for fusion
Wt SV5 F, HeV F or F protein mutants, in the presence or absence of the respective attachment proteins, were expressed via the pCAGGS system in BHK 21F cells using the Lipofectamine Plus reagent as described above. Syncytia were examined at 100× magnification 24–48 h post-transfection using a Nikon TS100 inverted phase-contrast microscope (Nikon Inc., Garden City, NY) and pictures taken using a Nikon Coolpix995 digital camera.
Expression of proteins at a reduced temperature
Wt SV5 F, HeV F or the CBF1 mutants were transfected using the Lipofectamine 2000 reagent (Invitrogen Life Technologies, Carlsbad, CA) per the manufacturer’s protocol in duplicate into BSR cells, a variant of BHK cells that stably expresses T7 polymerase and thus does not require vaccinia virus infection to drive transcription of genes using pGEM expression (35). For the SV5 F-expressing cells, after a 30 min starve and 30 min pulse with Tran[35S] (100 μCi/ml) at 37°C, one population was shifted to 30°C for a 3 h chase period, while the other cell population remained at 37°C. The HeV F-expressing cells were also divided into 30°C and 37°C populations, and labeled overnight with Tran[35S] (50 μCi/ml) followed by a two h chase. Immunoprecipitation and analysis were performed as described above.
Mutants which were proteolytically processed at 30°C were subcloned into the pCAGGS expression vector. After transfection using Lipofectamine Plus or Lipofectamine 2000 reagents, overnight metabolic labeling (without a chase period) and surface biotinylation were performed as described above.
RESULTS
Identification and mutagenesis of CBF1
Regions within the F1 subunit of paramyxovirus fusion proteins with well-defined functions include the fusion peptide, transmembrane domain and heptad repeat regions (Figure 1). However, no defined functions have been ascribed to the 250 amino acid region between heptad repeat A and B (HRA and HRB). To identify regions of amino acid conservation across the paramyxovirus family and to shed light upon domains of unknown function in the F protein, sequences from the F proteins of paramyxovirus family members, representing four genera, were aligned and analyzed. F protein amino acid sequences from the Rubulaviruses mumps virus (36) and SV5 (37), the Avulavirus Newcastle Disease virus (NDV) (38), the Respirovirus Sendai virus (39), and the Henipaviruses Nipah virus (4) and Hendra virus (40) were analyzed by Block Maker, utilizing both the GIBBs and MOTIF programs (41). Three regions were identified when either the MOTIF program or the GIBBs sampler were used in the analysis. The first identified block was in the fusion peptide/heptad repeat A region, known to play critical roles in F-mediated membrane fusion. The two other identified blocks of conservation were in the undefined F2 subunit (Gardner and Dutch, manuscript submitted) and the large intervening region between the heptad repeats of F1, which we designated as the Conserved Block in F1 (CBF1) (Figure 1). A protein BLAST search identified no matches to the CBF1 region outside of paramyxovirus fusion proteins, indicating that this conserved stretch of amino acids is unique to F proteins. A small number of mutations in the large, undefined region between the heptad repeats have been shown to induce folding defects (42, 43). However, none of these residues are located in CBF1. Since sequence conservation was identified in both the fusion peptide and HRA, and both have important functions in F-mediated membrane fusion, we hypothesized that CBF1 may also be conserved due to a critical role in the initial folding or biological activity of paramyxovirus F proteins.
Site-directed mutagenesis was utilized to mutate to alanine seven conserved residues within CBF1 in the fusion proteins of the disparate paramyxoviruses SV5 (G374A, N379A, I394A, Q396A, V402A, I405A and D406A) and Hendra virus (G380A, N385A, I400A, Q402A, L408A, I411A and D412A) in order to determine the function for this conserved region in paramyxovirus F proteins (Figure 1). The only conserved residue which differs between the two F proteins is V402A in SV5 F and the corresponding L408A in Hendra F. However, these two residues are considered highly similar and one of them is present at this position in all paramyxovirus F proteins aligned.
Proteolytic processing of the CBF1 mutants
We first examined expression and proteolytic processing of the wt and mutant F proteins, to assess proper folding and intracellular transport. The SV5 F protein undergoes proteolytic processing by the cellular protease furin after transport from the ER through to the TGN, while Hendra F protein processing is mediated by the endosomal protease cathepsin L after endocytosis of the uncleaved precursor from the cell surface (13, 16). Thus, proteolytic processing of both SV5 F and Hendra F requires exit from the ER following protein folding, with subsequent proper intracellular trafficking. Transient expression was performed using the recombinant vaccinia-T7 polymerase (vTF7-3) system. Mock-transfected cells (empty plasmid vector) were used as a negative control. Overnight labeling with Tran[35S], immunoprecipitation and visualization via reducing SDS-PAGE and the STORM phosphoimaging system revealed that the mutant F0 precursor proteins of the SV5 F (Figure 2A) and Hendra virus F mutants (Figure 2B) were only minimally cleaved into F1 and F2, compared to a much higher level of processing for wild-type F, suggesting a defect in initial protein folding and/or transport. The only exception to this is the HeV F G380A mutant, which was processed in a similar manner to wt HeV F (Figure 2B). It should be noted that there is an additional glycine at amino acid 379 in HeV F, absent in SV5 F, which could compensate for the loss in flexibility at G380A.
Figure 2.
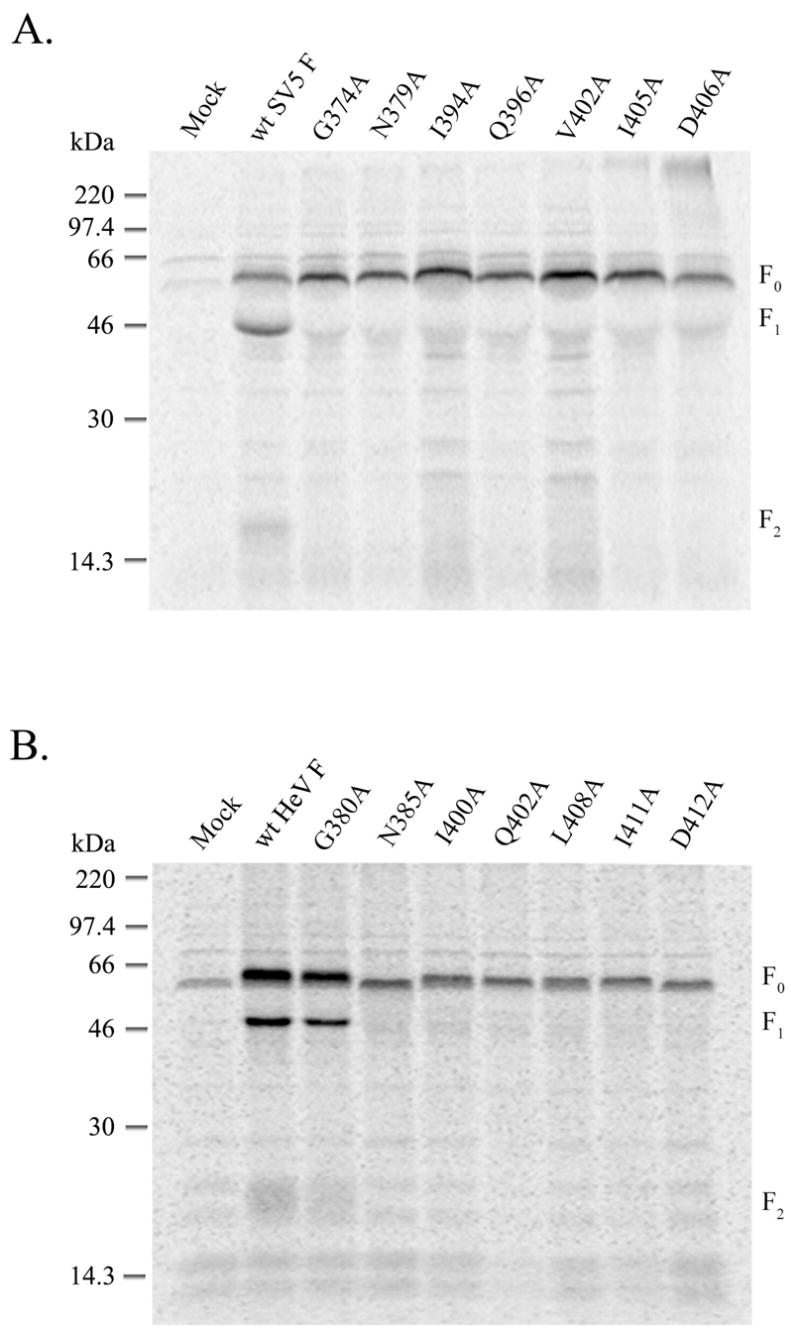
Processing of the CBF1 mutants. Wt SV5 F (A) and HeV F (B) and the CBF1 mutants were expressed in HeLa T4 or CV-1 cells, respectively, by use of the vTF7-3 system. Three h post-transfection, cells were labeled with Tran[35S] overnight and chased for 2h. Samples were immunoprecipitated, separated on a 15% SDS-polyacrylamide gel in the presence of a reducing agent and analyzed by use of the Storm or Typhoon system.
Surface expression of F proteins
Since the majority of the CBF1 mutations produced F proteins that were not properly proteolytically processed, we next examined the surface expression of these mutants to determine if they were indeed defective in transport to the cell surface. SV5 F proteins were transiently expressed using the vTF7-3 system and examined by flow cytometry using a monoclonal antibody to F1 and FITC-conjugated secondary antibody. Examination of the point mutations of the seven conserved residues in CBF1 from SV5 F indicates that these mutations lead to a defect in transport to the cell surface, as the surface expression of the mutant F proteins was comparable to the negative controls (Table 1). This confirms that mutations in SV5 F lead to a transport defect consistent with defects in initial F protein folding.
Table 1.
Surface expression of the SV5 CBF1 mutantsa
| Mutation | %Positiveb | M.F.I.c |
|---|---|---|
| Control (2°Ab only) | 0.495 | 12.5 |
| Mock | 0.862 | 17.7 |
| Wild-type SV5 F | 100 | 100 |
| G374A | 7.53 | 17.4 |
| N379A | 0.977 | 19.1 |
| I394A | 4.93 | 21.1 |
| Q396A | 16.1 | 21.8 |
| V402A | 16.3 | 23.4 |
| I405A | 2.60 | 14.4 |
| D406A | 3.27 | 20.2 |
Results are representative of three separate experiments and normalized to percent wild-type SV5 F, set at 100%.
Percent cells positive for F protein expression, determined by flow cytometry as described in Experimental Procedures.
M.F.I., mean fluorescent intensity, determined by flow cytometry.
Flow cytometry was not performed on Hendra virus F proteins due to lack of an appropriate antibody. Instead, HeV F protein surface populations were analyzed by biotinylation (Figure 3). F proteins were expressed in BSR cells via the vTF7-3 system. Cells were labeled overnight with Tran[35S], chased for 2 h in cold media and then biotinylated as described in Experimental Procedures. Similar to the SV5 CBF1 mutants, the HeV F CBF1 mutants were not expressed on the cell surface, confirming a defect in protein folding and transport (Figure 3). As expected, the only exception to this was the HeV F G380A mutant, which had been shown to be properly proteolytically processed (Figure 2). HeV F G380A displayed surface expression of the cleaved F1 protein at ~ 60% of wild-type F1 (representative of several experiments). However, the amount of F0 was also reduced, suggesting that lowered expression on the cell surface is due to a slight folding defect (i.e. less protein made and transported to the surface), rather than due to inefficient endosomal trafficking, proteolytic processing by cathepsin L and recycling to the cell surface.
Figure 3.
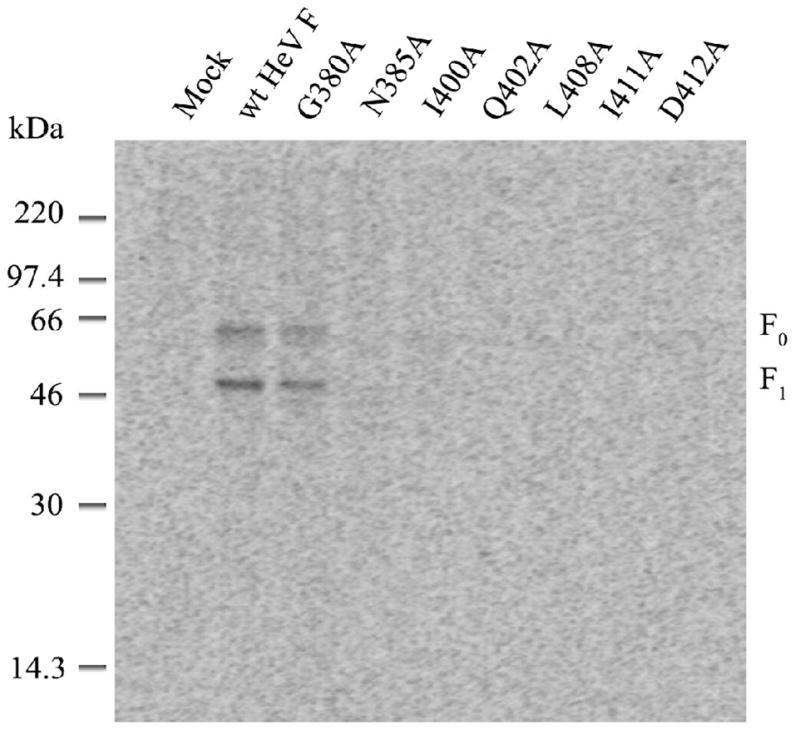
Surface expression of biotinylated HeV F proteins. Wt HeV F and the CBF1 mutants were expressed in BSR cells. Cells were labeled overnight with Tran[35S] and chased for 2h. Samples were biotinylated and immunoprecipitated as total and surface populations as described in Experimental Procedures. Samples were separated on a 15% SDS-polyacrylamide gel under reducing conditions and analyzed by use of the Storm or Typhoon system.
Fusion promotion by HeV F G380A
The fusogenically active form of the F protein exists as a proteolytically cleaved trimer on the cell surface. Since the majority of CBF1 mutations resulted in misfolded F proteins which were neither proteolytically processed nor expressed on the cell surface, these mutants were not assayed for fusion promotion activity. However, HeV F mutant G380A was expressed on the cell surface in a cleaved form at levels comparable to the wt protein, and this mutant was therefore examined for its ability to induce the formation of giant multi-nucleated cells termed syncytia. To examine well-defined syncytia in the absence of vaccinia virus infection, HeV F G380A was expressed using the pCAGGS plasmid, which allows for high-level expression from a chicken actin promoter (44). Expression assays confirmed that HeV F G380A was proteolytically processed after expression in this system (data not shown). Membrane fusion promoted by the Hendra virus F protein requires expression of the attachment protein G (45). Therefore, expression of HeV F alone, without co-expression of HeV G, was used as a negative control for syncytium formation. HeV F G380A did display the formation of syncytia in transfected BHK 21F cells, although they were slightly smaller and in fewer numbers than those seen for wt HeV F (Figure 4). The reduced fusion seen for G380A was not due to a reduced surface expression of HeV F G380A, as this mutant was shown to be expressed on the cell surface at slightly higher than wild-type HeV F levels in the pCAGGS system (data not shown), suggesting a slight fusion defect for this mutant.
Figure 4.

Syncytia assay for cell-cell fusion. Wt HeV F alone, F + G and G380A + G were expressed via the pCAGGS vector in BHK 21 F cells. Twenty-nine hours post-transfection, pictures were taken at 100× magnification and cells examined for the formation of giant multi-nucleated cells, or syncytia (indicated with arrows).
Oligomerization of CBF1 mutants
Surface expression and processing studies suggested that mutations in CBF1 result in a defect in protein folding. Since oligomerization of F0 into a homotrimer is known to occur in the endoplasmic reticulum prior to cleavage and transport to the cell surface (46), studies on the oligomerization of wild-type SV5 F and Hendra virus F and mutant F proteins were performed. HeLa T4 cells transiently expressed the F proteins via the vTF7-3 system and were briefly labeled with Tran[35S]. Nonidet P40 (NP40) was used to permeabolize the cell membrane, and DTSSP was used to chemically cross-link F with its binding partners. In the absence of cross-linker, the wt and mutant F proteins were primarily monomeric, with some dimer, trimer and higher molecular weight forms detected. Wild-type SV5 and HeV F proteins formed discrete trimers in the presence of cross-linker, with SV5 F more efficiently cross-linked to a trimer than HeV F (Figures 5A and 5B). The addition of cross-linker stabilizes interactions with F protein monomers within the length of the spacer arm, which results in a trimeric form for wild-type paramyxovirus F proteins. However, no corresponding trimer was seen for the mutant proteins except the processed, fusogenically active HeV F G380A (Figure 5B). Instead, cross-linking of the majority of the CBF1 mutants resulted in a smear at the top of the gels, suggesting that rather than stabilizing a discrete trimeric form of F, addition of cross-linker resulted in stabilization of high molecular weight aggregates. These data indicate that CBF1 is critical for proper folding and oligomerization of the paramyxovirus fusion proteins.
Figure 5.
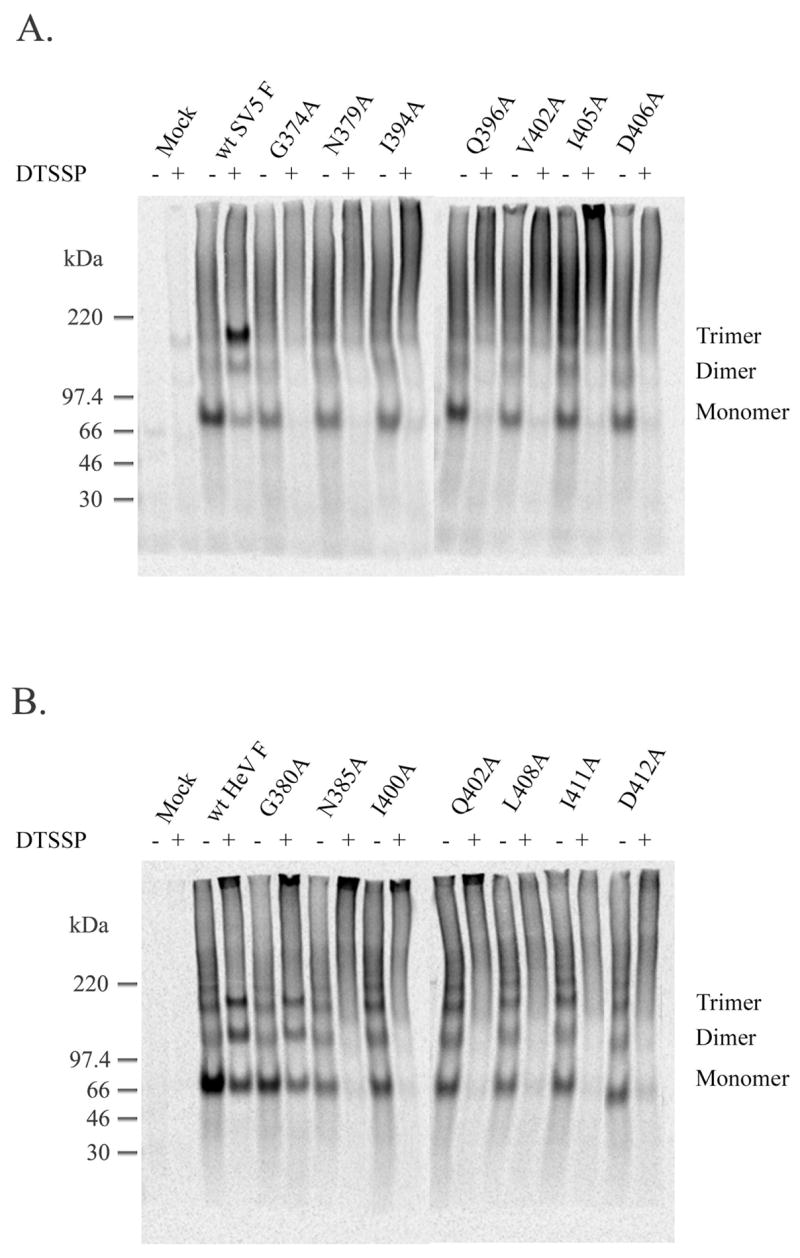
Oligomerization of the CBF1 mutants. Wt SV5 F (A) or HeV F (B) and the CBF1 mutants were expressed in HeLa T4 cells by use of the vTF7-3 system. Three h post-transfection, cells were starved for 30 min, pulsed with Tran[35S] for 30 min and chased for 1 h. Chemical cross-linking was performed on cell suspensions in the presence of 0.2% NP-40 and 1mM DTSSP. Samples were immunoprecipitated, analyzed on a 3.5% polyacrylamide gel in the absence of a reducing agent and visualized by use of the Storm or Typhoon system.
Importance of CBF1 in disulfide bond formation
Iwata et al. (25) demonstrated that the F protein of the paramyxovirus Sendai virus possesses ten conserved cysteine residues, with two participating in an intermolecular disulfide bond between F1 and F2 and the other eight in intramolecular disulfide bonds within F1. Three of these eight cysteines are located within CBF1, with a fourth just C-terminal to this conserved block. The proposed disulfide bonds between these four cysteines were confirmed by the recently published prefusogenic structure of PIV5/SV5 F (26). We tested whether the presence of individual or all four cysteines is critical for proper folding of paramyxovirus F proteins. SV5 F C380S, C385S, C387S and C410S and the quadruple mutant 4CysS were constructed and expressed as above. Removal of any of these individual cysteines disrupted an important intramolecular disulfide bond and left an unpaired cysteine, resulting in multiple sizes of the precursor F0 on a non-reducing gel. A more compact form is seen for wild-type SV5 F0 (Figure 6A). All mutations resulted in F proteins which did not oligomerize into homotrimers (Figure 6B) and thus were not properly processed (Figure 6C). These four cysteines located within or near CBF1 likely participate in critical intramolecular disulfide bond formation (25, 26), important in proper folding of paramyxovirus F proteins.
Figure 6.
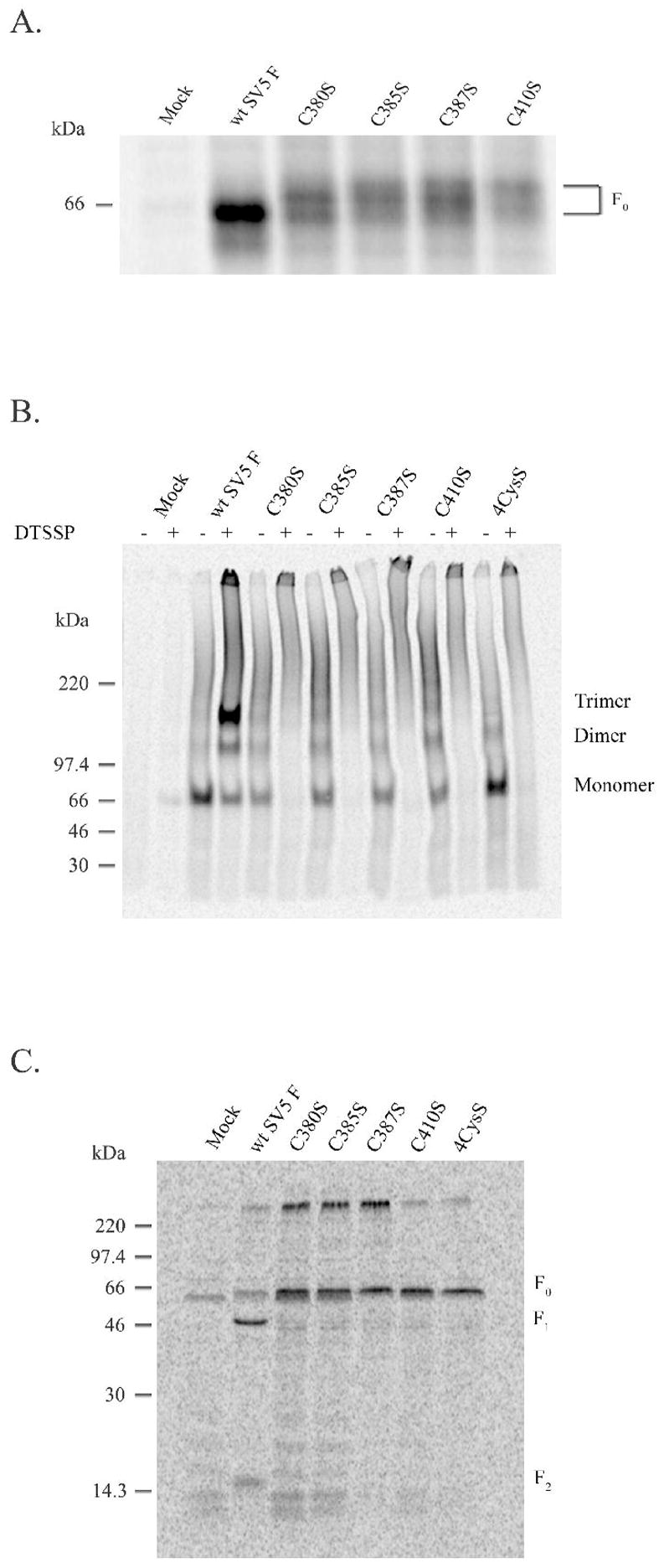
Expression of SV5 F cysteine mutants. A) Wt SV5 F and the Cys mutants were expressed in HeLa T4 cells by use of the vTF7-3 system. Three h post-transfection, cells were starved and labeled with Tran[35S] for 30 min followed by a 3 h chase. Samples were immunoprecipitated, separated on a 7.5% SDS-polyacrylamide gel in the absence of a reducing agent and analyzed as above. B) Wt SV5 F and the Cys mutants were expressed in HeLa T4 cells by use of the vTF7-3 system. Three h post-transfection, cells were starved for 30 min, pulsed with Tran[35S] for 30 min and chased for 1 h. Chemical cross-linking was performed on cell suspensions in the presence of 0.2% NP-40 and 1mM DTSSP. Samples were immunoprecipitated and analyzed on a 3.5% polyacrylamide gel in the absence of a reducing agent. C) Wt SV5 F and the Cys mutants were expressed in CV-1 cells by use of the vTF7-3 system. Three h post-transfection, cells were labeled overnight followed by a 2 h chase. Samples were immunoprecipitated, separated on a 15% SDS-polyacrylamide gel in the presence reducing agent and analyzed as above.
Expression of CBF1 mutants at a reduced temperature
Some mutations that result in misfolded proteins at non-permissive temperatures can permit properly folded proteins when expressed at reduced temperatures. For example, the temperature-sensitive vesicular stomatitis virus (VSV) G mutant ts045 folds improperly at a non-permissive 39°C, yet this folding defect can be rescued when the mutant is expressed at 32°C (47). Similarly, the E1 envelope protein of the Sindbis virus ts23 mutant undergoes proper folding and maturation when expressed at 28°C (48). Presumably, a lower temperature slows the folding process and allows for proper associations with ER folding chaperones. We therefore assessed proteolytic processing of the CBF1 mutants upon expression at a lowered temperature. In addition, to ensure that expression utilizing the vaccinia virus vTF7-3 expression system had not affected results, the wt F and mutant proteins were transiently expressed in BSR cells, which stably express the T7 polymerase, and thus drive expression from the T7 promoter in the absence of virus (35). The expression of CBF1 mutants at the physiological 37°C gave extremely low to undetectable levels of proteolytic processing for SV5 (Figure 7A) and Hendra virus (data not shown), indicating that the defects in folding and/or trafficking to allow processing were not due to the expression system utilized. Expression of wt SV5 F at 30°C allowed for proper proteolytic processing, though at a slightly reduced rate (Figure 7B), suggesting slowed movement and maturation through the secretory pathway of a properly folded protein at the reduced temperature. Expression at 30°C resulted in greatly increased processing of a number of the SV5 CBF1 mutant proteins (Figure 7B), with percent cleaved F [F2/(F2+F0)*100] ranging from 34% wt SV5 F cleaved for G374A up to 54% wt F cleaved for V402A. Numbers represent the averages from three separate experiments. However, while the wt HeV F protein was efficiently expressed and processed at the lowered temperature, the HeV CBF1 mutants (except G380A) were still processing-defective (data not shown).
Figure 7.

Expression of the SV5 CBF1 mutants at a reduced temperature. Wt SV5 F and the CBF1 mutants were expressed in BSR cells. After a 30 min starve and 30 min pulse period, one population of SV5 F mutants remained at 37°C for a 3 h chase period (A), while the other population was shifted to 30°C (B). Samples were immunoprecipitated and analyzed as above. Mutants that were processed at 30°C were expressed in Vero cells via the pCAGGS vector and metabolically labeled overnight at either 37°C (C) or 30°C (D), and surface biotinylation and analysis were performed as described above.
To assess the ability of the SV5 F mutants to promote fusion, mutant F proteins which were proteolytically processed at 30°C using pGEM expression (G374A, I394A, Q396A and V402A) were expressed using the pCAGGS plasmid. To confirm the pGEM results, wt SV5 F and the CBF1 mutants were expressed in Vero cells and assayed for proteolytic processing as well as surface expression using biotinylation. After expression and overnight metabolic labeling at both 37°C and 30°C, cell surface expression was examined. All of the CBF1 mutants were expressed in the cleaved form on the cell surface at 30°C, with the amount of cleaved F protein at 30°C versus that at 37°C increasing ~ 150 % for Q396A and almost 400% for V402A (Figures 7C and 7D).
Cell-cell fusion promotion at both 37°C and 30°C was examined using the syncytia assay. The strain of SV5 used in these studies has been shown to promote cell-cell fusion, albeit at reduced efficiency, after expression of the F protein in the absence of the normally required attachment protein HN (49–53). Wt SV5 F promoted syncytium formation at both temperatures (Figures 8A and 8B), although syncytia were much smaller and expression of HN was required at 30°C for detection of significant syncytia formation over background. G374A could promote membrane fusion when co-expressed with the attachment protein at the lowered temperature (Figure 8B), but no syncytia formation above background was observed at 37°C (data not shown), demonstrating that this mutation does not impair fusion if transport and proteolytic processing are rescued. Both I394A and Q396A were incapable of inducing syncytium formation at either temperature, indicating that some mutations in this region have negative impacts on both folding and transport, and on subsequent fusogenic activity. Interestingly, the mutant SV5 F V402A was found to be hyperfusogenic at both temperatures, forming extremely large syncytia that included the vast majority of the cells in the plate, in the presence or absence of HN (Figures 8A and 8B). Proteolytic processing for this mutant was barely detectable at 37°C (Figures 7A and 7C), and surface expression was significantly reduced from the wt protein (Table 1), indicating that this high level of fusion is promoted by only an extremely small level of surface-expressed, activated protein. These data suggest that the overall structure of SV5 F around this region may be more slightly more stable and tolerant to changes than HeV F, thus allowing F proteins with folding-defects to be properly folded, processed and trafficked to the cell surface, particularly after expression at a reduced temperature. These results also indicate that residues in CBF1 can modulate membrane fusion.
Figure 8.

Syncytium formation at a reduced temperature. Wt SV5 F and the CBF1 mutants were expressed alone or in the presence of the attachment protein HN in BHK 21 F cells via the pCAGGS vector. Forty-seven hours post-transfection, pictures were taken at 100× magnification and cells examined for the formation of syncytia.
DISCUSSION
SV5 (PIV5) and Hendra virus are two disparate members of the paramyxovirus family. We have shown that CBF1, a region conserved across the family, is critical for proper folding and oligomerization of both the SV5 and Hendra virus F protein. The conservation of amino acids throughout the family suggests that the critical role in protein folding is a conserved function of this region. The recently published prefusogenic structure of PIV5/SV5 F (26) indicates that CBF1 interacts with the fusion peptide. As seen in Figure 9A [adapted from (26)], CBF1 and flanking residues (purple) from one monomer of the F protein lie adjacent to the fusion peptide (orange) from another monomer, positioning consistent with our findings that this domain is critical for forming stable oligomers. The fusion peptide region is well-defined as the domain which inserts into a target cell membrane during the initiation of fusion. Since fusion peptide domains are extremely hydrophobic [(7) and Figure 9C, hydrophobic residues in green], they initially remain shielded from solvent within the F protein while the adjacent cleavage site remains accessible for proteolytic processing, as seen in the prefusogenic PIV5/SV5 F structure (26). Paramyxovirus F proteins are cleaved just N-terminal to the fusion peptide, thus making it the new N-terminus of the F1 ectodomain. We hypothesize that the conservation of CBF1 is due to a critical role in burying the fusion peptide in the precursor F0 protein by its position on the exterior of the F protein globular head (Figure 9B). Thus, point mutation of completely conserved residues within this block results in misfolding of the F protein, likely due to both exposure of this extremely hydrophobic region and failure to form proper monomer-monomer contacts. The conservation across the family indicates a similar function of this region for all F proteins.
Figure 9.

CBF1 and adjacent residues modeled within the prefusogenic form of PIV5/SV5 F. A) Close-up view of CBF1 and interacting residues. Purple = CBF1 and adjacent residues; orange = fusion peptide; green = F103, the first residue of the fusion peptide; yellow = disulfide bond; red = completely conserved residues in CBF1. B) CBF1 (purple) in the context of the entire F ectodomain. C) Potential hydrophobic interactions between CBF1 and flanking residues (purple) and the fusion peptide domain (orange). Hydrophobic residues = green. [Figures were adapted from (26) using PyMOL 0.99 and color coded for clarification.]
CBF1 may also properly stabilize trimers to assist in proper folding of heptad repeat B (HRB), a region involved in membrane fusion promotion. CBF1 is immediately N-terminal to HRB (Figure 9B, purple residues and stalk region), separated only by an HRB linker region. During the process of cotranslational protein folding, CBF1 would be at least partially folded during HRB translation and insertion into the endoplasmic reticulum. If proper interactions between residues within CBF1 are perturbed, interactions between residues C-terminal may also be affected. In the prefusogenic structure of SV5 F, HRB monomers associate into a trimeric coiled-coil. However, it has been shown that HRB peptides alone in solution do not form coiled-coil structures (54–56), suggesting that they must be expressed along with the entire F protein ectodomain in order to obtain the correct homotrimeric interaction. Also, mutations made to the HRB region result in fusion rather than folding defects (57, 58), consistent with a role for HRB in fusion promotion and for CBF1 in F protein folding and oligomerization.
We have shown that mutation of completely conserved residues within CBF1, including four cysteines known to participate in intramolecular disulfide bonds, results in F proteins which do not form homotrimers. The formation of intramolecular disulfide bonds is known to occur early during cotranslational folding within the endoplasmic reticulum, and is essential for subsequent proper folding of many glycoproteins (46, 59). Mutation of corresponding cysteine residues in Sendai virus F (59) and RSV F (60) resulted in F proteins that did not transit through the secretory pathway and reach the cell surface, and thus were defective for fusion. These results indicate that disulfide bond formation plays an integral part in the critical folding function of CBF1, and that this function is conserved across the paramyxovirus family.
The observed folding defect for four of the SV5 CBF1 mutants can be partially rescued by expressing these mutants at a reduced temperature, but no rescue of the HeV F mutants was observed, suggesting this region is more stable in SV5 F than in Hendra F. A difference in stability could be necessary due to the different mechanisms of proteolytic processing which SV5 F and Hendra F undergo. Furin cleaves SV5 F in the TGN and recognizes multiple basic amino acids. Cathepsin L cleaves Hendra F in the endosomal pathway, yet it has no specific consensus sequence. It is possible that a more structurally accessible cleavage site and a less rigid fold surrounding the fusion peptide domain allows for cleavage of Hendra F by cathepsin L.
For the four SV5 F mutants whose folding could be partially rescued at 30°C, differences in the fusogenic activity of the cleaved, surface expressed molecules were observed. SV5 F I394A and SV5 F Q396A were unable to promote syncytia formation above background at 30°C (Figure 8B), even though cleaved protein was present on the cell surface (Figure 7D). In contrast, fusion-promotion capability was restored at 30°C for mutant G374A, indicating that this mutation affects folding, but not subsequent membrane fusion. Finally, the mutation V402A produced an F protein which exhibited an extreme hyperfusogenic phenotype at both 30°C and 37°C, in the presence or absence of the SV5 HN attachment protein (Figure 8). This mutant was minimally processed at 37°C, indicating that only a small percentage of the properly folded and processed V402A mutant is required to promote significantly high levels of cell-cell fusion. Several other SV5 F protein mutants have been described with large decreases in surface expression and processing, but high levels of fusion, including the mutants SV5 F L447F or L447W and SV5 F I449F and I449W within the HRB linker region (61) and SV5 F mutant I49F, which is located within the conserved block in the F2 subunit (Gardner and Dutch, manuscript submitted). We hypothesize that mutations such as V402A destabilize the F protein, leading to malfolding of much of the produced protein, but increased fusion promotion for the small percentage that is initially folded, processed and transported to the cell surface. V402 is located at one of the bends in the middle of the immunoglobulin-like, seven-stranded β-sheet that comprises CBF1 and flanking residues. A change in side-chain volume and hydrophobic interactions (Figure 9C) may significantly contribute to this phenotype, as the substitution of valine for alanine decreases the relative hydrophobicity of residue 402 by over 50%.
Our results demonstrate that mutation of residues within CBF1 results in defects in F protein folding and trimerization, which can be reversed for some mutants if conditions are altered to allow folding and transport to occur more slowly. In comparing the location of CBF1 in both the prefusogenic structure of SV5 F (26) and the known postfusogenic structures of NDV F (62) and HPIV3 F (63), the secondary and tertiary structures of CBF1 alone do not change. CBF1 remains in an immunoglobulin-like shape in the postfusogenic structure, consisting of seven β-strands with disulfide bonds intact. The region as a whole is displaced during the transition from the metastable to stable postfusion structure, exposing the fusion peptide to initiate membrane fusion and allowing for the heptad repeat B linker to swing HRB around to form the stable six-helix bundle with HRA. This stable fold of CBF1 is consistent with a role for it in initial protein folding, and suggests that observed changes in fusion result from CBF1 interactions with the fusion peptide region, rather than in CBF1 directly participating in the fusion process. Those mutations which displayed increased proteolytic processing at a reduced temperature but no cell-cell fusion may have increased the stability of CBF1–fusion peptide interactions. In contrast, SV5 F V402A may significantly destabilize hydrophobic interactions within this region and with the fusion peptide such that the fusion peptide is triggered to undergo the transition from the pre- to postfusion form much more readily than in wt SV5 F.
Acknowledgments
We thank Dr. Robert Lamb (HHMI, Northwestern University) for the pGEM2X-SV5 F, pCAGGS-SV5 F and pCAGGS-SV5 HN plasmids and SV5 F-specific antibodies, Dr. Lin-Fa Wang (Australian Animal Health Laboratory) for the Hendra virus F and G plasmids and Dr. Richard Randall (University of St. Andrews, St. Andrews, Scotland) for the F1a monoclonal antibody. We are grateful to Dr. David Rodgers (University of Kentucky) for assistance with figure construction. We also thank Dr. Trevor Creamer (University of Kentucky) and members of the Dutch lab for critically reviewing this manuscript.
ABBREVIATIONS
- SV5
simian virus 5
- parainfluenza virus 5
PIV5 (SV5)
- HeV
Hendra virus
- F
fusion protein
- CB
conserved block
- CBF1
conserved block in F1
- HRA/HRB
heptad repeat A/B
Footnotes
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases (AI-51517) to R.E.D. A.E.G. was supported in part by a predoctoral fellowship from the American Heart Association, Ohio Valley Affiliate (0415223B).
References
- 1.Murray K, Selleck P, Hooper P, Hyatt A, Gould A, Gleeson L, Westbury H, Hiley L, et al. A morbillivirus that caused fatal disease in horses and humans. Science. 1995;268:94–97. doi: 10.1126/science.7701348. [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan JD, Allworth AM, Paterson DL, Snow TM, Boots R, Gleeson LJ, Gould AR, Hyatt AD, Bradfield J. Fatal encephalitis due to novel paramyxovirus transmitted from horses. Lancet. 1997;349:93–95. doi: 10.1016/s0140-6736(96)06162-4. [DOI] [PubMed] [Google Scholar]
- 3.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gulber DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels R, Ling AE, Peters CJ, Anderson LJ, Mahy BW. Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 4.Harcourt BH, Tamin A, Ksiazek Tg, Rollin PE, Anderson LJ, Bellini WJ, Rota PA. Molecular characterization of Nipah virus, a newly emergent paramxyovirus. Virology. 2000;271:334–349. doi: 10.1006/viro.2000.0340. [DOI] [PubMed] [Google Scholar]
- 5.Wang LF, Yu M, Hansson E, Pritchard LI, Shiell B, Michalski WP, Eaton BT. The exceptionally large genome of Hendra virus: support for creation of a new genus within the family Paramyxoviridae. J Virol. 2000;74:9972–9979. doi: 10.1128/jvi.74.21.9972-9979.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus-cell and cell-cell fusion. Annu Rev Cell Develop Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 7.Lamb RA, Kolakofsky D. Paramyxoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott-Raven Press; New York: 2001. pp. 1305–1340. [Google Scholar]
- 8.Schowalter RM, Smith SE, Dutch RE. Characterization of Human Metapneumovirus F Protein-Promoted Membrane Fusion: Critical Roles for Proteolytic Processing and Low pH. J Virol. 2006;80:10931–10941. doi: 10.1128/JVI.01287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolt G, Pedersen IR. The Role of Subtilisin-like proprotein Convertases for Cleavage of the Measles Virus Fusion Glycoprotein in Different Cell Types. Virology. 1998;252:387–398. doi: 10.1006/viro.1998.9464. [DOI] [PubMed] [Google Scholar]
- 10.Ortmann D, Ohuchi M, Angliker H, Shaw E, Garten W, Klenk H-D. Proteolytic cleavage of wild type and mutants of the F protein of human parainfluenza virus type 3 by two subtilisin-like endoproteases, furin and KEX2. J Virol. 1994;68:2772–2776. doi: 10.1128/jvi.68.4.2772-2776.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garten W, Hallenberger S, Ortmann D, Schafer W, Vey M, Angliker H, Shaw E, Klenk HD. Processing of viral glycoproteins by the subtilisin-like endoprotease furin and its inhibition by specific peptidylchloroalkylketones. Biochimie. 1994;76:217–225. doi: 10.1016/0300-9084(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 12.Zimmer G, Budz L, Herrler G. Proteolytic activation of respiratory syncytial virus fusion protein. Cleavage at two furin consensus sequences. J Biol Chem. 2001;276:31642–31650. doi: 10.1074/jbc.M102633200. [DOI] [PubMed] [Google Scholar]
- 13.Pager CT, Dutch RE. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J Virol. 2005;79(20):12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pager CT, Craft WW, Jr, Patch J, Dutch RE. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology. 2006;346:251–257. doi: 10.1016/j.virol.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diederich S, Moll M, Klenk HD, Maisner A. The nipah virus fusion protein is cleaved within the endosomal compartment. J Biol Chem. 2005;280:29899–29903. doi: 10.1074/jbc.M504598200. [DOI] [PubMed] [Google Scholar]
- 16.Meulendyke KA, Wurth MA, McCann RO, Dutch RE. Endocytosis plays a critical role in proteolytic processing of the Hendra virus fusion protein. J Virol. 2005;79:12643–12649. doi: 10.1128/JVI.79.20.12643-12649.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asano K, Asano A. Why is a specific amino acid sequence of F glycoprotein required for the membrane fusion reaction between envelope of HVJ (Sendai virus) and target cell membranes? Biochem International. 1985;10:115–122. [PubMed] [Google Scholar]
- 18.Novick SL, Hoekstra D. Membrane penetration of Sendai virus glycoproteins during the early stage of fusion with liposomes as determined by hydrophobic affinity labeling. Proc Natl Acad Sci USA. 1988;85:7433–7437. doi: 10.1073/pnas.85.20.7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sergel-Germano T, McQuain C, Morrison T. Mutations in the fusion peptide and heptad repeat regions of the Newcastle disease virus fusion protein block fusion. J Virol. 1994;68:7654–7658. doi: 10.1128/jvi.68.11.7654-7658.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker KA, Dutch RE, Lamb RA, Jardetzky TS. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 21.Dutch RE, Jardetzky TS, Lamb RA. Virus Membrane Fusion Proteins: Biological Machines that Undergo a Metamorphosis. Bioscience Reports. 2000;20:597–612. doi: 10.1023/a:1010467106305. [DOI] [PubMed] [Google Scholar]
- 22.Horvath CM, Lamb RA. Studies on the fusion peptide of a paramyxovirus fusion glycoprotein: roles of conserved residues in cell fusion. J Virol. 1992;66:2443–2455. doi: 10.1128/jvi.66.4.2443-2455.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russell CJ, Jardetzky TS, Lamb RA. Conserved glycine residues in the fusion peptide of the paramyxovirus fusion protein regulate activation of the native state. J Virol. 2004;78:13727–13742. doi: 10.1128/JVI.78.24.13727-13742.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.West DS, Sheehan MS, Segeleon PK, Dutch RE. Role of the simian virus 5 fusion protein N-terminal coiled-coil domain in folding and promotion of membrane fusion. J Virol. 2005;79:1543–1551. doi: 10.1128/JVI.79.3.1543-1551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwata S, Schmidt AC, Titani K, Suzuki M, Kido H, Gotoh B, Hamaguchi M, Nagai N. Assignment of disulfide bridges in the fusion glycoprotein of Sendai virus. J Virol. 1994;68:3200–3206. doi: 10.1128/jvi.68.5.3200-3206.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craft WW, Jr, Dutch RE. Sequence motif upstream of the Hendra virus fusion protein cleavage site is not sufficient to promote efficient proteolytic processing. Virology. 2005;341:130–140. doi: 10.1016/j.virol.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Pager CT, Wurth MA, Dutch RE. Subcellular localization and calcium and pH requirements for proteolytic processing of the Hendra virus fusion protein. J Virol. 2004;78:9154–9163. doi: 10.1128/JVI.78.17.9154-9163.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randall RE, Young DF, Goswami KKA, Russell WC. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J Gen Virol. 1987;68:2769–2780. doi: 10.1099/0022-1317-68-11-2769. [DOI] [PubMed] [Google Scholar]
- 31.Rose JK, Bonagurio B, Whitt MA. A new cationic liposome reagent mediating nearly quantitative transfection of animal cells. BioTechniques. 1991;10:520–525. [PubMed] [Google Scholar]
- 32.Paterson RG, Lamb RA. Ability of the hydrophobic fusion-related external domain of a paramyxovirus F protein to act as a membrane anchor. Cell. 1987;48:441–452. doi: 10.1016/0092-8674(87)90195-4. [DOI] [PubMed] [Google Scholar]
- 33.Lamb RA, Etkind PR, Choppin PW. Evidence for a ninth influenza viral polypeptide. Virology. 1978;91:60–78. doi: 10.1016/0042-6822(78)90355-0. [DOI] [PubMed] [Google Scholar]
- 34.Russell R, Paterson RG, Lamb RA. Studies with cross-linking reagents on the oligomeric form of the paramyxovirus fusion protein. Virology. 1994;199:160–168. doi: 10.1006/viro.1994.1108. [DOI] [PubMed] [Google Scholar]
- 35.Buchholz UJ, Finke S, Conzelmann KK. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol. 1999;73:251–259. doi: 10.1128/jvi.73.1.251-259.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okazaki K, Tanabayashi K, Takeuchi K, Hishiyama M, Okazaki K, Yamada A. Molecular cloning and sequence analysis of the mumps virus gene encoding the L protein and the trailer sequence. Virology. 1992;188:926–930. doi: 10.1016/0042-6822(92)90555-4. [DOI] [PubMed] [Google Scholar]
- 37.Paterson RG, Harris TJR, Lamb RA. Fusion protein of the paramyxovirus simian virus 5: Nucleotide sequence of mRNA predicts a highly hydrophobic glycoprotein. Proc Natl Acad Sci USA. 1984;81:6706–6710. doi: 10.1073/pnas.81.21.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGinnes LW, Morrison TG. Nucleotide sequence of the gene encoding the Newcastle disease virus fusion protein and comparisons of paramyxovirus fusion protein sequences. Virus Res. 1986;5:343–356. doi: 10.1016/0168-1702(86)90028-6. [DOI] [PubMed] [Google Scholar]
- 39.Fujii Y, Kiyotani K, Yoshida T, Sakaguchi T. Conserved and nonconserved regions in the Sendai virus genome: evolution of a gene possessing overlapping reading frames. Virus Genes. 2001;22:47–52. doi: 10.1023/a:1008130318633. [DOI] [PubMed] [Google Scholar]
- 40.Gould AR. Comparison of the deduced matrix and fusion protein sequences of equine morbillivirus with cognate genes of the Paramyxoviridae. Virus Res. 1996;43:17–31. doi: 10.1016/0168-1702(96)01308-1. [DOI] [PubMed] [Google Scholar]
- 41.Henikoff S, Henikoff JG, Alford WJ, Pietrokovski S. Automated construction and graphical presentation of protein blocks from unaligned sequences. Gene. 1995;163:GC17–26. doi: 10.1016/0378-1119(95)00486-p. [DOI] [PubMed] [Google Scholar]
- 42.Tanabayashi K, Takeuchi K, Hishiyama M, Yamada A. Effect on fusion induction of point mutations introduced into the F protein of mumps virus. Virology. 1994;204:851–853. doi: 10.1006/viro.1994.1607. [DOI] [PubMed] [Google Scholar]
- 43.Sergel TA, McGinnes LW, Morrison TG. A single amino acid change in the Newcastle disease virus fusion protein alters the requirement for HN protein in fusion. J Virol. 2000;74:5101–5107. doi: 10.1128/jvi.74.11.5101-5107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants by a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 45.Bossart KN, Wang L-F, Eaton B, Broder CC. Functional Expression and Membrane Fusion Tropism of the Envelope Glycoproteins of Hendra Virus. Virology. 2001;290:121–135. doi: 10.1006/viro.2001.1158. [DOI] [PubMed] [Google Scholar]
- 46.Doms RW, Lamb RA, Rose JK, Helenius A. Folding and assembly of viral membrane proteins. Virology. 1993;193:545–562. doi: 10.1006/viro.1993.1164. [DOI] [PubMed] [Google Scholar]
- 47.de Silva AM, Balch WE, Helenius A. Quality control in the endoplasmic reticulum: Folding and misfolding of the vesicular stomatitis virus G protein in cells and in vitro. J Cell Biol. 1990;111:857–866. doi: 10.1083/jcb.111.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carleton M, Brown DT. Events in the endoplasmic reticulum abrogate the temperature sensitivity of Sindbis virus mutant ts23. J Virol. 1996;70:952–959. doi: 10.1128/jvi.70.2.952-959.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paterson RG, Hiebert SW, Lamb RA. Expression at the cell surface of biologically active fusion and hemagglutinin-neuraminidase proteins of the paramyxovirus simian virus 5 from cloned cDNA. Proc Natl Acad Sci USA. 1985;82:7520–7524. doi: 10.1073/pnas.82.22.7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horvath CM, Paterson RG, Shaughnessy MA, Wood R, Lamb RA. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J Virol. 1992;66:4564–4569. doi: 10.1128/jvi.66.7.4564-4569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward CD, Paterson RG, Lamb RA. Mutants of the paramyxovirus SV5 fusion protein: regulated and extensive syncytium formation. Virology. 1995;209:242–249. doi: 10.1006/viro.1995.1250. [DOI] [PubMed] [Google Scholar]
- 52.Dutch RE, Joshi SB, Lamb RA. Membrane fusion promoted by increasing surface densities of the paramyxovirus F and HN proteins: comparison of fusion reactions mediated by simian virus 5 F, human parainfluenza virus type 3 F, and influenza virus HA. J Virol. 1998;72:7745–7753. doi: 10.1128/jvi.72.10.7745-7753.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsurudome M, Ito M, Nishio M, Kawano M, Komada H, Ito Y. Hemagglutinin-neuraminidase-independent fusion activity of simian virus 5 fusion (F) protein: difference in conformation between fusogenic and nonfusogenic F proteins on the cell surface. J Virol. 2001;75:8999–9009. doi: 10.1128/JVI.75.19.8999-9009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joshi SB, Dutch RE, Lamb RA. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248:20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- 55.Matthews J, Young TF, Tucky SP, Mackay JP. The core of the respiratory syncytial virus fusion protein is a trimeric coiled coil. J Virol. 2000;74:5911–5920. doi: 10.1128/jvi.74.13.5911-5920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu M, Wang E, Liu Y, Cao D, Jin N, Zhang CW, Bartlam M, Rao Z, Tien P, Gao GF. Six-helix bundle assembly and characterization of heptad repeat regions from the F protein of Newcastle disease virus. J Gen Virol. 2002;83:623–629. doi: 10.1099/0022-1317-83-3-623. [DOI] [PubMed] [Google Scholar]
- 57.Reitter JN, Sergel T, Morrison TG. Mutational analysis of the leucine zipper motif in the Newcastle disease virus fusion protein. J Virol. 1995;69:5995–6004. doi: 10.1128/jvi.69.10.5995-6004.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGinnes LW, Sergel T, Chen H, Hamo L, Schwertz S, Li D, Morrison TG. Mutational analysis of the membrane proximal heptad repeat of the newcastle disease virus fusion protein. Virology. 2001;289:343–352. doi: 10.1006/viro.2001.1123. [DOI] [PubMed] [Google Scholar]
- 59.Segawa H, Kato M, Yamashita T, Taira H. The roles of individual cysteine residues of Sendai virus fusion protein in intracellular transport. J Biochem. 1998;123:1064–1072. doi: 10.1093/oxfordjournals.jbchem.a022044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Day ND, Branigan PJ, Liu C, Gutshall LL, Luo J, Melero JA, Sarisky RT, Del Vecchio AM. Contribution of cysteine residues in the extracellular domain of the F protein of human respiratory syncytial virus to its function. Virol J. 2006;3:34. doi: 10.1186/1743-422X-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. The Journal of cell biology. 2003;163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, Gorman JJ, McKimm-Breschkin J, Lawrence LJ, Tulloch PA, Smith BJ, Colman PM, Lawrence MC. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure (Camb) 2001;9:255–266. doi: 10.1016/s0969-2126(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 63.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci USA. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]