

Abstract
The basic helix-loop-helix transcriptional repressor twist1, as an antagonist of nuclear factor κB (NF-κB)–dependent cytokine expression, is involved in the regulation of inflammation-induced immunopathology. We show that twist1 is expressed by activated T helper (Th) 1 effector memory (EM) cells. Induction of twist1 in Th cells depended on NF-κB, nuclear factor of activated T cells (NFAT), and interleukin (IL)-12 signaling via signal transducer and activator of transcription (STAT) 4. Expression of twist1 was transient after T cell receptor engagement, and increased upon repeated stimulation of Th1 cells. Imprinting for enhanced twist1 expression was characteristic of repeatedly restimulated EM Th cells, and thus of the pathogenic memory Th cells characteristic of chronic inflammation. Th lymphocytes from the inflamed joint or gut tissue of patients with rheumatic diseases, Crohn's disease or ulcerative colitis expressed high levels of twist1. Expression of twist1 in Th1 lymphocytes limited the expression of the cytokines interferon-γ, IL-2, and tumor necrosis factor-α, and ameliorated Th1-mediated immunopathology in delayed-type hypersensitivity and antigen-induced arthritis.
Twist1 and the closely related twist2 (also known as dermo1) genes encode basic helix-loop-helix transcription factors involved in the formation of mesoderm in Drosophila melanogaster (1), cranial neural tube and limb morphogenesis in mice (2), metastasis of tumor cells (3), control of apoptosis, and expression of cytokine genes in inflammation (4, 5). Mice deficient for twist2 or haploinsufficient for both twist1 and twist2 succumb to severe systemic inflammation, demonstrating the central role of Twist proteins in the regulation of inflammation. Twist1 and twist2 are expressed by fibroblasts and macrophages, and expression is promoted by TNF-α and type I IFNs (4, 5). The involvement of Twist expression by fibroblasts and macrophages in the control of inflammation is probable, but has not been investigated in detail so far.
T helper 1 (Th1) lymphocytes are potent inducers of inflammation. This has been demonstrated by adoptive transfer of Th1 lymphocytes in murine models of Th1-associated inflammatory diseases such as diabetes (6), inflammatory bowel disease (7), and rheumatoid arthritis (8). In these models, the induction of inflammation in a particular tissue by Th1 lymphocytes is dependent on restimulation by the cognate antigen in that tissue. Induction of inflammation by Th1 cells is mediated by expression of the proinflammatory cytokines TNF-α and IFN-γ, the latter being a hallmark of Th1 differentiation. IFN-γ also induces expression of the chemokine receptor CXCR3 and its ligands CXCL9, -10, and -11, attracting Th1 cells specifically to inflamed tissue (9). Th cells with the capacity to recall IFN-γ expression, i.e., Th1 memory cells, are detectable in chronically inflamed tissue (10, 11). The role of Th1 cells in the development and maintenance of chronic inflammation is less clear. Anti–IFN-γ therapy has been shown to be beneficial in various Th1-associated inflammatory diseases (12, 13). However, a regulatory role for IFN-γ has also been demonstrated, based on the induction of inducible nitric oxide synthase (14) and of IL-12 in antigen-presenting cells, in turn increasing the IL-10 synthesis of Th1 cells (15). Furthermore, IFN-γ inhibits the differentiation of naive Th cells into proinflammatory Th17 cells (16, 17).
In this study, we demonstrate specific autoregulation of Th1 cells by the transcription factor twist1. Expression of twist1 in Th cells is induced by IL-12/STAT4, NF-κB, and NFAT, and thus is specific for Th1 cells. Th1 effector memory (EM) cells (18) show increased transient reexpression of twist1 upon T cell receptor engagement. Twist1 reduces the functionality of Th1 cells by attenuating expression of IL-2, TNF-α, and IFN-γ. Th cells isolated from chronically inflamed gut tissue of patients with ulcerative colitis (UC) or Crohn's disease (CD) and synovial fluid of patients with spondyloarthropathies or rheumatoid arthritis are imprinted to express high levels of twist1, which indicates a history of repeated restimulation and an involvement in the pathogenesis of the disease. In murine models of acute and chronic inflammation, i.e., delayed-type hypersensitivity (DTH) and antigen-induced arthritis, expression of twist1 by Th1 cells regulates Th1-mediated inflammation.
RESULTS
Twist1 is transiently expressed in repeatedly activated Th1 cells
To define transcriptional changes during Th1 memory cell differentiation, we compared the global gene expression of murine Th1 cells, which were activated with antigen once or repeatedly at 6-d intervals. Naive CD4+CD62Lhi T lymphocytes expressing the transgenic DO11.10 TCR specific for OVA were activated in vitro with splenic APCs and the cognate peptide OVA327-339 under conditions that induce functional differentiation into Th1 cells, i.e., addition of IL-12 and blocking antibodies specific for IL-4. The transcriptional profiles of once- and four-time–stimulated Th1 cells were compared using high-density DNA microarrays. Among the 17 genes differentially expressed by a factor of two or more was twist1. Expression of twist1 was up-regulated 38-fold in four-time– versus once-stimulated Th1 cells (Table S1, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1).
Twist1 expression in Th1, but not Th2 or Th17, cells was confirmed by real-time PCR (Fig. 1). Twist1 mRNA expression in resting Th1 cells correlated with their age in vitro and the number of restimulations they had experienced. Expression was further enhanced 3 h after polyclonal stimulation either with PMA and the Ca2+ ionophore ionomycin, or with CD3- and CD28-specific antibodies (Fig. 1 A). When determined 3 h after restimulation with anti-CD3/-CD28 antibodies, expression of twist1 was close to the detection limit in naive Th cells. Expression increased ∼15-fold during the first Th1-polarizing stimulation, another 10-fold during the second stimulation, and another 3-fold during the third stimulation, reaching the maximum level after the fourth stimulation and remaining stable thereafter (Fig. 1 A). With PMA/ionomycin restimulation, maximum levels of twist1 mRNA were reached after two rounds of stimulation. In Th1 cells, upon anti-CD3/-CD28 restimulation, twist1 mRNA expression was up-regulated within the first hour, reaching maximum levels after 3 h, and decreasing again thereafter (Fig. 1 B). Expression of twist1 mRNA was also detectable in restimulated Th2 cells, but its level remained 30-fold lower than in Th1 cells. In Th17 cells, the level of twist1 mRNA expression was even lower than in Th2 cells (Fig. 1 C).
Figure 1.

Twist1 expression is induced in Th1, but not Th2 or Th17 cells. CD4+CD62Lhi OVA-specific T lymphocytes were stimulated in vitro under Th1 or Th2, or under Th17-polarizing conditions. Functional polarization of Th1, Th2, and Th17 cells, i.e., the cytokine expression profile, was confirmed by intracellular immunofluorescence (Fig. S1). (A) Twist1 mRNA in resting cells 6 d after the last stimulation (Th1; circles) or after 3 h of restimulation with anti-CD3/CD28 and IL-2 (Th1; squares) or PMA/ionomycin and IL-2 (Th1; triangles: Th2; diamonds) was determined by RT-PCR and normalized to hypoxanthine guanine phosphoribosyl transferase (HPRT). (B) Kinetics of twist1 mRNA expression in 6-d-old (open bars) and 24-d-old (shaded bars) Th1 cells, after stimulation with anti-CD3/CD28 and IL-2 or IL-2 alone. (C) Twist1 transcript levels in 6-d-old Th1, Th2, and Th17 cells after reactivation with PMA/ionomycin. (D) Twist1 protein expression in resting (-) and 5 h PMA/ionomycin-restimulated (+) 6- and 24-d-old Th1 and Th2 cells. Control: α-Tubulin immunoblot (bottom). (E) Kinetics of Twist1 protein expression in 24-d-old Th1 cells, before and at the indicated time intervals of stimulation with anti-CD3/CD28 and IL-2. Data are representative of two experiments. Fig. S1 is available at http://www.jem.org/cgi/content/full/jem.20072468/DC1.
Twist1 protein was detectable in 6-d-old restimulated Th1 cells. In 24-d-old Th1 cells, expression was enhanced. In resting Th1 cells and in reactivated or resting Th2 cells, Twist1 was not detectable by immunoblotting (Fig. 1 D). In agreement with the expression of twist1 mRNA, Twist1 protein expression peaked 3 h after reactivation, and then ceased, with levels still detectable 48 h after restimulation (Fig. 1 E), but not at 6 d after restimulation.
Twist2 was not expressed in the Th1 and Th2 cells analyzed here, as determined by real-time PCR (unpublished data).
Control of twist1 expression in Th1 lymphocytes
The specific expression of twist1 by activated Th1 cells suggests that Th1-polarizing or Th1-specific signals are required for the induction of twist1 expression in Th cells. Indeed, IL-12/STAT4 signaling induced twist1 expression in a dose-dependent fashion (Fig. 2 A). IFN-γ and STAT1 signaling did not induce twist1 expression in the absence of IL-12 (Fig. 2 A). T-bet, a T box transcription factor sufficient to induce Th1 differentiation (19), is not required for induction of expression of twist1; induction of twist1 by IL-12 was comparable in T-bet–deficient and wild-type Th cells (Fig. 2 B). Also, ectopically expressed T-bet did not induce twist1 expression in the absence of IL-12 in activated Th cells (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1).
Figure 2.

Twist1 induction requires IL-12 signaling via STAT4, but not IFN-γ or T-bet. (A) CD62Lhi DO11.10 Th cells were stimulated for 5 d under Th1-polarizing conditions (5 ng/ml IL-12, anti-IL4), reduced IL-12 (1/25:200 pg/ml; 1/625:8 pg/ml, anti-IL4), in the absence of IL-12 (anti–IL-12, anti-IL4), in the presence of IFN-γ (10 ng/ml IFN-γ, anti-IL-12, anti-IL-4), or under Th2-polarizing conditions. Twist1 mRNA in Th cells activated for 3 h with PMA/ionomycin and IL-2 was quantified by RT-PCR. The amount of twist1 transcripts induced under Th1-polarizing conditions was set to 1. Data are presented as the mean ± the SD of at least three experiments. (B) CD62Lhi Th cells of T-bet−/− mice and syngenic BALB/c mice were stimulated with anti-CD3/CD28 and BALB/c APCs under Th1 (IL-12 and IFN-γ), or under Th2-polarizing conditions for 6 d. Data represent the mean ± the SD of three experiments. The amount of twist1 transcripts induced in activated wt Th1 cells was set to 1. (C) CD4+ cells of STAT4−/− and syngenic BALB/c mice were stimulated with anti-CD3/CD28, and BALB/c APCs under Th1 (IL-12 and IFN-γ) or under Th2-polarizing conditions for 5 d. Data represent the mean ± SD (four mice each). (D) The binding of STAT4 to the proximal promoter of twist1 was analyzed by ChIP. 6-d-old Th1 cells were restimulated with PMA/ionomycin in the presence of 10 ng/ml IL-12 for 3 h or left unstimulated. The immunoprecipitated DNA was quantified by RT-PCR using primers specific for the proximal twist1 promoter. The precipitated DNA was normalized to the amount of input DNA. The amount of twist1 transcripts precipitated in the presence of IL-12 was set to 1. Data represent the mean ± SD of three experiments. (E) Naive DO11.10 Th cells were stimulated for 5 d with APCs and OVA327-339 under Th1-polarizing conditions. Cells were restimulated under the same conditions (Th1), or in the presence of anti–IL-12. Twist1 transcripts were quantified on d 11. The amount of twist1 mRNA on d 5 was set to 1. Data represent mean ± SD of three experiments.
When CD4+ T lymphocytes of STAT4-deficient mice were stimulated in the presence of IL-12 and IFN-γ, no induction of twist1 expression was detectable (Fig. 2 C). Phylogenetic comparison of the proximal promoter of twist1 from man and mouse revealed a conserved STAT binding site at position −117 to −137 (Fig. 3 A). STAT4 did bind to the proximal promoter of twist1 in activated Th1 cells in the presence of IL-12 as shown by chromatin immunoprecipitation (ChIP; Fig. 2 D).
Figure 3.

Signaling through NFAT and NF-κB is required for induction of twist1 expression. (A) Comparison of the genomic sequence of the murine and the human proximal twist1 promoter (−150 to −100). The murine sequence is displayed with conserved bases in capital letters. Selected putative DNA-binding motifs are indicated. ISRE, IFN-stimulated response element. (B) Twist1 mRNA in 24-d-old Th1 cells restimulated for 3 h with PMA/ionomycin and IL-2 in the presence of serial dilutions of the NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC; circles; starting concentration 50 μM), the NFAT inhibitor BTP1 (squares; starting concentration 100 nM), or the NF-κB and NFAT inhibitor cyclosporine A (CsA; triangles; starting concentration 30 nM). (C) 24-d-old Th1 cells were restimulated in the presence of IL-2 for 3 h with IL-2 alone (unstimulated) or 10 ng/ml TNF-α, PMA, or PMA/ionomycin. The binding of NFAT1 (D) and the NF-κB subunit p65 (E) to the proximal promoter of twist1 was analyzed by ChIP. 18–24-d-old Th1 and Th2 cells either in the resting state (-) or after restimulation with PMA/ionomycin and IL-2 for 1 h (+) were used. The immunoprecipitated DNA was quantified by RT-PCR using primers specific for the proximal promoter. The precipitated DNA was normalized to the amount of input DNA. The amount of twist1 transcripts precipitated in activated Th1 cells was set to 1. Data represent mean ± SD of 3 experiments.
IL-12/STAT4 is required for the initial induction of twist1 expression and apparently imprints Th1 cells for reexpression of twist1 upon reactivation by antigen only. Upon restimulation of Th1 cells, cells cultured in the absence of IL-12 showed a 12-fold increase in the expression of twist1, as compared with a 16-fold increase in the presence of IL-12 (Fig. 2 E). IL-4/STAT6 signaling could not suppress twist1 expression in residual IFN-γ–expressing cells of Th2 cultures (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1), but also did not induce twist1 expression in activated Th2 cells, as described in Fig. 1.
Because twist1 is expressed transiently upon activation of Th1 cells, we investigated whether signals from the TCR are involved in twist1 expression control. Phylogenetically conserved binding sites for the TCR signal transducers NF-κB and NFAT are located in the twist1 promoter at positions −104 to −116 and −129 to −140 (Fig. 3 A). The NF-κB inhibitor pyrrolidine dithiocarbamate (20), the NFAT-specific inhibitory 3,5-bistrifluoromethyl pyrazole derivative BTP1 (21), and the calcineurin-inhibitor cyclosporine A (22) all blocked the PMA/ionomycin-induced expression of twist1 mRNA (Fig. 3 B), showing that both NFAT and NF-κB are required for induction of twist1 expression in Th1 cells. Accordingly, stimulation with either TNF-α or PMA alone, both activating NF-κB (20) but not NFAT, did not induce twist1 expression, whereas PMA in combination with the NFAT-activating ionomycin did (Fig. 3 C).
Using ChIP, the specific binding of NFAT1 and the transactivating NF-κB subunit p65 to the twist1 promoter of repeatedly stimulated Th1 cells was evident 1 h after reactivation (Fig. 3, D–E). In Th2 cells, no activation-induced binding of NFAT1 or NF-κB to the twist1 promoter was detectable, emphasizing that NFAT1 and NF-κB are required, but not sufficient, to induce transcription of twist1 in Th cells. For the initial expression of twist1 in Th1 cells, and its imprinting for reexpression in further restimulations, the concerted action of activated STAT4, NFAT1, and NF-κB is required. Imprinting of the twist1 gene for reexpression in Th1 cells is reflected by increased acetylation of histone H3 and trimethylated histone H3 at lysine 4, in both resting and reactivated Th1 cells, as compared with Th2 cells (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1).
Th cell–specific twist1 expression in vivo
The exclusive expression of twist1 in Th1 EM cells generated in vitro is reflected by the expression pattern of twist1 in Th cells isolated ex vivo. Activated CD4+CD62Lhi naive and CD4+CD62Llo memory cells isolated from the spleen and lymph nodes of healthy DO11.10 mice, kept under specific pathogen–free conditions, expressed low levels of twist1 (Fig. 4 A). In CD4+CD62Llo memory Th cells from lymph nodes of 4–6-mo-old nephritic lupus-prone MRL/lpr mice (23) twist1 mRNA was up-regulated by approximately twofold, as compared with CD4+CD62Llo Th cells from DO11.10 spleen (Fig. 4 A). Twist1 expression was enhanced by five- to eightfold in CD3+B220+ T cells from MRL/lpr mice, which represent chronically activated T lymphocytes (24, 25).
Figure 4.

Ex vivo isolated memory Th cells express twist1. Cells were sorted by flow cytometry and restimulated for 3 h with PMA/ionomycin (A–C). (A) Cells were isolated from the spleen and lymph nodes of 8–12-wk-old DO11.10 mice (squares, each representing a pool of 15 individual mice) and the pooled inguinal and mesenteric lymph nodes of 4–6-mo-old nephritic MRL/lpr mice (circles, 1–2 mice each). Of note: 90% of the CD4+CD62Lhi cells in MRL/lpr mice represented activated (CD44+) cells (not depicted). (B) Twist1 mRNA in peripheral human Th lymphocytes. The mean expression of twist1 mRNA normalized to ubiquitin ligase H5 in total CD3+CD4+ cells was set to 1. Subpopulations were defined according to expression of the following surface markers: naive (CD4+CD45RA+CCR7+), CM (CD4+CD45RA−CCR7+), EM (CD4+CD45RA−CCR7−) with each data point representing one individual healthy donor. (C) Twist1 transcripts in CD3+CD4+ cells purified from patient material: blood (total peripheral CD3+CD4+ cells from healthy donors, see B), colon (noninflamed colon tissue), UC, and CD (endoscopic biopsies from UC and CD patients, respectively), RA, ReA, PsA, and AS (synovial fluid from rheumatoid arthritis, reactive arthritis, psoriatic arthritis, and ankylosing spondylitis patients, respectively) with each dot representing one individual patient. Mean twist1 mRNA expression is displayed from patients who were repeatedly sampled.
In analogy to murine Th lymphocytes, human Th cells can express twist1. Twist1 expression was enhanced by threefold in EM (i.e., CD45RA−CCR7−) Th cells, and eightfold in “terminally” differentiated CD27− EM Th cells (26, 27), whereas naive (CD45RA+CCR7+) and central memory (CM; i.e., CD45RA−CCR7+) Th lymphocytes expressed less twist1 than unseparated Th cells. Activated Th1 EM cells, characterized by the expression of CCR5 (28), expressed twist1, whereas Th2 EM cells characterized by expression of chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTh2) (29) did not (Fig. 4 B).
Th cells isolated from inflamed tissue of patients with chronic inflammatory diseases are imprinted for high twist1 expression. Although highly variable, twist1 transcripts were increased by up to 400-fold, as compared with peripheral Th cells, in CD3+CD4+ cells isolated from the synovial fluid of inflamed joints of patients with rheumatoid arthritis or spondyloarthropathies, and in Th cells isolated from mucosal endoscopic biopsies and surgical specimens of patients suffering from CD or UC (Fig. 4 C and Table S2, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1). Twist1 mRNA expression in repeatedly sampled patients with persistent inflammation of colon or synovia remained in the same range over up to 18 mo (Table S2). Among T cells isolated from the inflamed tissue, only CD4+ Th cells showed enhanced twist1 expression. CD3+CD4− cells, i.e., cytotoxic (Tc) lymphocytes, expressed twist1 transcript levels lower than the reference value of total peripheral Th cells (unpublished data).
Functional modulation of Th1 cells by twist1
We analyzed the impact of twist1 on the function of Th1 EM cells by ectopic overexpression of twist1 in murine DO11.10 Th cells. A global view of twist1-induced modulation of gene expression in Th1 cells is provided in Fig. 5. Of the 14,000 genes analyzed for transcription, 58 were differentially expressed by a factor of 1.5 or more when comparing activated Th1 cells that express twist1 ectopically and those that do not (Fig. 5). These genes fall into 4 groups, with respect to their presumptive function: 17 genes are involved in cell activation and apoptosis, 11 genes are involved in cell adhesion and motility, 13 genes relate to the chemokine/cytokine repertoire of Th1 cells, and 17 genes are of metabolic or undefined relevance.
Figure 5.

Genes differentially expressed upon ectopic twist1 overexpression. Splenic DO11.10 cells were activated in vitro with the cognate peptide OVA327-339 in the presence of 1 ng/ml IL-12 and 1 ng/ml IL-2. On d 2, cells were infected with control retrovirus, or twist1-encoding virus. On d 5, cells were sorted according to expression of the viral marker gene gfp. Cells were restimulated for 4 h with PMA/ionomycin. The transcriptional profiles of duplicates of cultures were compared.
Of relevance for the effector function of Th1 EM cells, twist1 attenuated expression of the effector cytokine genes il-2, ifn-γ, and tnf-α by a factor of up to 1.6 (Fig. 5 and Table S3, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1). The moderate reduction of mRNA levels of effector cytokines had a drastic effect on protein expression regardless of the direction of Th cell differentiation. Ectopic twist1 expression reduced the frequencies of cytokine-expressing reactivated Th1 or Th2, or of Th cells that had been stimulated without addition of polarizing cytokines to the culture, to 40–50% of the controls (Fig. 6 and Fig. S5).
Figure 6.

Twist1 suppresses the expression of effector cytokines. DO11.10 Th cells were stimulated for 5 d under Th1- or Th2-polarizing conditions or without addition of cytokines. On d 2, cells were infected with control virus or twist1-encoding virus. The cells were restimulated on d 6 and stained for intracellular cytokine expression. (A) Representative histograms of cytokine expression in Th cells ectopically expressing twist1 (black line) and control cells (gray filled). The cells displayed were gated for expression of CD4 and the viral marker gene gfp. (B) Frequencies of cytokine-expressing cells among infected, i.e., GFP+CD4+ T cells relative to the noninfected CD4+ cells. Cells had been stimulated without addition of cytokines and infected with control virus (open bars) or twist1-encoding virus (filled bars). Data represent the mean ± SD of four independent experiments.
The molecular basis of the regulation of cytokine gene expression by twist1 could be either direct inhibition of NF-κB, as has been shown for COS cells (4), or binding of Twist1 to E-boxes of regulatory elements of the cytokine genes, blocking activating transcription factors, as has been shown for primary macrophages (5). Here, we show that ectopic twist1 expression in activated Th cells cannot inhibit activation-induced transcription of an NF-κB–reporter construct lacking E-boxes, i.e., Twist1 does not inhibit NF-κB directly (Fig. 7 A), in contrast to the constitutively active mutant form of inhibitor of NF-κB (I-κBαM), in an experimental situation where both Twist1 and I-κBαM are expressed at similar levels (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1), and both attenuate the expression of endogenous cytokine genes to the same degree (Fig. 7 B). This result implies that twist1 regulates gene expression of Th1 cells by binding to E-boxes of specific target genes, and not the entirety of NF-κB–regulated genes. NF-κB signaling for the generation and survival of Th1 memory cells (30, 31) is not inhibited by twist1, as is also evident from the high twist1 expression of repeatedly restimulated Th1 cells (Fig. 1).
Figure 7.

Inhibition of NF-κB–mediated signaling by twist1 is promoter-specific. DO11.10 Th cells were stimulated with OVA327-339, APCs, and 1 ng/ml IL-12. On d 2, cells were infected with control virus, twist1, or I-κBαM-encoding virus. On d 3, cells were nucleoporated with a mixture of a plasmid encoding Renilla luciferase, and a firefly luciferase reporter construct, driven by a NF-κB–responsive promoter (4xκB-luc). (A) Cells were restimulated with PMA/ionomycin for 6 h, sorted according to expression of the viral marker gene gfp, and luciferase signals were quantified in duplicates (mean ± SD). (B) The very same Th1 cultures were restimulated on d 5 and stained for intracellular cytokine expression. Frequencies of cytokine-expressing cells among infected, i.e., GFP+CD4+ T cells, are displayed. Data are representative of two experiments.
Twist1 regulates Th1-mediated inflammation
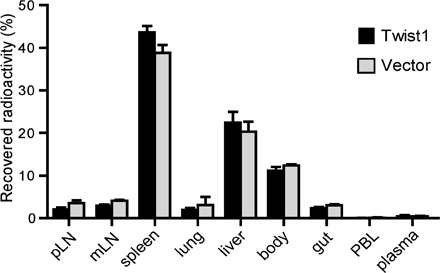
In a murine transfer model of OVA-specific DTH, the effect of twist1 expression on the inflammation induced by transferred Th1 cells was analyzed. OVA-specific 6-d-old Th1 cells ectopically overexpressing twist1 and control Th1 cells were transferred intravenously into naive BALB/c mice. After 1 d, OVA323-339 emulsified in IFA was injected into one footpad and the swelling of this footpad was monitored. Th1 cells overexpressing twist1 showed a significantly (P < 0.05) reduced induction of footpad swelling, starting from day 4 to 7, when compared with control Th1 cells (Fig. 8 A). For DTH mediated by Th1 cells the proinflammatory role of Th1-derived cytokines such as IFN-γ has been shown (32). Transferred Th cells overexpressing twist1, when reisolated from the draining lymph nodes of the host, expressed fourfold reduced levels of IFN-γ mRNA as compared with the control cells (Fig. 8 B). Homing of transferred Th1 cell populations to the inflamed tissue and the draining lymph nodes was not affected by twist1 overexpression, as shown by the transfer of radiolabeled cells (Fig. 8 C). The accumulation of ectopically twist1-expressing Th1 cells in the draining lymph nodes and the inflamed foot pads, as well as in other organs (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1), was comparable to that of transferred control Th1 cells.
Figure 8.

Ectopic twist1 overexpression controls DTH. (A) Naive DO11.10 Th cells were stimulated under Th1-polarizing conditions. On d 2, cells were infected with control virus (circles) or twist1-encoding virus (squares). On d 6, infected GFP+ cells were injected i.v. into BALB/c mice. The DTH response was induced by s.c. OVA323-339/IFA injection into the left footpad (filled symbols), and Δ footpad thickness (mean ± SD; n = 4, Mann-Whitney test, nonparametric) was determined thereafter. Injection of PBS/IFA served as control (open symbols). (B) Ex vivo IFN-γ mRNA expression in transferred GFP+ Th1 cells 24 h after DTH induction isolated from the draining popliteal lymph node (left foot). (C) To monitor the migratory capacity of the transferred cell populations, infected GFP+ Th cells were radiolabeled and injected i.v. into BALB/c mice 1 d after induction of the DTH response. 24 h later radioactivity recovered from indicated tissues was determined using a γ-counter (mean ± SD; n = 4).
Autoregulation of Th1-mediated inflammation by twist1 was also analyzed in a murine adoptive transfer model of antigen-induced arthritis (Fig. 9 A). In this model, we used the complementary genetic approach to knockdown twist1 expression in Th1 cells by RNA interference. Murine DO11.10 Th1 cells generated in vitro were infected with a retrovirus encoding a small hairpin RNA (shRNA) targeting twist1 (3) or a corresponding scrambled control shRNA. Twist1-specific shRNA reduced the level of activation-induced endogenous twist1 transcripts in Th1 cells to ∼30% of the control value (Fig. 9 B). Two-time restimulated Th1 cells expressing those shRNAs were intravenously injected into SCID mice. 1 d after cell transfer, arthritis was induced by injection of cationized OVA into the knee joint, and histological analysis was performed 3 wk after induction of arthritis, i.e., in the chronic phase of inflammation. Twist1 knockdown and control Th1 cells equally homed to and persisted in the spleen and mesenteric and draining lymph nodes (Fig. 9, C–E). However, twist1 knockdown in Th1 cells resulted in a significantly higher histological score of inflammation and tissue destruction (Fig. 9 F and Table S4, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1). In particular, infiltration of granulocytes and mononuclear cells into the inflamed tissue of the knee joint was drastically enhanced (Fig. 10 and Fig. S8), as were characteristics of chronic inflammation, i.e., prominent hyperplasia of the lining cells, pannus formation, increased vascularization, and hyperplasia of synovial fibroblasts in the sublining layer (Fig. 10 and Fig. S8). Increased numbers of Gr-1+ granulocytes at the surface of the synovium and focal accumulation of F4/80+ macrophages were observed after transfer of twist1 knockdown Th1 cells as compared with control Th1 cells (Fig. S8). Anti-TNF-α staining of joint sections revealed stronger TNF-α expression, especially in the lining cells and the sublining layer after transfer of Th1 cells expressing a twist1-targeting shRNA (Fig. 10). Presumably as an additional consequence of the higher inflammation in the mice receiving twist1 knockdown Th1 cells, more CD3+ T cells were found in the inflamed joints (Fig. 10).
Figure 9.

Twist1 knockdown increases inflammatory response in murine arthritis. (A) Experimental scheme. (B) Twist1 mRNA in 18-d-old Th1 expressing twist1-targeting shRNA or control shRNA restimulated with PMA/ionomycin. (C–E) Cell numbers of adoptively transferred GFP+ Th cells in spleen, mesenteric lymph nodes, and draining lymph nodes were analyzed on d 21 using FlowCount Beads. (F) Transfer of Th1 cells expressing a twist1-targeting shRNA leads to a significantly higher histological score in murine arthritis compared with control Th1 cells (d 21). Data are representative of two experiments.
Figure 10.

Twist1 knockdown leads to pronounced signs of chronic inflammation in murine arthritis. Representative hematoxylin/eosin staining of knee joint sections (d 21) showing hyperplasia of the lining cells and the subintimal layer with pannus formation (bar, 200 μm), more pronounced infiltration of CD3+ T cells (magnification of boxed areas in top row; bar, 100 μm) and stronger expression of TNF-α (red) especially of the enlarged lining and sublining layer (magnification of boxed areas in second row; bar, 50 μm) after transfer of Th1 cells expressing a twist1-targeting shRNA as compared with the control group. Data are representative of five mice each.
DISCUSSION
Control of inflammation critically depends on the twist genes. Genetic haploinsufficiency of both twist1 and twist2 results in fatal systemic inflammatory immunopathology in mice, which die before day 14 (4). In this study, we show that twist1 is expressed in Th1 EM cells. Its expression is induced by IL-12 via STAT4 and TCR signaling, activating NFAT and NF-κB. Expression of twist1 follows TCR stimulation transiently and increases upon repeated stimulation. Thus, imprinting for enhanced twist1 expression is a hallmark of repeatedly restimulated Th1 memory cells.
The proximal promoter of twist1 contains phylogenetically conserved binding sites for NFAT, NF-κB, and STAT proteins. Both NFAT and NF-κB have to bind to the promoter of twist1 in Th cells to induce expression, i.e., twist1 is expressed only by activated Th cells. In the initial activation of naive Th cells, NFAT and NF-κB cannot induce transcription of twist1 on their own, but require concerted binding of activated STAT4 to the promoter of twist1. Of all costimulatory signals involved in the lineage differentiation of murine Th1, Th2, or Th17 cells, only IL-12 was able to induce transcription of twist1. Neither IL-4, in Th2 polarization, nor IL-6, TGF-β, and IL-23 in Th17 polarization, could induce expression of twist1. Of the Th1-polarizing signals, IL-12/STAT4, but not IFN-γ/STAT1, is required to induce twist1 expression. T-bet is not involved. STAT4 is required only in the original stimulation, to imprint the gene for reexpression. This imprinting is evident from the increased reexpression upon stimulation with TCR signals alone, in the absence of IL-12, and it is reflected in the acetylation and trimethylation of H3 histones at the twist1 promoter region, as is shown here. The requirement of NFAT for induction of twist1 expression in Th1 cells distinguishes control of twist1 expression in Th cells from its control in fibroblasts, where TNF-α–induced activation of NF-κB is sufficient to induce expression (4). In activated macrophages, type I IFNs induce the expression of twist1 (5), and they express both twist genes, i.e., twist1 and twist2, whereas activated Th1 cells exclusively express twist1. In Th lymphocytes, twist1 expression is not induced by type I IFNs. Addition of IFN-α during the primary activation of Th cells in the absence of IL-12 did not suffice to induce twist1 expression (Fig. S9, available at http://www.jem.org/cgi/content/full/jem.20072468/DC1). These findings provide evidence that expression of the antiinflammatory twist genes is controlled by type I IFNs and TNF-α in fibroblasts and macrophages in innate immune responses (4, 5), and is controlled by antigen and IL-12 in Th cells in adaptive immune responses; i.e., twist expression is induced by those cytokines controlling the induction of inflammatory immune responses.
In this study, we have identified twist1 as a gene that is differentially expressed by Th1 cells versus Th2 cells, comparing the transcriptomes of once and repeatedly restimulated murine Th1 and Th2 cells generated in vitro from bona fide naive Th cells. Expression of twist1 increases upon repeated restimulation in vitro in Th1 cells, whereas it is not induced even in repeatedly restimulated Th2 or Th17 cells. Accordingly, in Th cells isolated ex vivo, twist1 expression is restricted to a subset of memory cells. Among murine CD4+CD62Llo memory-phenotype Th cells that were isolated from spleen or lymph nodes of naive DO11.10 mice, expression of twist1 is low. In CD3+B220+ T cells isolated from nephritic MRL/lpr mice, which represent chronically activated T cells (24, 25), twist1 expression is up-regulated five- to eightfold compared with DO11.10 memory-phenotype Th cells. This is moderate when compared with the ∼20-fold up-regulation upon repeated restimulation of Th1 cells observed in vitro, and may reflect the heterogeneous composition of the cell populations analyzed. As expected from the phylogenetic conservation of the twist1 promoter, twist1 is also expressed by activated human Th cells. It is low in peripheral human naive Th cells and in CCR7+ CM Th cells. Twist1 expression is enhanced in CCR7− EM Th cells, in particular in the “terminally differentiated” CCR7−CD27− EM cells (26, 27). Among CCR7− EM cells, twist1 is expressed less by CRTh2+ Th cells, which have been shown to be mostly Th2 cells (29). CCR7−CCR5+ EM cells, which contain Th1 EM and Th17 EM cells (33) have up-regulated activation-induced expression of twist1, as we show here. Because at least Th17 cells generated in vitro do not express twist1, the expression in CCR7−CCR5+ Th EM cells is probably caused by Th1 EM cells. Thus, the phenotype of human peripheral twist1-expressing Th cells is that of repeatedly restimulated Th1 EM cells. Remarkably, expression of twist1 is highly up-regulated in CD3+CD4+ T cells isolated from inflamed tissues of patients with chronic inflammation of joints or gut. Repeated biopsies from individual patients show persistent twist1 mRNA levels over time, i.e., the persistence of chronically reactivated Th1 cells, despite state-of-the-art therapeutic treatment. Their imprinting for increased twist1 expression, maintained over time in individual patients, indicates a history of repeated restimulation and an endogenous regulation of proinflammatory effector functions under the control of NFAT and NF-κB.
Twist1 itself is a transcriptional repressor binding to E-boxes in the regulatory regions of target genes (34). For COS cells, it has been shown that ectopically expressed twist1 can interact directly with the p65 subunit of NF-κB and inhibit its function (4). In contrast, for primary murine macrophages, twist1 has been shown to block transcription of target genes by binding to E-boxes within the promoter (5). In this study, we show that in Th1 cells, twist1 cannot block NF-κB–driven transcription of a reporter gene construct that lacks E-boxes in the promoter, suggesting that twist1 acts through binding to E-boxes, as in macrophages. In accordance, ectopically expressed twist1 only regulates the expression of a restricted set of 58 genes in 1-wk-old Th1 cells by a factor of at least 1.5. Expression of 29 genes is up-regulated, and that of another 29 genes down-regulated. Apart from several genes of unknown and metabolic function, these genes are involved in survival, effector function, and motility of the cells. With respect to cytokine expression, twist1 reduces activation-induced expression of TNF-α, IL-2, and IFN-γ by Th1 memory cells by >50% at the protein level. Apart from cytokine expression, twist1 also decreases expression of proinflammatory chemokines and chemokine receptors, and attenuates cytokine receptor signaling by up-regulating SOCS-1 and -2, the IL-1 decoy receptor, and by down-regulating Jak2, the kinase involved in IL-12 signaling. Twist1 in Th1 cells thus acts as an endogenous regulator limiting the proinflammatory potential of Th1 cells in the continuous presence of antigen, i.e., repeated activation of NFAT and NF-κB.
The key role of twist1 expression by Th1 cells for the self-limitation of Th1-induced inflammation is evident from the present analysis of murine models of inflammation. Ectopic overexpression of twist1 in 6-d-old Th1 cells, which still had a low endogenous expression level, drastically reduced their pathogenic contribution to DTH. Conversely, shRNA-mediated knockdown of twist1 in Th1 cells significantly enhanced their potential to induce chronic inflammation in a murine model of antigen-induced arthritis. In this model, OVA-specific Th1 cells, when adoptively transferred into SCID mice and challenged by intraarticular OVA, induce a chronic inflammation of the joint. Adoptive transfer of Th1 cells versus Th1 cells with a shRNA-mediated knockdown of twist1 expression demonstrates the relevance of twist1 expression by Th1 cells for the control of immunopathology. When analyzed on day 21 after transfer, mice with a knockdown of twist1 expression in Th1 cells showed increased hyperplasia of the lining cells and synovial fibroblasts in the sublining layer, pannus formation, and vascularization. Numbers of Gr-1+ granulocytes and F4/80+ macrophages, as well as TNF-α expression of lining and sublining cells, were increased in the Th1 twist1 knockdown situation.
Why is expression of twist1 up-regulated gradually? One explanation might be that a regulatory gene is not expressed in initial Th1 activations, to allow full initial reactivity of the Th1 cells. Accordingly, ectopic twist1 expression in Th1 cells right from the beginning impairs their function in acute inflammation of DTH. In repeated restimulations, reflecting a lack of clearance of the antigen and increased risk of immunopathology, regulation of Th1 may help to limit immunopathology, as we show in the chronic inflammation of the antigen-induced arthritis model by knockdown of twist1, but twist1-expressing Th1 cells are still capable of driving inflammation.
Apart from its role in limiting immunopathology in chronic inflammation, twist1 and the genes controlled by it could be regarded as biomarkers for pathogenic EM cells. Twist1 is not imprinted for enhanced expression in naive and CM CCR7+ Th cells. Even CCR7−CCR5+ Th1 EM cells will only express a low level of twist1 upon restimulation compared with Th cells from chronically inflamed tissue. Initial clinical studies aiming at a complete depletion of CD4+ T lymphocytes in patients with chronic inflammatory diseases suggested a clinical benefit, but the studies had to be terminated because of the severe side effects of systemic depletion of Th lymphocytes (35–37). Targeting of CD4- and CD3-expressing cells with nondepleting antibodies and neutralization of Th-related effector cytokines have demonstrated clinical efficacy (12, 13, 15, 38–40), but all of them impair protective as well as pathogenic T cell memory. Targeting of twist1-expressing Th cells, by means of either addressing twist1 or a gene regulated by it, seems more promising, because it is more specific for repeatedly restimulated Th1 memory cells involved in chronic inflammation. It remains to be shown whether these cells in chronic inflammatory diseases are the critical Th cells capable of driving inflammation, as we show in this study for the experimental model of antigen-induced arthritis.
MATERIALS AND METHODS
Mice and reagents.
MRLlpr/lpr, BALB/c, C57BL/6, SCID, STAT4-deficient mice (Stat4tm1Gr), and OVA-TCRtg/tg DO11.10 mice were purchased from The Jackson Laboratory or were bred under specific pathogen–free conditions in our animal facility. T-bet–deficient mice were a gift from J. Penninger (Institute for Molecular Biotechnology of the Austria Academy of Sciences, Vienna, Austria). All animal experiments were performed in accordance with institutional, state, and federal guidelines (Landesamt Für Gesundheit und Soziales, Berlin, Germany). Reagents were purchased from Sigma-Aldrich unless otherwise stated. BTP1 was synthesized by M. Paetzel (Humboldt University of Berlin, Berlin, Germany). Cyclosporin A was purchased from Calbiochem.
Patients.
Endoscopic mucosal and surgical mucosal specimens were obtained from UC (n = 9) and CD (n = 7) patients. UC and CD were diagnosed according to established clinical, endoscopic, radiological, and pathological criteria. All UC and CD patients displayed moderately to severely active disease according to the Truelove and Witts Severity Index (41) and the Harvey Bradshaw Severity Index (42), respectively. Control samples were obtained from patients (n = 4) undergoing colonectomy because of colon cancer. Mucosal control specimens used in the study were from the macroscopically noninvolved tissue distant from any detectable lesion. Synovial fluid was obtained from patients suffering from rheumatic diseases who had active synovitis with effusion. Patients with rheumatoid arthritis (RA; n = 4) fulfilled the American College of Rheumatology 1987 classification criteria for RA, patients with ankylosing spondylitis (AS; n = 5) fulfilled the modified New York criteria (1984), and patients with psoriatic arthritis (n = 3) or reactive arthritis (n = 3) fulfilled the European Spondyloarthropathy Study Group (ESSG) criteria for SpA. Clinical characteristics of the patients are listed in Table S2. All experiments were approved by the local ethics committee (Charite Ethikkommision), and all patients gave informed consent.
Isolation of human lymphocytes.
PBMCs from buffy coats from healthy donors, and synovial fluid mononuclear cells were isolated by density gradient centrifugation (Lymphocyte separation medium; PAA). Pooled intraepithelial leukocytes and lamina propria leukocytes were obtained from mucosal specimens by treatment with collagenase type IV, followed by passage through a sieve. Mononuclear cells were collected from the 40–70% interphase of a discontinuous percoll (Pharmacia) gradient.
Flow cytometry.
The following antibodies directed against murine antigens were either purified from hybridoma supernatants and conjugated in-house or purchased as indicated: anti-CD3 (145-2C11), anti-CD4 (GK1.5), anti-CD8 (53–6.7), anti-CD11c (N418), anti-CD44 (IM7), anti-CD62L (MEL14), anti-DO11.10 OVA-TCR (KJ1.26), anti-B220 (RA3.6B2), anti-IL2 (JES6-5H4; Caltag Laboratories), anti-IL-4 (11B11; BD Biosciences), anti-IFN-γ (AN18.17.24), and anti-TNF-α (MP6-XT22; Caltag). Antibodies recognizing human antigens were obtained from BD Biosciences unless stated otherwise: anti-CD3 (OKT3; in-house conjugate), anti-CD4 (TT1; in-house conjugate), anti-CD27 (L128), anti-CD45RA (HI100), anti-CRTh2 (BM16), and anti-CCR5 (2D7/CCR5). Cells were counted by using FlowCount Beads (Beckman Coulter). Cytometric analysis was performed with FACSCalibur using CellQuest (BD Biosciences) and FCS Express (De Novo) software. Cells were separated by fluorescence-activated cell sorting (FACSAria and FACSDiva; BD Biosciences).
Cell culture.
Naive CD4+CD62L+ lymphocytes from 6–8-wk-old DO11.10 mice were isolated and polarized under Th1 or Th2 conditions, as previously described (15). Irradiated (30 Gy) BALB/c splenocytes were used as APCs at a ratio of 5:1 and the cognate peptide OVA323-339 (provided by R. Volkmer-Engert, Humboldt University of Berlin, Berlin, Germany) was added at 0.5 μM. Alternatively, plates were coated with 3 μg/ml anti-CD3 (145-2C11) in PBS, and CD4+ cells were plated at a density of 2 × 106 cells/ml in medium plus 1 μg/ml soluble anti-CD28 (37.51). For Th17 differentiation, cells were stimulated in the presence of 1 ng/ml TGF-β (R&D Systems), 20 ng/ml IL-6 (R&D Systems), and 20 ng/ml IL-23 (R&D Systems), as well as 10 μg/ml anti-IL-4 and 10 μg/ml anti–IFN-γ. Irradiated IL-12 p35−/− splenocytes were used as APCs. Every 6 d, viable Th cells were harvested and restimulated under the original conditions, except that 10 ng/ml murine IL-2 (R&D Systems) was added to the Th1 and Th2 cultures.
Mitogenic restimulation and intracellular cytokine staining.
Cells were restimulated with 10 ng/ml PMA, and 1 μg/ml ionomycin or with plate-bound anti-CD3 (10 μg/ml), and soluble anti-CD28 (1 μg/ml). For restimulation of murine T cells, 10 ng/ml IL-2 was added. For intracellular staining of cytokines, T cells were stimulated for 2 h with PMA/ionomycin and an additional 3 h with 5 μg/ml of brefeldin A. Cells were fixed with 2% formaldehyde in PBS for 15 min at room temperature and permeabilized with 0.5% wt/vol saponin.
ChIP.
ChIP was performed as previously described (15) using anti-NFAT1 (AB1-209; ImmunoGlobe Antikoerpertechnik), anti-p65 (C-20; Santa Cruz Biotechnology, Inc.), anti-STAT4 (C-20; Santa Cruz Biotechnology, Inc.), anti–acetyl-histone 3 (#06–599; Millipore) or anti–trimethyl-K4-histone 3 (#07–473; Millipore), followed by incubation with Protein A microbeads. The relative amount of precipitated DNA was calculated with EΔCp (input - immunoprecipitate). The following primers were used to amplify the proximal twist1 promoter: (−150 forward) 5′-GGGCTGGAAAGAGGAAACTT-3′; (+4 reverse) 5′-CGCGAGGTGTCTGAGAGTT-3′.
Th1-mediated DTH model.
The OVA-specific DTH model was performed as described elsewhere (43). In brief, 5 × 105 Th1 cells were injected i.v. into BALB/c mice, and 24 h later, the DTH response was induced by s.c. injection of 250 ng OVA323-339/IFA into the left footpad. PBS/IFA, injected into the right footpad, served as a control. The footpad thickness measured before the injection of the antigen was subtracted from the footpad thickness measured during the DTH response. The homing of adoptively transferred Th1 cells was performed as previously described (44). In brief, Th cells were labeled with 51Chromium (Amersham Buchler) at 37°C (2 × 107 cells/ml; 20 μCi/ml) in fresh medium. Removal of dead cells was done by gradient centrifugation (17.1% isotonic Nycodenz). Labeled cells were co-adoptively transferred into recipient animals at 1 d after the DTH induction. 24 h later, indicated tissues were removed and differential measurement of recovered radioactivity was done on a γ counter (Wallac Counter).
Antigen-induced arthritis.
2 × 106 12-d-old GFP+ Th1 cells were transferred i.v. into naive SCID mice. 1 d later, arthritis was induced by intraarticular injection of 60 μg cationized OVA into one knee joint. The contralateral knee joint was left untreated. 21 d later, mice were killed and knee joints were fixed in 10% formaldehyde, decalcified in saturated EDTA solution, and embedded in paraffin. Knee joint sections were stained with hematoxylin/eosin and scored for exudates, granulocyte infiltration, hyperplasia, fibroblast proliferation/mononuclear cell infiltration, periarticular mononuclear cell infiltration (each scoring 0–3), bone/cartilage destruction (scoring 0–4), and an additional score of 1 for visible fibrin deposition and periarticular granulocyte infiltration, resulting in a maximum score of 21.
Immunohistochemistry.
Formalin-fixed paraffin-embedded tissue was subjected to a heat-induced epitope retrieval step and stained with anti-CD3 (#N1580; Dako), TNF-α (PeproTech), anti-Gr-1 (RB6-8C5; eBioscience), or F4/80 (eBioscience). For detection biotinylated donkey anti–rat or donkey anti–goat (Dianova) secondary antibodies were used followed by the streptavidin AP kit (K5005; Dako). Or sections were incubated with goat anti-TNF-α antibody followed by Alexa Fluor 555-conjugated anti–goat antibody (Invitrogen) and incubated with rabbit polyclonal anti-CD3 followed by Alexa Fluor 488–conjugated anti–rabbit antibody (Invitrogen). Nuclei were counterstained with DAPI (Roche), and slides were mounted in Fluoromount-G (SouthernBiotech). Images were acquired using a fluorescence microscope (AxioImager Z1) equipped with a charge-coupled device camera (AxioCam MRm) and processed with Axiovision software (Carl Zeiss, Inc.).
Immunoblot.
Immunoblot was performed with a monoclonal Twist-specific antibody (αTwiMab-1) (45) conjugated in-house to digoxygenin (Roche Diagnostics), anti–tubulin-α (DM1A; Calbiochem), anti-T-bet (4B10; Santa Cruz Biotechnology, Inc.), or anti-IκBα (Cell Signaling Technology), followed by incubation with horseradish peroxidase–coupled anti-digoxigenin FAB-fragments (Roche) or anti–mouse or anti–rabbit (Santa Cruz Biotechnology, Inc.) secondary antibodies. Individual bands were visualized with enhanced chemiluminescence (GE Healthcare) and the Intelligent Dark Box System LAS-3000 (Fujifilm).
Retroviral expression vectors and retroviral infection.
The twist1-targeting shRNA vector was generated by amplification of the EF1α-promotor and GFP from pLVTHM, provided by D. Trono (Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) followed ligation into pQCXIX (Clontech Laboratories, Inc.) with XbaI and EcoRV. The twist1 target sequence corresponds to Twist-siRNA3 (5′-AAGCTGAGCAAGATTCAGACC-3′), as published by Yang et al. (3). A corresponding scrambled sequence was used as control. The DNA oligonucleotides were subcloned using HpaI and XhoI into pLL3.7, which was provided by L. Van Parijs (Massachusetts Institute of Technology, Cambridge, MA). The fragment containing the murine U6-promotor and the shRNA-encoding sequence was amplified by PCR, introducing an additional 5′-XhoI site and ligated into the SalI site of pQXIX-gfp located in the inactivated 3′-LTR. For retroviral overexpression the vector GFP-RV (46) was used, provided by K.M. Murphy (Howard Hughes Medical Institute, St. Louis, Missouri). Murine t-bet (cDNA generated from Th1-cells), murine twist1 (IMAGE cDNA clone; AccessionID: BC033434-NCBI), and constitutively active IκBαM (pCMV-IκBαM; Clontech Laboratories, Inc.) were amplified, introducing BglII- and XhoI-compatible restriction sites and ligated into the vector upstream of the internal ribosome entry site-gfp cassette. Sequences of primers and oligonucleotides for shRNA expression are listed in Table S5. Retroviral stocks were obtained by calcium phosphate cotransfection of HEK293 cells with the retrovirus packaging plasmids pECO and pCGP. The medium was replaced after 4 h, and viral supernatants were collected 24–48 h later. Th cells were infected 40 h after activation by 60 min centrifugation at 700 g at 30°C with viral supernatant and 8 μg/ml polybrene, followed by replacement of the viral supernatant with the former culture supernatant.
Luciferase reporter assay.
NF-κB activity was monitored using the pNF-κB-Luc vector (Clontech Laboratories, Inc.). Transfection efficiency was controlled by cotransfecting pRL-TK (Promega). Murine T cells were electroporated with the reporter constructs using Mouse T cell kit (AMAXA) and Nucleofector I (AMAXA). Luciferase activity was quantified with Dual Luciferase Assay kit (Promega) and a luminometer (Moonlight 3096; BD Biosciences).
Microarray experiments.
Total RNA was extracted using Trizol reagent (Invitrogen), 10 μg was reverse-transcribed, followed by cDNA extraction with a PhaseLock gel (Eppendorf), and precipitated with ethanol and ammonium acetate. Biotinylated cRNA was transcribed with the MEGAscript high-yield transcription kit (Ambion), fragmented, and the hybridization cocktail was prepared according to Affymetrix protocols (15 μg fragmented biotin-labeled cRNA spiked with Eukaryotic Hybridization control). The Murine Genome U74A version 2, and 430A version 2 GeneChip arrays (Affymetrix) were hybridized at 45°C for 16 h, stained with streptavidin-phycoerythrin using the Affymetrix GeneChip Fluidics Workstation 400, and scanned on a Hewlett-Packard Gene Array Scanner (MGU74Av2 arrays) or on an Affymetrix GeneChip Scanner 3000 (MG430Av2 arrays). Data were analyzed using the Microarray Suite 5.0 software (Affymetrix). Microarrays were globally normalized and scaled to a trimmed mean expression value of 200. All arrays were compared with each other, and a relational database was generated using Microsoft Access software, including the following parameters: expression heights, call for presence of transcripts, P value for presence or absence of transcripts, log2 value of fold change and 95% confidence intervals, call for the significance of differentially expression, and the P value for that call. For each transcript the significance of differential expression between the groups of arrays was calculated using strict Bonferroni-corrected Welch t tests. Significantly differentially expressed genes were filtered according to the following criteria: mean fold change ≥2 or ≥1.5; difference of means ≥200; P value ≤0.05; and immunoglobulin genes were excluded. Data are deposited at http://www.ncbi.nlm.nih.gov/geo/ (AccessionID SuperSeries: GSE11556).
Real-time PCR.
Real-time PCR was performed as previously described (15). For normalization of murine and human cDNA the transcripts for the housekeeping genes hypoxanthine guanine phosphoribosyl transferase and ubiquitin ligase H5 (UbcH5) were quantified, respectively. Relative expression was calculated as follows: EtΔCp target gene (reference - sample)/EhΔCp housekeeping gene (reference - sample), where Cp represents the crossing point and E represents the reaction efficiency, determined by serial dilution of DNA. Primer sequences are listed in Table S5 (available at http://www.jem.org/cgi/content/full/jem.20072468/DC1).
In silico genomic DNA analysis.
The genomic sequences for the twist1 locus of Mus musculus and Homo sapiens were obtained from UCSC Genome Bioinformatics (http://genome.ucsc.edu) and submitted to Matinspector analysis at http://www.genomatix.de/matinspector.html.
Statistics.
Statistical significance in animal models (DTH and antigen-induced arthritis) was calculated using the two-tailed Mann–Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
Online supplemental material.
Fig. S1 shows representative cytokine profiles of ex vivo–polarized Th1, Th2, and Th17 cells. Fig. S2 displays the result of ectopic T-bet expression on twist1 mRNA induction, as well as an immunoblot to confirm T-bet overexpression. Fig. S3 shows cytokine data and mRNA profiles of cells cultured in the simultaneous presence of IL-12 and IL-4 compared with Th1 cells. The ChIP for acetylated and trimethylated histone 3 binding to the twist1 promoter are shown in Fig. S4. The dot plots comparing cytokine expression in twist1 overexpressing and control Th cells are shown in Fig. S5. Immunoblots confirming comparable protein expression levels of Twist1 and IκB-αM upon retroviral overexpression are shown in Fig. S6. Fig. S7 displays in vivo migration capacity of twist1 overexpressing and control Th1 cells to remaining organs despite footpad and draining lymph nodes in the DTH response. Fig. S8 shows additional immunohistochemical stainings of the inflammatory response in murine arthritis. In Fig. S9, the effect of type I IFNs on twist1 induction is shown. Genes differentially expressed in once- versus four-time–stimulated Th1 cells are shown in Table S1. Clinical characteristics of patients in the study are listed in Table S2. Genes differentially expressed upon ectopic twist1 overexpression are listed in Table S3. Table S4 shows detailed results of the histological scoring in murine arthritis. Table S5 shows primer sequences. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20072468/DC1.
Supplementary Material
Acknowledgments
We thank Simone Spieckermann for excellent technical assistance in immunohistochemistry experiments. We thank R. Brothwick and C. Raulfs for critical reading; S. Pade for collection of clinical samples; D. Trono for provision of the pLVTHM vector; L. Van Parijs for pLL3.7; and K.M. Murphy for GFP-RV.
This work was supported by the German Ministry of Education and Research (BMBF grant 1KW9913 and 0313068), within the National Genome Research Network (NGFN 1-2, BMBF grant 01GS0110, 01GS0160, 01GS0413), by the German Science Foundation (SFB 421, SFB 633) and by the European Community Grant MUGEN LSHG-CT-2005-005203. U. Niesner was a recipient of a doctoral fellowship from Boehringer Ingelheim Fonds.
The authors have no conflicting financial interests.
Abbreviations used: CD, Crohn's disease; ChIP, chromatin immunoprecipitation; CM, central memory; DTH, delayed-type hypersensitivity; EM, effector memory; shRNA, small hairpin RNA; UC, ulcerative colitis.
U. Niesner and I. Albrecht contributed equally to this paper.
References
- 1.Thisse, B., M. el Messal, and F. Perrin-Schmitt. 1987. The twist gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 15:3439–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen, Z.F., and R.R. Behringer. 1995. Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 9:686–699. [DOI] [PubMed] [Google Scholar]
- 3.Yang, J., S.A. Mani, J.L. Donaher, S. Ramaswamy, R.A. Itzykson, C. Come, P. Savagner, I. Gitelman, A. Richardson, and R.A. Weinberg. 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 117:927–939. [DOI] [PubMed] [Google Scholar]
- 4.Sosic, D., J.A. Richardson, K. Yu, D.M. Ornitz, and E.N. Olson. 2003. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 112:169–180. [DOI] [PubMed] [Google Scholar]
- 5.Sharif, M.N., D. Sosic, C.V. Rothlin, E. Kelly, G. Lemke, E.N. Olson, and L.B. Ivashkiv. 2006. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med. 203:1891–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katz, J.D., C. Benoist, and D. Mathis. 1995. T helper cell subsets in insulin-dependent diabetes. Science. 268:1185–1188. [DOI] [PubMed] [Google Scholar]
- 7.Iqbal, N., J.R. Oliver, F.H. Wagner, A.S. Lazenby, C.O. Elson, and C.T. Weaver. 2002. T helper 1 and T helper 2 cells are pathogenic in an antigen-specific model of colitis. J. Exp. Med. 195:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maffia, P., J.M. Brewer, J.A. Gracie, A. Ianaro, B.P. Leung, P.J. Mitchell, K.M. Smith, I.B. McInnes, and P. Garside. 2004. Inducing experimental arthritis and breaking self-tolerance to joint-specific antigens with trackable, ovalbumin-specific T cells. J. Immunol. 173:151–156. [DOI] [PubMed] [Google Scholar]
- 9.Rotondi, M., E. Lazzeri, P. Romagnani, and M. Serio. 2003. Role for interferon-gamma inducible chemokines in endocrine autoimmunity: an expanding field. J. Endocrinol. Invest. 26:177–180. [DOI] [PubMed] [Google Scholar]
- 10.Morita, Y., M. Yamamura, M. Kawashima, S. Harada, K. Tsuji, K. Shibuya, K. Maruyama, and H. Makino. 1998. Flow cytometric single-cell analysis of cytokine production by CD4+ T cells in synovial tissue and peripheral blood from patients with rheumatoid arthritis. Arthritis Rheum. 41:1669–1676. [DOI] [PubMed] [Google Scholar]
- 11.Fuss, I.J., M. Neurath, M. Boirivant, J.S. Klein, C. de la Motte, S.A. Strong, C. Fiocchi, and W. Strober. 1996. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 157:1261–1270. [PubMed] [Google Scholar]
- 12.Skurkovich, B., and S. Skurkovich. 2006. Inhibition of IFN-gamma as a method of treatment of various autoimmune diseases, including skin diseases. Ernst Schering Res. Found. Workshop. 56:1–27. [DOI] [PubMed] [Google Scholar]
- 13.Hommes, D.W., T.L. Mikhajlova, S. Stoinov, D. Stimac, B. Vucelic, J. Lonovics, M. Zakuciova, G. D'Haens, G. Van Assche, S. Ba, et al. 2006. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn's disease. Gut. 55:1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Veen, R.C., T.A. Dietlin, and F.M. Hofman. 2003. Tissue expression of inducible nitric oxide synthase requires IFN-gamma production by infiltrating splenic T cells: more evidence for immunosuppression by nitric oxide. J. Neuroimmunol. 145:86–90. [DOI] [PubMed] [Google Scholar]
- 15.Chang, H.D., C. Helbig, L. Tykocinski, S. Kreher, U. Niesner, and A. Radbruch. 2007. Expression of IL-10 in Th memory lymphocytes is conditional on IL-12 or IL-4, unless the IL-10 gene is imprinted by GATA-3. Eur. J. Immunol. 37:801–817. [DOI] [PubMed] [Google Scholar]
- 16.Veldhoen, M., R.J. Hocking, C.J. Atkins, R.M. Locksley, and B. Stockinger. 2006. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 24:179–189. [DOI] [PubMed] [Google Scholar]
- 17.Harrington, L.E., R.D. Hatton, P.R. Mangan, H. Turner, T.L. Murphy, K.M. Murphy, and C.T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 18.Sallusto, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 401:708–712. [DOI] [PubMed] [Google Scholar]
- 19.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 20.Sun, S.C., P.A. Ganchi, D.W. Ballard, and W.C. Greene. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 259:1912–1915. [DOI] [PubMed] [Google Scholar]
- 21.Trevillyan, J.M., X.G. Chiou, Y.W. Chen, S.J. Ballaron, M.P. Sheets, M.L. Smith, P.E. Wiedeman, U. Warrior, J. Wilkins, E.J. Gubbins, et al. 2001. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J. Biol. Chem. 276:48118–48126. [DOI] [PubMed] [Google Scholar]
- 22.Marienfeld, R., M. Neumann, S. Chuvpilo, C. Escher, B. Kneitz, A. Avots, A. Schimpl, and E. Serfling. 1997. Cyclosporin A interferes with the inducible degradation of NF-kappa B inhibitors, but not with the processing of p105/NF-kappa B1 in T cells. Eur. J. Immunol. 27:1601–1609. [DOI] [PubMed] [Google Scholar]
- 23.Singer, G.G., A.C. Carrera, A. Marshak-Rothstein, C. Martinez, and A.K. Abbas. 1994. Apoptosis, Fas and systemic autoimmunity: the MRL-lpr/lpr model. Curr. Opin. Immunol. 6:913–920. [DOI] [PubMed] [Google Scholar]
- 24.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 25.Mixter, P.F., J.Q. Russell, G.J. Morrissette, C. Charland, D. Aleman-Hoey, and R.C. Budd. 1999. A model for the origin of TCR-alphabeta+ CD4-CD8- B220+ cells based on high affinity TCR signals. J. Immunol. 162:5747–5756. [PubMed] [Google Scholar]
- 26.De Jong, R., M. Brouwer, B. Hooibrink, T. Van der Pouw-Kraan, F. Miedema, and R.A. Van Lier. 1992. The CD27- subset of peripheral blood memory CD4+ lymphocytes contains functionally differentiated T lymphocytes that develop by persistent antigenic stimulation in vivo. Eur. J. Immunol. 22:993–999. [DOI] [PubMed] [Google Scholar]
- 27.Fritsch, R.D., X. Shen, G.P. Sims, K.S. Hathcock, R.J. Hodes, and P.E. Lipsky. 2005. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J. Immunol. 175:6489–6497. [DOI] [PubMed] [Google Scholar]
- 28.Bonecchi, R., G. Bianchi, P.P. Bordignon, D. D'Ambrosio, R. Lang, A. Borsatti, S. Sozzani, P. Allavena, P.A. Gray, A. Mantovani, and F. Sinigaglia. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata, K., K. Tanaka, K. Ogawa, K. Kemmotsu, T. Imai, O. Yoshie, H. Abe, K. Tada, M. Nakamura, K. Sugamura, and S. Takano. 1999. Selective expression of a novel surface molecule by human Th2 cells in vivo. J. Immunol. 162:1278–1286. [PubMed] [Google Scholar]
- 30.Wan, Y.Y., and J. DeGregori. 2003. The survival of antigen-stimulated T cells requires NFkappaB-mediated inhibition of p73 expression. Immunity. 18:331–342. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt-Supprian, M., G. Courtois, J. Tian, A.J. Coyle, A. Israel, K. Rajewsky, and M. Pasparakis. 2003. Mature T cells depend on signaling through the IKK complex. Immunity. 19:377–389. [DOI] [PubMed] [Google Scholar]
- 32.Fong, T.A., and T.R. Mosmann. 1989. The role of IFN-gamma in delayed-type hypersensitivity mediated by Th1 clones. J. Immunol. 143:2887–2893. [PubMed] [Google Scholar]
- 33.Annunziato, F., L. Cosmi, V. Santarlasci, L. Maggi, F. Liotta, B. Mazzinghi, E. Parente, L. Filì, S. Ferri, F. Frosali, et al. 2007. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204:1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murre, C., P.S. McCaw, and D. Baltimore. 1989. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 56:777–783. [DOI] [PubMed] [Google Scholar]
- 35.Choy, E.H., C. Pitzalis, A. Cauli, J.A. Bijl, A. Schantz, J. Woody, G.H. Kingsley, and G.S. Panayi. 1996. Percentage of anti-CD4 monoclonal antibody-coated lymphocytes in the rheumatoid joint is associated with clinical improvement. Implications for the development of immunotherapeutic dosing regimens. Arthritis Rheum. 39:52–56. [DOI] [PubMed] [Google Scholar]
- 36.Emmrich, F., G. Horneff, W. Becker, W. Luke, A. Potocnik, U. Kanzy, J.R. Kalden, and G. Burmester. 1991. An anti-CD4 antibody for treatment of chronic inflammatory arthritis. Agents Actions Suppl. 32:165–170. [DOI] [PubMed] [Google Scholar]
- 37.Horneff, G., G.R. Burmester, F. Emmrich, and J.R. Kalden. 1991. Treatment of rheumatoid arthritis with an anti-CD4 monoclonal antibody. Arthritis Rheum. 34:129–140. [DOI] [PubMed] [Google Scholar]
- 38.Nepom, G.T. 2002. Therapy of autoimmune diseases: clinical trials and new biologics. Curr. Opin. Immunol. 14:812–815. [DOI] [PubMed] [Google Scholar]
- 39.Panaccione, R., J.G. Ferraz, and P. Beck. 2005. Advances in medical therapy of inflammatory bowel disease. Curr. Opin. Pharmacol. 5:566–572. [DOI] [PubMed] [Google Scholar]
- 40.Baumgart, D.C., and A.U. Dignass. 2004. Current biological therapies for inflammatory bowel disease. Curr. Pharm. Des. 10:4127–4147. [DOI] [PubMed] [Google Scholar]
- 41.Truelove, S.C., and L.J. Witts. 1955. Cortisone in ulcerative colitis; final report on a therapeutic trial. BMJ. 2:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harvey, R.F., and J.M. Bradshaw. 1980. A simple index of Crohn's-disease activity. Lancet. 1:514. [DOI] [PubMed] [Google Scholar]
- 43.Feuerer, M., K. Eulenburg, C. Loddenkemper, A. Hamann, and J. Huehn. 2006. Self-limitation of Th1-mediated inflammation by IFN-gamma. J. Immunol. 176:2857–2863. [DOI] [PubMed] [Google Scholar]
- 44.Siegmund, K., M. Feuerer, C. Siewert, S. Ghani, U. Haubold, A. Dankof, V. Krenn, M.P. Schon, A. Scheffold, J.B. Lowe, A. Hamann, U. Syrbe, and J. Huehn. 2005. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood. 106:3097–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gitelman, I. 1997. Twist protein in mouse embryogenesis. Dev. Biol. 189:205–214. [DOI] [PubMed] [Google Scholar]
- 46.Afkarian, M., J.R. Sedy, J. Yang, N.G. Jacobson, N. Cereb, S.Y. Yang, T.L. Murphy, and K.M. Murphy. 2002. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat. Immunol. 3:549–557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}