

Abstract
Huntington's disease (HD) is an inherited neurodegenerative disorder characterized by both neurological and systemic abnormalities. We examined the peripheral immune system and found widespread evidence of innate immune activation detectable in plasma throughout the course of HD. Interleukin 6 levels were increased in HD gene carriers with a mean of 16 years before the predicted onset of clinical symptoms. To our knowledge, this is the earliest plasma abnormality identified in HD. Monocytes from HD subjects expressed mutant huntingtin and were pathologically hyperactive in response to stimulation, suggesting that the mutant protein triggers a cell-autonomous immune activation. A similar pattern was seen in macrophages and microglia from HD mouse models, and the cerebrospinal fluid and striatum of HD patients exhibited abnormal immune activation, suggesting that immune dysfunction plays a role in brain pathology. Collectively, our data suggest parallel central nervous system and peripheral pathogenic pathways of immune activation in HD.
Huntington's disease (HD) is an incurable, autosomal dominantly inherited neurodegenerative condition caused by a CAG repeat expansion in the gene encoding huntingtin. The mutant protein causes neuronal dysfunction and death resulting in the cardinal disease features of movement disorder, cognitive decline, and psychiatric symptoms (1). Huntingtin is expressed ubiquitously (2), and HD includes several abnormalities outside the central nervous system (CNS), including up-regulation of immune proteins (3–6). The interactions between CNS pathology and changes detectable in peripheral tissues in HD are poorly understood but may be of importance in measuring or slowing disease progression. We previously demonstrated evidence of immune activation in peripheral plasma in manifest HD using proteomic profiling (6), but no significant differences between controls and premanifest mutation carriers have previously been shown.
The nature of the immune activation in HD remains incompletely explored. It is not known whether the innate or adaptive arm of the immune system, or both, is activated in HD, and the alterations in each immunomodulatory cytokine at each disease stage are unknown. The cause of the immune activation peripherally is also unknown. IL-6, which triggers the acute phase response, is produced primarily by monocytes and lymphocytes, but this could be due to dysfunction of these cells caused by expression of mutant huntingtin (i.e., a cell-autonomous effect) or in response to inflammation-triggering events outside these cells, such as huntingtin-induced tissue damage or the mutant protein itself being interpreted as an antigen (i.e., non–cell-autonomous pathways). Of interest in this respect is the finding that the IκB kinase/NF-κB signaling pathway that triggers IL-6 release is up-regulated by mutant huntingtin, and this may contribute to neurotoxicity (7).
Critically, the relationship between peripheral inflammation and CNS pathology in HD is unknown. Certain inflammatory proteins, such as complement proteins and clusterin, are also up-regulated both peripherally and in the brain in HD (6, 8, 9). In vivo imaging, in vitro and postmortem studies have shown that microglia, the CNS counterpart of macrophages, are activated in premanifest (10) and manifest HD (11), that microglial activation correlates with disease severity (12), and that mutant huntingtin is expressed in microglia (13). Thus, inflammation is an established, though incompletely understood, feature of HD with likely pathogenic importance. Inflammatory changes in the CNS and peripheral tissues in HD may be due to independent effects of mutant huntingtin in both compartments, causing analogous derangements centrally and peripherally; or inflammatory activation may begin peripherally and spread to the CNS, or vice versa, through the passage of immunomodulatory molecules across the blood–brain barrier.
Insights into CNS and peripheral immune system interactions in HD may provide new biomarkers and improve knowledge of key pathogenic mechanisms, possibly leading to novel therapeutic approaches. The present work seeks to elucidate further the nature of the peripheral inflammatory activation in HD through quantification of levels of key inflammatory and immunomodulatory molecules in human plasma, and serum from three different mouse models of HD. To investigate possible links between peripheral inflammation and neuronal dysfunction, we examined correlations between individual inflammatory molecules and clinical features of HD. We used targeted transcription profiling to examine expression of key immunomodulatory proteins in the HD striatum to determine whether the inflammatory activation seen peripherally is mirrored in the CNS. We examined expression of huntingtin in its WT and mutant forms in human monocytes to investigate the possibility that the immune activation is due to a disease-related cell-autonomous dysfunction of these cells. We confirmed this with functional studies of human monocytes, and macrophages and microglia from HD mouse models, demonstrating that there is disease-related dysfunction of CNS and peripheral inflammatory cells in HD.
RESULTS
We collected 194 plasma samples from HD mutation carriers ranging from premanifest to moderate HD and from control subjects (Table S1, available at http://www.jem.org/cgi/content/full/jem.20080178/DC1) and quantified levels of key inflammatory and immunomodulatory molecules using multiplex sandwich ELISAs and single radial immunodiffusion assays. We found an altered profile of cytokine levels in HD patients (Fig. 1). The most striking increases across subject groups from controls to progressing disease were in IL-6 and IL-8 (P < 0.0001 in each case). In addition, IL-4, IL-10, and TNF-α levels increased significantly with disease progression (Fig. 1 B). Moreover, IL-6 levels were significantly increased in premanifest subjects with an estimated mean of 16 yr until motor onset (Fig. 1 A) (14). Interestingly, the cytokines that were increased earliest in the disease course (IL-6 and IL-8) are involved in the innate immune response (15). IL-10 and IL-4, antiinflammatory cytokines involved in the adaptive immune response, increased significantly in moderate stage disease. There was no difference in levels of Igs (IgG, IgA, or IgM) at any disease stage (Fig. 2), arguing against widespread activation of the adaptive humoral immune system.
Figure 1.

Altered immune profile peripherally in HD. (A) Multiplex ELISA quantification of cytokine levels in plasma from HD patients (premanifest, early and moderate HD stages) compared with control subjects. Graphs show mean concentrations with standard error bars. Significant differences between individual groups are shown (ANOVA with post-hoc Tukey HSD test). (B) The overall trend for increasing levels of cytokines across all groups, analyzed using linear regression, was highly significant for IL-6 and IL-8 and significant for IL-4, IL-10, TNF-α, and IL-5. R-values (partial correlation coefficients) are corrected for age and sex. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Figure 2.

Plasma Ig levels are unchanged in HD. Quantification of plasma IgG, IgA, and IgM by single radial immunodiffusion assays revealed no difference in Ig levels across disease stages, arguing against widespread activation of the adaptive immune system. Graphs show mean concentration standard error bars.
We then examined correlations between these cytokine changes and clinical characteristics. Plasma IL-8 levels increased markedly with disease progression (Fig. 1) and correlated with clinical measures of HD. Levels of IL-8 correlated positively with worsening disease, as demonstrated by unified HD rating scale (UHDRS) (16) chorea scores (R = −0.37; P < 0.01) and total motor scores (R = 0.27; P < 0.05), as well as negatively with total functional capacity (TFC) scores where lower scores indicate more severe disease (R = −0.28; P < 0.05) (Fig. 3 A). TNF-α levels in plasma correlated with UHDRS chorea scores (R = 0.28; P < 0.01) and UHDRS motor scores (R = 0.26; P < 0.05) (Fig. 3 B); a negative correlation with TFC was seen that approached statistical significance (R = −0.20; P = 0.07).
Figure 3.

Correlations between plasma cytokine levels and clinical severity scores in premanifest and manifest HD gene carriers. Levels of (A) IL-8 and (B) TNF-α correlated with worsening disease as demonstrated by increasing UHDRS chorea score and UHDRS total motor score and decreasing total functional capacity score.
Objective markers of HD progression are needed to facilitate the conduct of clinical trials of disease-modifying therapies in HD (17). We used stepwise logistic regression analysis to construct receiver operating characteristic (ROC) curves that demonstrated a strong ability of combinations of plasma cytokine levels to discriminate between disease groups (Fig. 4). We found that a combination of IL-6, IL-10, and IL-5 best discriminated between premanifest HD and controls with an area under the curve (AUC) of 0.81. A combination of IL-6, IL-8, and IL-10 best discriminated all HD expansion carriers (premanifest and manifest) from controls (AUC 0.82). IL-6, IL-8, and IL-10 together best discriminated between premanifest and manifest HD patients (AUC 0.85).
Figure 4.

ROC curves demonstrating the ability of different combinations of plasma cytokine levels to discriminate between subject groups. In a ROC curve plot, the “true positive” diagnosis rate (sensitivity) is plotted against the “false positive” diagnosis rate (1-specificity) for a test with a binary outcome. The AUC summarizes the discrimination of the test, i.e., its ability to classify cases correctly. A perfect test would have an AUC of 1; a worthless test would have an AUC of 0.5. AUC values may be classified as follows: 0.9–1, excellent; 0.8–0.9, good; 0.7–0.8, fair; 0.6–0.7, poor; 0.5–0.6, fail (reference 36). For the present analysis, optimum combinations were identified by stepwise logistic regression analysis using a threshold of P = 0.05 for each cytokine removed from the model. A combination of IL-6, IL-10, and IL-5 best discriminated between controls and premanifest HD; a combination of IL-6, IL-8, and IL-10 best discriminated manifest from premanifest HD; and a combination of IL-6, IL-8, and IL-10 best discriminated controls from HD gene carriers (both premanifest and manifest).
IL-6 triggers the acute phase response and is produced primarily by monocytes/macrophages and lymphocytes (15). To define the possible source of peripheral cytokines, we purified monocytes from whole blood from HD patients and control subjects by flow cytometric and magnetic sorting. Using RT–quantitative PCR (QPCR), we found monocytes from HD patients to express mutant huntingtin (Fig. 5, A and B).
Figure 5.

Human monocytes express WT and mutant huntingtin. (A) RT-QPCR studies of human monocytes obtained by flow cytometry demonstrated huntingtin expression in 100% of monocyte samples tested from controls (n = 2) and HD patients (n = 3). Expression ratios are relative to B2M. Graph shows mean expression ratios with standard error bars. (B) PCR amplification of CAG repeat tracts from huntingtin mRNA reveals that HD monocytes express both WT and mutant huntingtin, supporting the possibility of cell-autonomous dysfunction resulting in immune activation. WT and mutant CAG repeat lengths are shown. +, p4G6E4.0 plasmid expressing HTT exon 1 with 18 CAG repeats.
We then stimulated isolated monocytes with LPS and found that monocytes from premanifest HD mutation carriers behave abnormally, displaying excess IL-6 production compared with cells from control subjects (Fig. 6 A). Mutant huntingtin thus appears to produce functional overactivity of monocytes. We stimulated isolated macrophages from the yeast artificial chromosome (YAC)128 mouse model of HD with LPS. Echoing the results seen in human HD monocytes, macrophages from the YAC128 responded with enhanced secretion of IL-6 in response to stimulation compared with macrophages from WT mice (Fig. 6 B). To test whether the presence of mutant huntingtin per se is sufficient to produce dysfunction, we then examined the response to LPS stimulation in macrophages from the YAC18 mouse, which differs from the YAC128 only in the length of the polyglutamine stretch. Excessive IL-6 release was not seen in YAC18 cells (Fig. 6 C). To determine whether microglia, the CNS equivalent of monocytes/macrophages, are also dysfunctional in HD, we performed LPS stimulation of microglia isolated from the widely used R6/2 transgenic mouse model of HD (18). HD microglia, too, demonstrated hyperactivity in response to stimulation (Fig. 6 D).
Figure 6.

HD monocytes, macrophages, and microglia are overactive when stimulated. (A) No IL-6 was detectable in the supernatant of monocytes from control (n = 9) or premanifest HD subjects (n = 8) in the unstimulated state or after priming with IFN-γ. Monocytes stimulated by the addition of both IFN-γ and 2 μg/ml LPS expressed IL-6, but expression levels were significantly higher from HD monocytes. (B) Alveolar macrophages from the YAC128 HD mouse model have similarly altered function when stimulated. YAC128 macrophages stimulated by the addition of both IFN-γ and 100 ng/ml LPS expressed significantly more IL-6. n = 3 WT and 4 YAC128. (C) Macrophages from YAC18 mice, which differ from YAC128 cells only in the number of CAG repeats, behaved no differently from WT cells (P = 0.231; n = 4 per genotype) in response to stimulation at the same LPS concentration, suggesting that the hyperactivity in the YAC128 is due to mutant huntingtin. (D) Microglia isolated from neonatal R6/2 mice are also hyperactive when stimulated by 10 ng/ml LPS (n = 4 per group). Graphs show mean concentrations with standard error bars. ND, not detected. Unpaired t tests: *, P < 0.05; **, P < 0.01.
We investigated expression of inflammatory transcripts in postmortem human striatal tissue using RT-PCR and found markedly increased expression of IL-6, IL-8, and TNF-α in HD (Fig. 7), mirroring the key changes seen in plasma much earlier in the disease.
Figure 7.

Altered expression of inflammatory transcripts in postmortem HD striatal tissue. Levels of IL-6, IL-8, and TNF-α RNA were significantly higher in the striatum of HD patients than in control striatum. Graphs show means with standard error bars. n = 6 controls and 17 HD patients (see Table S3). Unpaired t tests: *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
To investigate the relationship between central and peripheral inflammatory processes, we measured IL-6 and IL-8 in matched plasma and cerebrospinal fluid (CSF) samples from HD patients and controls using ELISA. CSF and plasma levels of IL-6 and IL-8 correlated closely (Fig. 8; R = 0.74 and R = 0.66, respectively; P < 0.0001 for both).
Figure 8.

Correlations between matched CSF and plasma levels of IL-6 and IL-8. ELISA-quantified levels in CSF and matched plasma samples correlated strongly for both IL-6 and IL-8.
We used multiplex ELISA to determine whether peripheral immune activation is present in serum in HD mouse models and found increased levels of several cytokines in the R6/2 transgenic mouse and the full-length knock-in model of HD (HdhQ150/Q150) (19). In 12-wk R6/2 mice, IL-6, IL-10, IL-1β, and IL-12p70 were significantly increased (Fig. 9 A). In 22-mo knock-in Hdh150Q/150Q mice, IL-6, IL-10, and IL-12p70 were significantly elevated (Fig. 9 B). In the YAC128 mouse model of HD at 12 mo of age, we saw similar elevations in serum IL-6 and mKC, a mouse functional homologue of IL-8 (20) (Fig. 9 C). 12-mo YAC128 animals are phenotypically equivalent to early HD (21), and these animals therefore model the human patients whose plasma was studied.
Figure 9.

Mouse models of HD recapitulate features of human immune dysfunction. Serum levels of cytokines, measured by multiplex ELISA, are elevated in both (A) R6/2 and (B) HdhQ150Q/Q150 knock-in mouse models (n = 9 per genotype) at end-stage. (C) In 12-mo YAC128 animals (equivalent to early human disease), serum IL-6 and mKC, a mouse functional homologue of IL-8, are significantly increased (n = 3 WT and 4 YAC128). Graphs show mean levels with standard error bars. Unpaired t tests: *, P < 0.05; **, P < 0.01.
DISCUSSION
Collectively, our data show that immune activation in HD is widespread and detectable in peripheral plasma across disease stages. Key cytokines of the innate immune system are up-regulated both centrally and peripherally, and robust changes even take place in premanifest HD mutation carriers many years before the onset of motor abnormalities. To our knowledge, the elevated IL-6 level seen in premanifest subjects with a mean of 16 yr until predicted clinical onset represents the earliest plasma abnormality identified to date in HD. The peripheral changes correlate well with clinical variables and are accompanied by alterations in striatal gene expression.
Although cytokines such as IL-4 and IL-10 are increased later in the disease, normal Ig levels throughout the disease course suggest that there is no generalized activation of the adaptive immune response. Whereas IL-6 and IL-8 production are triggered by NF-κB activation (7, 22), IL-4 and IL-10 act to down-regulate NF-κB (15). The late involvement of these cytokines may reflect an adaptive response to chronic immune activation, possibly involving altered interactions between monocytes/macrophages and Th2 cells.
The correlation we show between plasma and CSF levels of IL-6 and IL-8 links the central and peripheral immune activation in HD. We show that IL-6 and IL-8 are increased in plasma and the striatum. These cytokines are not thought to cross the healthy blood–brain barrier in the acute setting (23, 24), and studies of brain penetration of specific molecules in HD have not identified alterations of the blood–brain barrier (e.g., reference 25). Mutant huntingtin therefore probably induces parallel dysfunction in both compartments (Fig. 10). The immune dysfunction we demonstrate in monocytes from premanifest HD gene carriers may reflect similar central changes in HD microglia and therefore act as a window onto central disease pathogenesis at this very early stage in the disease process. Given the dramatic changes seen in striatal cytokine expression and the chronic nature of these changes, passage of cytokines from the CNS into blood is a possibility we cannot exclude, although cytokines are rapidly broken down (26) and would likely be subject to considerable dilution in plasma. However, the presence of primary abnormalities of both peripheral and central cytokine-producing cells (monocytes/macrophages and microglia, respectively) suggests that parallel disease-related derangements in the periphery and CNS are the simplest explanation for these CSF–plasma correlations.
Figure 10.

Immune activation, induced by mutant huntingtin, occurs both peripherally and centrally in HD. A cell-autonomous effect of the mutant protein may be responsible for the innate immune response. The NF-κB signaling pathway that triggers IL-6 release is known to be up-regulated by mutant huntingtin (reference 7), and microglia-derived toxicity can influence disease progression (references 7 and 28). We find that the innate immune response detectable in plasma very early in the disease is strongly linked to disease progression and recapitulated in HD striatum, that human monocytes express mutant huntingtin, and that monocytes, macrophages, and microglia overexpress IL-6 when stimulated. Early innate immune activation could be a target in the development of disease-modifying therapies.
Microglia have previously been implicated in the pathogenesis of HD (10–12) and are increasingly seen as key players in the pathogenesis of neurodegenerative diseases (27). The increased cytokine production we observe in the CNS and peripherally could be caused by dysfunction of microglia and their counterparts, monocytes and macrophages. We show that monocytes from HD patients express mutant huntingtin. Further, we demonstrate that microglia, monocytes, and macrophages in HD are all hyperactive when stimulated. The NF-κB signaling pathway that triggers IL-6 release is known to be up-regulated by mutant huntingtin (7), and the microglial kynurenine monooxygenase pathway has been identified as a potential therapeutic target in HD, establishing immune dysfunction as a possible pathogenic pathway in HD (28).
The functional overactivity of macrophages from 12-mo YAC128 mice, corresponding to early human HD, was not seen in macrophages from YAC18 mice, which are identical to YAC128 animals except for the length of the polyglutamine tract. This suggests that the presence of mutant huntingtin alone is sufficient to cause derangement of monocytes/macrophages.
Interestingly, although IL-6 is undetectable in the supernatant of isolated human monocytes, levels of IL-6 in plasma are not zero in control subjects and HD patients. The same is true of mouse macrophages and serum. Thus, there is low-level background cytokine production in vivo, perhaps due to normal cell turnover or environmental immune challenges, that is absent in vitro. A primary dysfunction of monocytes is sufficient to explain the increased plasma cytokine levels in HD, as hyperactive HD monocytes will respond excessively to this physiological background stimulation. However, it is possible that HD also causes differences in the level of background immune stimulation, which could contribute to the later up-regulation of the adaptive immune response cytokines IL-4 and IL-10.
There is a need for markers of progression (“state biomarkers”) in HD and other neurodegenerative diseases (29). Our results suggest that inflammatory changes detected in peripheral plasma may be biologically relevant and mirror the neurodegenerative process occurring in the CNS (Fig. 10). Indeed, combined peripheral markers of inflammation were recently suggested to be biomarkers for diagnosis and progression in Alzheimer's disease (29). Remarkably, peripheral inflammatory changes may also reveal early pathogenic events in HD, occurring more than 15 yr before the onset of neurological manifestations. The inflammatory changes seen in patients are echoed in mouse models of HD. Importantly, they may therefore provide translational biomarkers for the use of HD mouse models in the development of therapeutic interventions. Finally, the mechanism of early innate immune activation in HD warrants further study as a potential source of targets for disease-modifying therapies.
MATERIALS AND METHODS
Ethical approval.
All human experiments were performed in accordance with the declaration of Helsinki and approved by University of British Columbia (UBC) Clinical Research Ethics Board (Canadian patients) or University College London (UCL)/UCL Hospitals Joint Research Ethics Committee (UK patients), as appropriate. All subjects gave informed written consent. All animal experiments were performed in accordance with relevant legislation and approved by local and national regulatory authorities (Institutional Review Board of the University of Washington, King's College London Animal Care and Use Committee, or UBC Committee on Animal Care and the Canadian Council on Animal Care, as appropriate).
Collection and processing of human plasma samples.
Blood samples were obtained from control subjects and genetically diagnosed HD patients and processed as described previously (6). Subjects with inflammatory or infective conditions were excluded. A subset of subjects was assessed on the UHDRS (16) by a neurologist experienced in assessment of HD patients. Subjects' demographic and clinical data are given in Table S1.
Collection of matched CSF and blood samples.
CSF donors were recruited through the UBC HD Medical Clinic. 20 HD patients and 10 control subjects, age-matched and lacking the HD mutation, were recruited (Table S1). Mutation-positive subjects were staged early or moderate according to independence score. CSF and matched blood samples were obtained within 1 h and plasma was extracted as described previously (6).
Collection of mouse serum samples.
For the present experiments, HdhQ150/Q150 knock-in and R6/2 exon 1 models that develop comparable and widespread molecular phenotypes (19) were used. R6/2 (18) and HdhQ150/Q150 mice (original nomenclature, CHL2) (30) were bred and serum samples were collected as described previously (19). All animals had unlimited access to water and breeding chow (Special Diet Services) under a 12-h light–12-h dark cycle. YAC128 mice were maintained on the FVB/N strain background (21). Numbers and ages of animals are shown in Table S4 (available at http://www.jem.org/cgi/content/full/jem.20080178/DC1).
Serum and plasma analyses.
Cytokine levels were quantified using Meso Scale Discovery (MSD) assays as per the manufacturer's protocol and analyzed on a SECTOR 2400 instrument (MSD). The operator was unaware of the disease state of each sample during processing, and statistical analysis was performed independently. Serum levels of Igs (IgG, IgM, and IgA) were determined by single radial immunodiffusion assays (The Binding Site Ltd) according to the manufacturer's protocol.
Human monocyte huntingtin expression study.
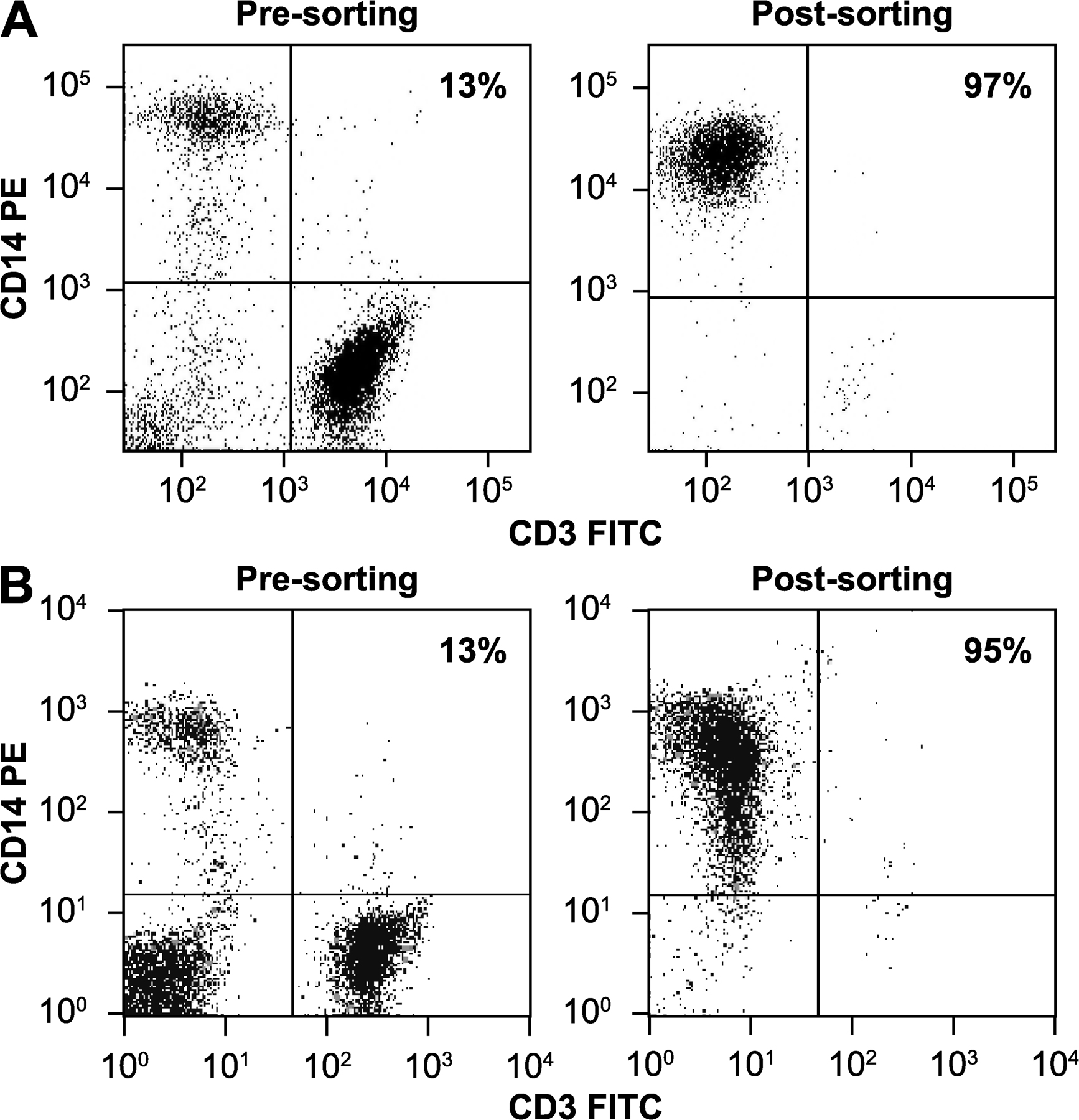
Whole blood was collected from HD patients, and controls were matched for age and sex (Table S5, available at http://www.jem.org/cgi/content/full/jem.20080178/DC1). Leukocytes were isolated by density gradient centrifugation over Lymphoprep solution (Axis-Shield). Monocytes were obtained by flow cytometry. In brief, mononuclear cell suspensions were labeled with anti-CD45 FITC and anti-CD14 PE (Becton Dickinson), and viable monocytes were sorted flow cytometrically by immunophenotype (CD45+/CD14+) and forward angle light scatter signals (FACSAria high speed cell sorter; Becton Dickinson) to at least 95% purity (Fig. S1 A).
RNA was prepared from pellets of 5 × 106 cells using an RNeasy mini kit (QIAGEN) according to the manufacturer's instructions. Quality and quantity of RNA was assessed using the RNA nanochip method on a BioAnalyzer (Agilent Technologies). RT of 1 μg of total RNA was performed in 50 mM KCl, 10 mM Tris–HCl, pH 9.0, 0.1% Triton X-100, 6.5 mM MgCl2, 10 mM DTT, 1 mM dNTPs, 10 ng/μl random hexamers with 0.35 U/μl RNasin (Promega), and MMLV reverse transcription (Invitrogen) for 10 min at 23°C and then 40 min at 37°C. The RT reaction was diluted 10-fold in nuclease-free water (Sigma-Aldrich), and 5 μl was used in a 25-μl reaction containing Precision Mastermix (PrimerDesign), 300 nM primers, and a 200-nM probe. Cycling conditions were as follows: 2 min at 50°C, 15 min at 95°C, 44 cycles (1 min at 94°C, 1 min at 60°C) using the Opticon 2 real-time PCR machine (MJ Research). The threshold used for the analysis was set at 0.05, and reactions were performed in triplicate for each sample. Primer and probe sequences are listed in Table S6 (available at http://www.jem.org/cgi/content/full/jem.20080178/DC1). Expression of huntingtin was calculated using 2−ΔΔCT with β-2-microglobulin (B2M) as the reference (31). Positive control samples with known B2M expression levels produced consistent results under these experimental conditions.
CAG repeats were measured in RNA using an ABI3730 automated sequencer, and all instruments and materials were obtained from Applied Biosystems unless indicated. The p4G6E4.0 plasmid, which expresses exon 1 of huntingtin with 18 CAG repeats, was used as a positive control. PCR was performed in AM buffer, 10% DMSO, 200 μM dNTPs, 10 ng/μl of primer with 0.5 U/μl Taq polymerase (PerkinElmer). Cycling conditions were 90 s at 94°C, 25 cycles (30 s at 94°C, 30 s at 68°C, 90 s at 72°C), and 10 min at 72°C. The FAM-tagged PCR product (1 μl) together with MegaBACE ET900 (GE Healthcare) internal size standard (0.04 μl) were denatured at 94°C for 5 min in 9 μl of HiDi formamide. The run conditions were as follows: capillary size, 36 cm, Polymer-PoP-7. The run module was oven temperature, 66°C; buffer temperature, 35°C; prerun voltage, 15 kV; prerun time, 180 s; injection voltage, 3 kV; injection time, 20 s; first readout time, 200 msec; second readout time, 200 msec; run voltage, 10 kV; voltage number of steps, 10; voltage step interval, 20 s; voltage tolerance, 0.6 kV; current stability, 10 μA; ramp delay, 1 s; data delay, 120 s; run time, 2,700 s. Data analysis was performed using the plate manager application GeneMapper v5.2-3730XL.
Functional study of human monocytes.
Whole blood was collected in heparin (CP Pharmaceuticals). Leukocytes were isolated by density gradient centrifugation over Histopaque 1077 solution (Sigma-Aldrich). Monocytes were obtained by magnetic sorting to increase yield and minimize handling time. Mononuclear cell suspensions were labeled with anti-CD14 microbeads and sorted through magnetic cell separation columns (Miltenyi Biotec) to at least 95% purity (Fig. S1 B). Monocytes were counted, and 5 × 105 cells per well seeded into 24-well culture plates in RPMI culture medium supplemented with 5% FBS, 2 mM l-glutamine, and 1% penicillin/streptomycin (Invitrogen). Cells were incubated for 16 h before stimulation. The medium was then changed to fresh cell culture medium with or without 10 ng/ml IFN-γ (R&D Systems). For LPS stimulation, 2 μg/ml LPS was added to the medium (Sigma-Aldrich). After 24 h, supernatants were harvested from two separate wells for each subject/condition. The cells remaining were lysed in 50 mM Tris, pH 8, 150 mmol NaCl, 0.5% sodium deoxycholate, and 0.5% Triton X-100 and assayed for total protein concentration using a protein assay kit according to the manufacturer's instructions (Bio-Rad Laboratories). IL-6 concentrations in supernatants were determined using the MSD assay and adjusted for total protein concentration.
Functional study of tissue macrophages.
Alveolar macrophages were isolated from 12-mo-old WT, YAC18, and YAC128 mice, all maintained on a pure FVB/N strain background. The YAC128 (line 53) mouse line expresses high levels of full-length human huntingtin with ∼128 polyglutamine repeats and is a well-established model of HD. These mice develop an age-dependent phenotype similar to that seen in HD patients, including cognitive deficits, motor dysfunction, and selective neurodegeneration; 12-mo mice are equivalent to early human HD (21). YAC18 mice (line 212) express transgenic human WT huntingtin and do not exhibit any disease phenotype relative to their WT littermates. They differ from YAC128 mice only in the length of the polyglutamine tract.
Animals were killed using 10 mg avertine via i.p. injection. Blood was drawn from the inferior vena cava, and serum samples were obtained by two-stage centrifugation. Alveolar macrophages were extracted by intratracheal infusion of ice-cold PBS (Invitrogen) followed by centrifugation and resuspension of extracted cells. Cells were counted and seeded at 1.5 × 105 cells/ml onto 96-well gelatin-coated plates and incubated in culture media containing 5% medium (RPMI 1640 [Invitrogen], 5% FBS [Cansera], and 1% penicillin/streptomycin [Invitrogen]). After 24 h this was changed to 1% medium (RPMI 1640, 1% FBS, and 1% penicillin/streptomycin). Functional studies were performed after an additional 24 h. The medium was changed to fresh 1% medium or 1% medium containing 10 μg/L IFN-γ (R&D Systems) with or without 100 μg/L control standard endotoxin (Associates of Cape Cod). After 24 h, IL-6 concentrations were measured in supernatants from two independent wells from each animal for each condition, using a commercial mouse IL-6 ELISA kit according to the manufacturer's instructions (eBioscience). Numbers of animals used in each experiment are given in Table S4.
Functional study of microglia.
Mixed primary glial cultures were prepared from single brains of R6/2 mice (B6CBA-Tg(HDexon1)62Gpb/3J; Jackson ImmunoResearch Laboratories) as described previously (32). In brief, 4-d-old mice were decapitated. The brains were removed and submerged in ice-cold Hank's saline. The meninges and blood vessels were removed before the tissue was trypsinized, carefully dissociated with a 5-ml pipette, resuspended, and filtered twice (100-μm diameter Falcon filter; Becton Dickinson) before seeding the cells in 5 ml of medium per flask (one brain/flask). Cells were cultured in poly-ornithine–coated 25-cm2 flasks in DME and supplemented with 10% FBS (D10F).
Littermate heterozygote R6/2 and WT mice were used. Each brain was processed and cultured individually to prevent cross-contamination between animals. Genotype and CAG repeat length were determined by PCR from tail samples taken at the time of CNS culture preparation (Laragen). Once astrocytes reached confluence (5–7 d), D10F medium was supplemented with 2 ng/ml GM-CSF. Microglial cells were collected, pooled according to genotype, and seeded in 96-well Primaria plates (2.5 × 104 cells in 250 μl D10F per well; Becton Dickinson). Cultures were >95% pure as assessed by CD11b immunostaining. For each experiment, WT and R6/2 microglial cells were processed in parallel.
24 h after plating, the cells were serum starved (MSFM, 0.2 ng/ml GM-CSF) for an additional 24 h. They were then stimulated with 10 U IFN-γ ± 10 ng/ml LPS or carrier control. After 24 h of stimulation, supernatant was collected and stored at −80°C for further analysis.
IL-6 concentration was measured by Luminex bead array system (QIAGEN). 60 μl of supernatant of three representative experiments (n = 4 for each condition) was thawed and processed using the BioPlex platform (Bio-Rad Laboratories).
Striatal gene expression study.
Total RNA was isolated and purified from striatal samples obtained from The New Zealand Neurological Foundation Human Brain Bank and the New York Brain Bank at Columbia University (6 controls and 17 patients with pathological grades as shown in Table S3, which is available at http://www.jem.org/cgi/content/full/jem.20080178/DC1) with the RNeasy mini kit (QIAGEN). mRNA was transcribed into cDNA with SuperScript III (Invitrogen). RT-QPCR was performed in triplicate with target-specific Roche Universal Library probes (FAM) and Roche universal master mix (Roche Diagnostics), and analyzed with an ABI PRISM 7500 RT-QPCR System. Mitochondrial ribosomal protein S35 (MRPS35) expression was determined by duplex PCR (VIC) in the same sample to normalize target expression to a housekeeping gene. MRPS35 was chosen based on gene array data showing it not to be regulated in HD striatum (Strand, A., personal communication). The target/MRPS35 ratio was used to compare the relative target expression using the modified ΔΔCT method (33). Purity of mRNA was checked by performing QPCR without prior RT. Stability of expression of the housekeeping gene MRPS35 was confirmed by comparison with a second housekeeping gene, hypoxanthine-guanine phosphoribosyltransferase.
Statistical analysis.
For the human plasma cytokine and Ig data, intergroup differences were identified by one-way ANOVA with post-hoc Tukey HSD testing to allow for multiple comparisons. Linear regression analysis using coded variables for each subject group (1, control; 2, premanifest; 3, early; 4, moderate), using age and sex as covariates, was used to identify significant change with advancing disease (6).
Calculations of estimated time to onset in premanifest subjects were made using the age- and CAG-dependent conditional onset probability formula of Langbehn et al. (14).
Correlations with clinical variables were examined using linear regression analysis and partial correlations. Because the distribution of UHDRS and TFC data were not Gaussian, bootstrapping with 1,000 replications was used to enable linear regression analysis and the use of age as a covariate. This analysis inevitably involved the use of multiple statistical tests, but because the associations under investigation were of independent scientific interest, p-values were not corrected for multiple comparisons (34).
To examine for the ability of combinations of plasma cytokines to distinguish between different subject groups, stepwise logistic regression was used (35). The model tested the six cytokines with statistically significant correlations across disease stage in order of diminishing R-value (IL-6, IL-8, IL-4, IL-10, TNF-α, and IL-5; see Fig. 1 B); variables were removed from the model when P < 0.05 for the logistic regression. The comparisons assessed were as follows: controls versus premanifest HD; controls versus all HD expansion–positive subjects; and premanifest versus manifest HD.
Unpaired two-tailed t tests were used to identify significantly different serum levels for each cytokine in mouse serum and to compare mRNA levels (expressed as 2−ΔΔCT) between controls and HD patients in the two gene expression studies. Unpaired one-tailed t tests were used to test the hypotheses that monocytes, macrophages, and microglia produce more IL-6 than WT animals when stimulated with LPS.
Online supplemental material.
Fig. S1 shows representative flow cytometry plots demonstrating purity of cells obtained by flow cytometry and magnetic sorting. Table S1 shows the characteristics of subjects in each human biofluid study. Table S2 shows plasma cytokine levels by disease stage measured by multiplex ELISA assay. Table S3 shows characteristics of subjects in the postmortem striatal expression study. Table S4 shows details of animals used for mouse experiments. Table S5 shows details of the subjects whose blood was used for the monocyte huntingtin expression study. Table S6 shows the primers used for the human monocyte expression study. The online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20080178/DC1.
Supplementary Material
Acknowledgments
We thank the patients and controls who donated samples; the staff of the multidisciplinary HD clinics in London and Vancouver; Professor Richard Faull of Auckland University and the New York Brain Bank at Columbia University for supplying human postmortem samples; Ms. Janet North for flow cytometry assistance; Dr. Martin R. Stämpfli (McMaster University, Hamilton, ON) for assistance with serum assays; and Professor Chris Frost for statistical advice.
This study was financially supported by CHDI (previously the High Q Foundation), New York, and also in part by the Medical Research Council (UK), the Wellcome Trust (66270), the Canadian Institutes for Health Research and the Huntington Society of Canada. It was undertaken in part at UCH/UCLH, which received a proportion of funding from the UK Department of Health's NIHR Biomedical Research Centres funding scheme. M.R. Hayden is a Killam University Professor and holds a Canada Research Chair in Human Genetics.
The authors have no conflicting financial interests.
Abbreviations used: AUC, area under the curve; B2M, β-2-microglobulin; CNS, central nervous system; CSF, cerebrospinal fluid; HD, Huntington's disease; QPCR, quantitative PCR; ROC, receiver operating characteristic; TFC, total functional capacity; UHDRS, unified HD rating scale; YAC, yeast artificial chromosome.
M. Björkqvist and E.J. Wild contributed equally to this paper.
References
- 1.Bates, G., P.S. Harper, and L. Jones, editors. 2002. Huntington's Disease. Oxford University Press, Oxford. 558 pp.
- 2.Sathasivam, K., C. Hobbs, M. Turmaine, L. Mangiarini, A. Mahal, F. Bertaux, E.E. Wanker, P. Doherty, S.W. Davies, and G.P. Bates. 1999. Formation of polyglutamine inclusions in non-CNS tissue. Hum. Mol. Genet. 8:813–822. [DOI] [PubMed] [Google Scholar]
- 3.Van Raamsdonk, J.M., Z. Murphy, D.M. Selva, R. Hamidizadeh, J. Pearson, A. Petersen, M. Bjorkqvist, C. Muir, I.R. Mackenzie, G.L. Hammond, et al. 2007. Testicular degeneration in Huntington disease. Neurobiol. Dis. 26:512–520. [DOI] [PubMed] [Google Scholar]
- 4.Robbins, A.O., A.K. Ho, and R.A. Barker. 2006. Weight changes in Huntington's disease. Eur. J. Neurol. 13:e7. [DOI] [PubMed] [Google Scholar]
- 5.Bjorkqvist, M., A. Petersen, K. Bacos, J. Isaacs, P. Norlen, J. Gil, N. Popovic, F. Sundler, G.P. Bates, S.J. Tabrizi, et al. 2006. Progressive alterations in the hypothalamic-pituitary-adrenal axis in the R6/2 transgenic mouse model of Huntington's disease. Hum. Mol. Genet. 15:1713–1721. [DOI] [PubMed] [Google Scholar]
- 6.Dalrymple, A., E.J. Wild, R. Joubert, K. Sathasivam, M. Bjorkqvist, A. Petersen, G.S. Jackson, J.D. Isaacs, M. Kristiansen, G.P. Bates, et al. 2007. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J. Proteome Res. 6:2833–2840. [DOI] [PubMed] [Google Scholar]
- 7.Khoshnan, A., J. Ko, E.E. Watkin, L.A. Paige, P.H. Reinhart, and P.H. Patterson. 2004. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. 24:7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gasque, P., M. Fontaine, and B.P. Morgan. 1995. Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J. Immunol. 154:4726–4733. [PubMed] [Google Scholar]
- 9.Hodges, A., A.D. Strand, A.K. Aragaki, A. Kuhn, T. Sengstag, G. Hughes, L.A. Elliston, C. Hartog, D.R. Goldstein, D. Thu, et al. 2006. Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 15:965–977. [DOI] [PubMed] [Google Scholar]
- 10.Tai, Y.F., N. Pavese, A. Gerhard, S.J. Tabrizi, R.A. Barker, D.J. Brooks, and P. Piccini. 2007. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. 130:1759–1766. [DOI] [PubMed] [Google Scholar]
- 11.Sapp, E., K.B. Kegel, N. Aronin, T. Hashikawa, Y. Uchiyama, K. Tohyama, P.G. Bhide, J.P. Vonsattel, and M. DiFiglia. 2001. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 60:161–172. [DOI] [PubMed] [Google Scholar]
- 12.Pavese, N., A. Gerhard, Y.F. Tai, A.K. Ho, F. Turkheimer, R.A. Barker, D.J. Brooks, and P. Piccini. 2006. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 66:1638–1643. [DOI] [PubMed] [Google Scholar]
- 13.Shin, J.-Y., Z.-H. Fang, Z.-X. Yu, C.-E. Wang, S.-H. Li, and X.-J. Li. 2005. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 171:1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langbehn, D.R., R.R. Brinkman, D. Falush, J.S. Paulsen, and M.R. Hayden. 2004. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin. Genet. 65:267–277. [DOI] [PubMed] [Google Scholar]
- 15.Kindt, T.J., R.A. Goldsby, B.A. Osborne, and J. Kuby. 2006. Kuby Immunology. W.H. Freeman, New York. 608 pp.
- 16.The Huntington Study Group. 1996. Unified Huntington's disease rating scale: reliability and consistency. Mov. Disord. 11:136–142. [DOI] [PubMed] [Google Scholar]
- 17.Wild, E.J., and S.J. Tabrizi. 2006. Predict-HD and the future of therapeutic trials. Lancet Neurol. 5:724–725. [DOI] [PubMed] [Google Scholar]
- 18.Mangiarini, L., K. Sathasivam, M. Seller, B. Cozens, A. Harper, C. Hetherington, M. Lawton, Y. Trottier, H. Lehrach, S.W. Davies, and G.P. Bates. 1996. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 87:493–506. [DOI] [PubMed] [Google Scholar]
- 19.Woodman, B., R. Butler, C. Landles, M.K. Lupton, J. Tse, E. Hockly, H. Moffitt, K. Sathasivam, and G.P. Bates. 2007. The HdhQ150/Q150 knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 72:83–97. [DOI] [PubMed] [Google Scholar]
- 20.Bozic, C.R., L.F. Kolakowski Jr., N.P. Gerard, C. Garcia-Rodriguez, C. von Uexkull-Guldenband, M.J. Conklyn, R. Breslow, H.J. Showell, and C. Gerard. 1995. Expression and biologic characterization of the murine chemokine KC. J. Immunol. 154:6048–6057. [PubMed] [Google Scholar]
- 21.Slow, E.J., J. van Raamsdonk, D. Rogers, S.H. Coleman, R.K. Graham, Y. Deng, R. Oh, N. Bissada, S.M. Hossain, Y.Z. Yang, et al. 2003. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12:1555–1567. [DOI] [PubMed] [Google Scholar]
- 22.Fietta, A.M., M. Morosini, F. Meloni, A.M. Bianco, and E. Pozzi. 2002. Pharmacological analysis of signal transduction pathways required for mycobacterium tuberculosis-induced IL-8 and MCP-1 production in human peripheral monocytes. Cytokine. 19:242–249. [PubMed] [Google Scholar]
- 23.Steensberg, A., M.K. Dalsgaard, N.H. Secher, and B.K. Pedersen. 2006. Cerebrospinal fluid IL-6, HSP72, and TNF-alpha in exercising humans. Brain Behav. Immun. 20:585–589. [DOI] [PubMed] [Google Scholar]
- 24.Billiau, A.D., P. Witters, B. Ceulemans, A. Kasran, C. Wouters, and L. Lagae. 2007. Intravenous immunoglobulins in refractory childhood-onset epilepsy: effects on seizure frequency, EEG activity, and cerebrospinal fluid cytokine profile. Epilepsia. 48:1739–1749. [DOI] [PubMed] [Google Scholar]
- 25.Hersch, S.M., S. Gevorkian, K. Marder, C. Moskowitz, A. Feigin, M. Cox, P. Como, C. Zimmerman, M. Lin, L. Zhang, et al. 2006. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology. 66:250–252. [DOI] [PubMed] [Google Scholar]
- 26.Pan, W., and A.J. Kastin. 2007. Adipokines and the blood-brain barrier. Peptides. 28:1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lobsiger, C.S., and D.W. Cleveland. 2007. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 10:1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giorgini, F., P. Guidetti, Q. Nguyen, S.C. Bennett, and P.J. Muchowski. 2005. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 37:526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray, S., M. Britschgi, C. Herbert, Y. Takeda-Uchimura, A. Boxer, K. Blennow, L.F. Friedman, D.R. Galasko, M. Jutel, A. Karydas, et al. 2007. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat. Med. 13:1359–1362. [DOI] [PubMed] [Google Scholar]
- 30.Lin, C.H., S. Tallaksen-Greene, W.M. Chien, J.A. Cearley, W.S. Jackson, A.B. Crouse, S. Ren, X.J. Li, R.L. Albin, and P.J. Detloff. 2001. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 10:137–144. [DOI] [PubMed] [Google Scholar]
- 31.Gabert, J., E. Beillard, V.H. van der Velden, W. Bi, D. Grimwade, N. Pallisgaard, G. Barbany, G. Cazzaniga, J.M. Cayuela, H. Cave, et al. 2003. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia-a Europe Against Cancer program. Leukemia. 17:2318–2357. [DOI] [PubMed] [Google Scholar]
- 32.Weydt, P., E.C. Yuen, B.R. Ransom, and T. Moller. 2004. Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia. 48:179–182. [DOI] [PubMed] [Google Scholar]
- 33.Pfaffl, M.W. 2001. Validities of mRNA quantification using recombinant RNA and recombinant DNA external calibration curves in real-time RT-PCR. Biotechnol. Lett. 23:275. [Google Scholar]
- 34.Savitz, D.A., and A.F. Olshan. 1995. Multiple comparisons and related issues in the interpretation of epidemiologic data. Am. J. Epidemiol. 142:904–908. [DOI] [PubMed] [Google Scholar]
- 35.Teunissen, C.E., D. Lutjohann, K. von Bergmann, F. Verhey, F. Vreeling, A. Wauters, E. Bosmans, H. Bosma, M.P.J. van Boxtel, M. Maes, et al. 2003. Combination of serum markers related to several mechanisms in Alzheimer's disease. Neurobiol. Aging. 24:893–902. [DOI] [PubMed] [Google Scholar]
- 36.Hanley, J.A., and B.J. McNeil. 1982. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 143:29–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}