

Abstract
Blood coagulation factor VIII (fVIII) is activated by thrombin to form an A1/A2/A3-C1-C2 heterotrimer, which functions as a cofactor for factor IXa during intrinsic pathway factor X activation. Human fVIIIa decays rapidly because of first-order dissociation of the A2 subunit, which may function to regulate the coagulation mechanism. The three fVIII A domains each consist of two cupredoxin-like subdomains. Substitution of the COOH-terminal A1 subdomain of porcine fVIIIa, which decays more slowly than human fVIIIa, reduces the dissociation rate constant for fVIIIa decay. Examination of a human fVIII A1-A2-A3 homology model (Pemberton, S., et al., Blood 89: 2413 - 2421, 1997) revealed a possible interaction between Q316 in the FG helix of the COOH-terminal A1 subdomain and M539 in the FG helix of the NH2-terminal A2 subdomain, which are sites where human and porcine fVIII differ. Decays of purified recombinant human and porcine fVIIIa and the human fVIIIa mutants Q316H, M539L and Q316H/M539L were compared at 23 °C and 37 °C. The decay rates of the Q316H and Q316H/M539L mutants, but not the M539L mutant, were significantly slower than human fVIIIa. These results indicate that the FG helix of the COOH-terminal A1 cupredoxin-like subdomain of fVIII may be under selective pressure by the requirements of hemostatic balance.
Factor VIII (fVIII1) contains a sequence of domains designated A1-A2-B-ap-A3-C1-C2 and is activated by thrombin to form a 160-kDa A1/A2/A3-C1-C2 heterotrimer (1-3). FVIIIa is a cofactor for factor IXa during the activation of factor X in the intrinsic pathway of blood coagulation. At pH 7.4, the activity of human fVIIIa at its physiological concentration (∼1 nM) spontaneously decays to undetectable levels with a half-life of ∼2 min at 23 °C (4-7) as a result of A2 subunit dissociation (2;3;8). A2 subunit dissociation appears to be an important regulatory feature of the blood coagulation mechanism because mutations in the fVIII gene that increase the rate of A2 subunit dissociation produce hemophilia A (9). The rate constant for A2 subunit dissociation of human fVIIIa is ∼3-fold greater than that of porcine fVIIIa at 23 °C (10). The differential A2 subunit dissociation rates are due to amino acid differences in the A1 subunit rather than the A2 subunit because substitution of the porcine A1 subunit into human fVIIIa reproduces the porcine fVIIIa decay rate (7).
The three fVIII A domains are homologous to corresponding A domains in factor V and ceruloplasmin (11). Each A domain contains two ∼20 kDa subdomains that are homologous to copper-binding cupredoxins such as plastocyanin and azurin (12). Substitution of the porcine COOH-terminal A1 subdomain for the corresponding human subdomain produces an A2 subunit dissociation rate constant that is distinguishable from porcine fVIIIa (7). Additionally, substitution of the NH2-terminal A1 subdomain of porcine fVIII produces a dissociation rate constant that is intermediate between human and porcine fVIIIa, suggesting a possible cooperative interaction between the two A1 subdomains. In this study, we compared the amino acid sequences of the COOH-terminal A1 and NH2-terminal A2 subdomains of human and porcine fVIII and identified Q316 as a possible site involved in human fVIIIa A2 subunit dissociation. Additionally, M539 in the A2 domain was identified as a candidate residue that interacts with Q316. Decay rate analysis of the human-to-porcine fVIIIa mutants Q316H and Q316H/M539L are consistent with involvement of Q316 in the regulation of fVIIIa decay.
Experimental Procedures
Materials
HEPES (N-2-hydroxyethylpiperazine-N′-2-ethane-sulfonic acid) and oligonucleotides were was purchased from Invitrogen (Carlsbad, CA). Type III-E egg yolk L-α-phosphatidylcholine and bovine brain L-α-phosphatidyl-L-serine were purchased from Avanti Polar Lipids (Alabaster, AL). Small unilamellar phosphatidylcholine/phosphatidylserine (PCPS) (75/25, w/w) vesicles were prepared using an Avanti Polar Lipids Mini-Extruder kit according to instructions supplied by the manufacturer. SP-Sepharose, Source Q, mono S and mono Q chromatography resins were purchased from GE Healthcare Life Sciences. Tween-80 was purchased from Pierce (Rockford, IL). Recombinant desulfatohirudin was a generous gift from Dr. R.B. Wallis, Ciba-Geigy Pharmaceuticals. Human thrombin was a gift from Dr. Sriram Krishnaswamy, University of Pennsylvania. Human factors IXaβ and X were purified as described previously (13;14). Spectrazyme Xa (Methoxycarbonyl-D-cyclohexylglycyl-gylcyl-arginine-p-nitroanilide acetate) was purchased from American Diagnostica (Stamford, CT)
Construction and Expression of FVIII cDNAs
The construction of cDNA constructs encoding B domain-deleted (BDD) human fVIII and porcine fVIII in the mammalian expression vector ReNeo has been described previously (15;16). The human fVIII mutants Q316H and M539L were constructed by splicing-by-overlap extension mutagenesis (SOE) using the BDD human fVIII ReNeo plasmid for the initial PCR reactions (17) according to previously published procedures (18-20). For the Q316H mutant, a mutation was made at nucleotide 1006 in the human fVIII cDNA, converting a CAA codon to a CAC codon. The first PCR used oligonucleotides corresponding to human fVIII cDNA nucleotides 538-563 and 1023-985 as the forward and mutagenic reverse primers (Table 1). The second PCR used oligonucleotides corresponding to nucleotides 985-1023 and 1466-1441 as the mutagenic forward and reverse primers. The SOE reaction used fragments from the PCRs as templates and the 538-563 and 1466-1441 oligonucleotides as primers. The SOE product and BDD human fVIII/ReNeo ligation fragments were generated using Spe I and Mlu I. For the M539L mutant, mutations were made at nucleotides 1672 and 1674 in the human fVIII cDNA, converting an ATG codon to a CTA codon. The first PCR used oligonucleotides corresponding to human fVIII cDNA nucleotides 1150-1194 and 1692-1654 as the forward and mutagenic reverse primers. The second PCR used oligonucleotides corresponding to nucleotides 1654-1692 and 2413-2393 as the mutagenic forward and reverse primers. The SOE reaction used fragments from the PCRs as templates and the 1150-1194 and 2413-2393 oligonucleotides as primers. The SOE product and BDD human fVIII/ReNeo ligation fragments were generated using Mlu I and BsiW I. The Q316H/M539L mutant was made by digesting the Q316H and M539L plasmids with Mlu I and BsiW I and ligating the products. PCR-generated sequences were confirmed by DNA sequencing. Expression of stably integrated fVIII cDNAs from baby hamster kidney-derived (BHK-M) cells was performed as described previously (15). Geneticin-resistant clones were screened for fVIII coagulant activity in serum-free medium. The highest producing clone of each construct was used for preparative scale expression of fVIII.
Table 1. Oligonucleotide PCR Primers.
| Sequence numbera | Oligonucleotide |
|---|---|
| 538 - 563 | 5′-CATGTGGACCTGGTAAAAGACTTGAA-3′ |
| 1023 - 985 | 5′-AGCTTCCATGCCATCATGGTGGTGGGAAGAGATATGACA-3′ |
| 985 - 1023 | 5′-TGTCATATCTCTTCCCACCACCATGATGGCATGGAAGCT-3′ |
| 1466 - 1441 | 5′-CTTGCTTGATTCTTAAATATAATCAA-3′ |
| 1150 - 1194 | 5′-TACTCTAGTTTCGTTAATCTAGAGAGAGATCTAGCTTC-3′ |
| 1692 - 1654 | 5′-GAAGCTAGATCTCTCTCTAGATTAACGAAACTAGAGTA-3′ |
| 1654 - 1692 | 5′-TACTCTAGTTTCGTTAATCTAGAGAGAGATCTAGCTTC-3′ |
| 2413 - 2393 | 5′-TCTTTTGAAAGCTGCGGGGG-3′ |
Human fVIII cDNA (GenBank Accession number NM000132). Position 1 corresponds to the start of the initial ATG codon in the signal peptide.
Purification of recombinant fVIII
BDD human fVIII, porcine fVIII and the human fVIII mutants Q316H, M539L and Q316H/M539L were purified from cell culture medium using SP-Sepharose and Source Q ion-exchange chromatography as described previously (15) except that Q316H underwent an additional mono S chromatography step. All preparations were at least 90% pure as judged by SDS-PAGE and consisted predominantly of a heavy chain (A1-A2)/light chain (ap-A3-C1-C2) heterodimer with a small amount of unprocessed single chain fVIII. The concentrations of the purified constructs was calculated using a molar extinction coefficient at 280 nm of 260,000 M-1cm-1, based on their predicted tyrosine, tryptophan and cysteine compositions (21), which did not differ among the proteins.
Measurement of the kinetics of fVIIIa decay
A chromogenic substrate assay for fVIIIa activity was used based on factor X activation by an enzymatic complex consisting of factor IXa, PCPS, calcium, and limiting amounts of fVIIIa as described previously (7;10). For measurements of fVIIIa decay at 23 °C, fVIII (1 nM) was reacted with 100 nM thrombin at 23 °C for 30 s in 0.15 M NaCl, 0.02 M HEPES, 5 mM CaCl2, 0.01% Tween-80, pH 7.4 (Buffer A), followed by addition of desulfatohirudin (150 nM) to inhibit thrombin. At various times, fVIII activation mixtures were diluted to a total fVIIIa concentration of 0.2 nM into 2 nM factor IXa/ 20 μM PCPS in Buffer A at 23 °C. Factor X then was added immediately to a final concentration of 300 nM and factor Xa formation was measured using the chromogenic substrate Spectrazyme Xa as described previously (22) using a VersaMax kinetic plate reader (Molecular Devices, Sunnyvale, CA). For measurements of fVIIIa decay at 37 °C, 20 nM fVIII was reacted with 100 nM thrombin at 23 °C in Buffer A for 15 s, followed by to dilution 1 nM in Buffer A containing 8 nM desulfatohirudin at 37 °C for various times and then assayed described above. Buffer pH was adjusted to correct for the variation of the pKa of HEPES with temperature as described previously (7).
Analysis of the kinetics of fVIIIa decay
Decay kinetics of fVIIIa were analyzed using a model describing reversible binding of the A2 subunit to the A1/A2/A3-C1-C2 dimer as described previously (7;10): The dissociation and association rate constants, k and k′, respectively, are related by the dissociation constant, Kd = k/k′. The dissociation constants for human and porcine fVIIIa at 23 °C are ∼160 nM and ∼ 90 nM respectively (7). Thus, when fVIII is activated at its physiological concentration (∼1 nM), the equilibrium lies far towards the dissociated state, resulting in a single exponential decay.
Under these conditions, the fraction of fVIIIa remaining as a function of time, f(t),
is given by
where ao is the initial concentration of fVIIIa and a(t) is the concentration of dissociated product at time t. Because fVIIIa activity is measured indirectly by measuring factor X activation, ao cannot be measured directly, but rather is estimated by extrapolation to the ordinate of the line formed from plotting the logarithm of the initial velocity of factor X activation versus time for the 1 and 3 min points. Estimates and intra-experimental 95% confidence limits for k were obtained by fitting values of f(t) as a function of ao and t using weighted nonlinear least-squares regression analysis and Student's t distribution as described previously (7).
Results
FVIII, factor V and ceruloplasmin each contain three orthologous A domains. Each A domain consists of two cupredoxin-like subdomains. A homology model of the fVIII A domains has been constructed based on a crystal structure of ceruloplasmin (23). Comparison of the crystal structures of ceruloplasmin and a proteolytic fragment of bovine factor Va consisting of the A1/A3-C1-C2 domains has revealed that the Cα backbone of the A1 and A3 domains is highly conserved (12;24). Additionally, the A domains of both structures contain a conserved pseudo-three fold axis of symmetry. These results indicate that the fVIII homology model may be useful for predicting amino acid side chains involved in fVIIIa subunit interactions. The pseudo-three fold axis of symmetry of fVIII, shown schematically in Figure 1A, indicates that NH2-terminal subdomains interface with COOH-terminal subdomains of neighboring domains. Each cupredoxin-like subdomain has a β-sheet structure, made up of 7 parallel or antiparallel strands, which is orthologous to plastocyanin and other members of the cupredoxin family (Fig 1B).
Fig. 1. FVIII domain and subdomain structure.

(A) The arrangement of the NH2- and COOH-terminal cupredoxin-like subdomains of the three A domains is shown, along with the B, C1 and C2 domains. (B) β-strand structure of cupredoxin-like molecules (37). The two β-sheets are formed by strands A, C and E and strands G, F, and D, respectively.
To test the hypothesis that the difference between human and porcine fVIIIa decay rates is due to amino acid differences in the A1-COOH and/or A2-NH2 subdomains, the fVIII homology model was used to identify neighboring groups in the two subdomains. To attempt to avoid excluding possible important interactions, all inter-domain groups within 6 Å were defined as neighboring groups. This analysis identified N280 and Q316 in the A1-COOH subdomain and F536 and M539 in the A2-NH2 subdomain as the only neighboring residues in which human and porcine fVIII differ (Table 2). Additionally, residue P25, which is a histidine in porcine fVIII, in the extended loop between the A and B stands of A1-NH2 subdomain, was neighbored by several residues in the A2-NH2 subdomain. N280 is in a loop between strands D and E in the A1-COOH subdomain (Fig. 2), but is exposed (solvent accessibility 51%) making it an unlikely candidate for interacting with the A2 domain. Q316 is in an α-helix between the F and G strands of the A1-COOH subdomain and is buried (solvent accessibility 13%) by virtue of an interaction with the α-helix between strands F and G in the A2-NH2 subdomains, where F536 and M539 are located. Because of the possible interaction of the FG helices in the two subdomains, residues 316 and 539 were selected for mutagenesis to the corresponding porcine residues. The human-to-porcine mutant cDNAs encoding Q316H, M539L and Q316H/M539L human fVIII were constructed and the corresponding expressed proteins were purified as described in “Experimental Procedures”.
Table 2. Neighboring amino acids in the A1-COOH and A2-NH2 subdomains of the human fVIII homology modela.
| Subdomain | |
|---|---|
| A1-COOH | A2-NH2 |
| F270 | Y476, P477, H478, G479 |
| E272 | Y476, F536 |
| G273 | Y476, D482 |
| T275 | G479, D519, R531 |
| F276 | E518 |
| N280 | T522, S524 |
| H281 | S524, D525, T522 |
| R282 | D519, G520, P521, T522, L529, L531 |
| A284 | H478, G479 |
| S285 | Y532 |
| M301 | E518 |
| D302 | D482, V483 |
| L303 | V483 |
| F306 | D482, V483 |
| F309 | F536, M539 |
| H311 | H478, F536, M539, L543 |
| I312 | A644 |
| S313 | M539, E540, L543, A544 |
| Q316 | M539, E540, L543 |
| H317 | E540, R583 |
| P333 | E518 |
| L335 | T481, T516, E518, D519 |
| R336 | T514, T516, V517, E518 |
Structural analysis of the fVIII homology model (23) was performed using DeepView (www.expasy.org/spdbv), version 3.7 (36). All inter-subdomain groups within 6 Å of each other are listed.
Shown in bold are residues that differ between human and porcine fVIII.
Fig. 2. Alignment of the A1-COOH and A2-NH2 subdomains of human and porcine fVIII.

A1-COOH and A2-NH2 subdomains are defined as residues 191-336 and 373-556, respectively, using human fVIII amino acid sequence numbering (38). Residues 337-372 (ar) correspond to an acidic region that is COOH-terminal of an activated protein C recognition site at R336. The ar segment is not paralogous to the fVIII A2 or A3 domains or orthologous to factor V or ceruloplasmin. Asterisks denote identical amino acids. Residues Q316 and M539 are marked by the closed circles. A, B, C, D, E, F and G represent β-sheets in the fVIII homology model (see Fig. 1B).
Decay Kinetics of FVIIIa Constructs
After the activation of fVIII at physiological concentrations (∼1 nM) by thrombin, the kinetics of fVIIIa decay are governed by the rate constant, k, for the dissociation of the A2 subunit from the A1/A3-C1-C2 dimer as shown in Scheme I (7;10). The decay kinetics of human fVIIIa, porcine fVIIIa and human fVIIIa mutants Q316H, M539L and Q316H/M539L were measured at 23 °C and 37 °C using a chromogenic substrate assay of the intrinsic fXase complex under conditions in which fVIIIa was the limiting function component as described in “Experimental Procedures”. FVIIIa decay rates were fit to a single exponential decay model using weighted nonlinear least-squares regression analysis. The experiment was performed twice for each construct and the results are summarized in Fig. 3 and Tables II. Good reproducibility of the estimates of the dissociation rate constant, k, were obtained as judged by inter-experimental values that were within the 95% confidence limits obtained from intra-experimental regression analysis in most cases. The decay of porcine fVIIIa was ∼3-fold slower than human fVIIIa at 23 °C and ∼2-fold slower at 37 °C, which is consistent with earlier results using plasma-derived or recombinant human and porcine fVIIIa (7;10). The decay rates at both temperatures of the Q316H and the Q316H/M539L mutants were significantly slower than human fVIIIa, but faster than porcine fVIIIa. In contrast, the decay of the M539L mutant was indistinguishable from human fVIIIa.
Scheme I.

Fig. 3. Decay rates of fVIIIa constructs.


Decays of human fVIIIa (●), porcine fVIIIa (■) and human fVIIIa mutants Q316H (○), M539L (▼) and Q316H/M539L (□) at 23 °C (A) or 37 °C (B) were measured at a fVIIIa starting concentration of 1 nM as described in “Experimental Procedures”. The curves are weighted nonlinear least-squares fits to a single exponential decay with the fitted parameters shown in Tables II, Experiment 1.
Discussion
The results of this study indicate that the interaction of the A2 subunit with the A1/A3-C1-C2 dimer in human fVIIIa involves Q316 in the FG helix region (Fig. 2) of the COOH-terminal cupredoxin-like A1 subdomain (Fig. 4). This residue was the only candidate residue identified by molecular modeling in the COOH-terminal A1 subdomain that might account for the differential decay rates of human and porcine fVIIIa (Table 2). Q316 is predicted to interact with M539 (Figure 4), where the corresponding porcine residue is a leucine. However, the M539L fVIIIa mutant decayed similarly to human fVIIIa unless combined with the Q316H mutation (Fig. 3 and Table 3). This result was not unexpected because substitution of the entire porcine A2 domain for the corresponding human domain produces a fVIIIa decay rate similar to human fVIIIa. The porcine Q316H substitution at this site in human fVIIIa produced a decay rate intermediate between porcine and human fVIIIa. This indicates that, although this site is involved in the A1 – A2 interactions, at least one other site is involved in the differential human-porcine fVIIIa decay rate. N280 was the only other candidate site identified in the COOH-terminal A1 subdomain by distance criteria, but is solvent exposed and not buried at the A1 – A2 interface. There presumably are structural rearrangements involving A domain interactions in the activation of factor VIII that may not be predicted by the homology model. Additionally, differences between human and porcine fVIII at site(s) remote from the A1 – A2 interface may drive subunit interactions, which would not be predicted using the homology model.
Fig. 4. Model of the interface of the fVIII COOH-terminal A1 and the NH2-terminal A2 subdomains.
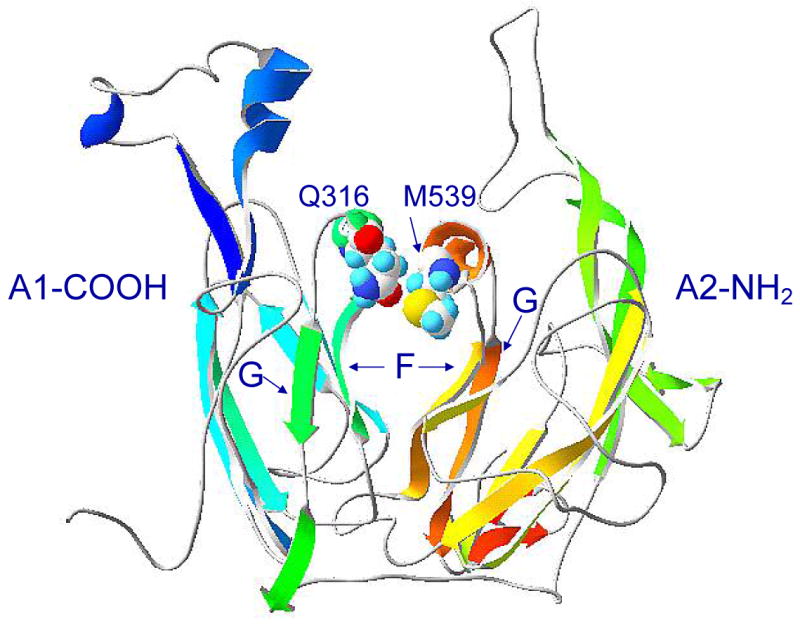
F and G represent the β-strands flanking the FG helices in the two subdomains.
Table 3. FVIIIa dissociation rate constants.
| k (min-1)a | |||
|---|---|---|---|
| Construct | Experiment | 23 °C | 37 °C |
| Human | 1 | 0.360 ± 0.063 | 0.539 ± 0.074 |
| 2 | 0.368 ± 0.051 | 0.788 ± 0.202 | |
| Porcine | 1 | 0.152 ± 0.024 | 0.329 ± 0.033 |
| 2 | 0.122 ± 0.012 | 0.366 ± 0.042 | |
| Q316H | 1 | 0.217 ± 0.045 | 0.400 ± 0.069 |
| 2 | 0.224 ± 0.044 | 0.432 ± 0.093 | |
| M539L | 1 | 0.406 ± 0.065 | 0.641 ± 0.084 |
| 2 | 0.378 ± 0.058 | 0.739 ± 0.143 | |
| Q316H/M539L | 1 | 0.275 ± 0.038 | 0.468 ± 0.042 |
| 2 | 0.277 ± 0.018 | 0.477 ± 0.051 | |
Parameter estimates and 95% confidence limits from weighted nonlinear regression analysis as described in “Experimental Procedures”.
The FG helices in the COOH-terminal A1 and NH2-terminal A2 subdomains consist of residues 314-317 and 539-544, respectively. Missense mutations have not been reported in the A1 subdomain FG helix. However, a missense mutation at R541 has been identified that produces mild hemophilia A (25). Additionally, there are several D542 mutations that produce severe hemophilia (26-29).
Several mutations in the fVIII gene result in abnormally fast A2 subunit dissociation and hemophilia A, including A1 mutations A284E and S289L at the A1-A2 and A1-A3 interfaces, respectively, A2 mutation R531H at the A1-A2 interface and A2 mutations N694I and R698L at the A2-A3 interface (9;30;31). The fast dissociation rates associated with the N694I and R698L mutations indicate that the A3 domain of fVIII interacts with the A2 subunit in fVIIIa. Substitution of the porcine A3 domain into human fVIIIa produces a decay rate similar to human fVIIIa (7). These results indicate that A3 residues important for the interaction with the A2 domain are conserved in human and porcine fVIII.
Elevated levels of fVIII are associated with an increased risk of arterial and venous thrombosis ((32), review). Although abnormally fast fVIIIa decay is associated with a bleeding diathesis, slow fVIIIa decay could tip hemostatic balance towards thrombosis. Like porcine fVIIIa, decay of murine fVIIIa is considerably slower than human fVIIIa (33). The relatively fast human A2 subunit dissociation rate may be an evolutionary adaptation associated with the development of a bipedal gait, an upright posture and venous pooling in the lower extremities. The acquisition of a bipedal gait, which separates humans from other mammals, including all other primates, is considered the signal event in human evolution, representing a preadaptation to tool making and the expansion of brain size.
Amino acid sequence variation between orthologous genes is due to random fixation of mutations with no fitness significance, purifying selection to eliminate deleterious mutations, or, more rarely, positive selection to retain advantageous mutations. Several proteins have been proposed to be under positive selection, including the primate Rh blood group, primate lysozymes, and numerous viral proteins (34). However, positive selection is difficult to detect because usually few amino acid sites are involved and must be detected against the large background of neutral and negative selection (35). Phylogenetic analysis of the fVIII gene combined with characterization of the decay rates of orthologous fVIIIa molecules will be necessary to test the hypothesis that A2 subunit dissociation is under positive selective pressure.
Acknowledgments
We thank Rachel T. Barrow for excellent technical assistance.
Abbreviations and Textual Footnotes
- fVIII
factor VIII
- fVIIIa
thrombin-activated fVIII
- BDD
B domain –deleted
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid)
- PCPS
phosphatidylcholine/phosphatidylserine
Footnotes
This work was supported by a grant from the National Institutes of Health (R01-HL40921, P.L.), by a Bayer Hemophilia Award to P.L. and by Hemophilia of Georgia, Inc.
References
- 1.Lollar P, Parker CG. Subunit structure of thrombin-activated porcine factor VIII. Biochemistry. 1989;28:666–674. doi: 10.1021/bi00428a038. [DOI] [PubMed] [Google Scholar]
- 2.Lollar P, Parker CG. pH-dependent denaturation of thrombin-activated porcine factor VIII. J Biol Chem. 1990;265:1688–1692. [PubMed] [Google Scholar]
- 3.Fay PJ, Haidaris PJ, Smudzin TM. Human factor VIIIa subunit structure: reconstitution of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991;266:8957–8962. [PubMed] [Google Scholar]
- 4.Hultin MB, Jesty J. The activation and inactivation of human factor VIII by thrombin: effect of inhibitors of thrombin. Blood. 1981;57:476–482. [PubMed] [Google Scholar]
- 5.Lollar P, Knutson GJ, Fass DN. Stabilization of thrombin-activated porcine factor VIII:C by factor IXa and phospholipid. Blood. 1984;63:1303–1308. [PubMed] [Google Scholar]
- 6.Lollar P, Knutson GJ, Fass DN. Activation of porcine factor VIII:C by thrombin and factor Xa. Biochemistry. 1985;24:8056–8064. doi: 10.1021/bi00348a033. [DOI] [PubMed] [Google Scholar]
- 7.Parker ET, Doering CB, Lollar P. A1 subunit-mediated regulation of thrombin-activated factor VIII A2 subunit dissociation. J Biol Chem. 2006;281:13922–13930. doi: 10.1074/jbc.M513124200. [DOI] [PubMed] [Google Scholar]
- 8.Lollar P, Parker ET. Structural basis for the decreased procoagulant activity of human factor VIII compared to the porcine homolog. J Biol Chem. 1991;266:12481–12486. [PubMed] [Google Scholar]
- 9.Pipe SW, Eickhorst AN, McKinley SH, Saenko EL, Kaufman RJ. Mild hemophilia A caused by increased rate of factor VIII A2 subunit dissociation: evidence for nonproteolytic inactivation of factor VIIIa in vivo. Blood. 1999;93:176–183. [PubMed] [Google Scholar]
- 10.Lollar P, Parker ET, Fay PJ. Coagulant properties of hybrid human/porcine factor VIII molecules. J Biol Chem. 1992;267:23652–23657. [PubMed] [Google Scholar]
- 11.Church WR, Jernigan RL, Toole JJ, Hewick RM, Knopf J, Knutson GJ, Nesheim ME, Mann KG, Fass DN. Coagulation factors V and VIII and ceruloplasmin constitute a family of structurally related proteins. Proc Natl Acad Sci U S A. 1984;81:6934–6937. doi: 10.1073/pnas.81.22.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziatseva I, Zaitsev V, Card G, Moshkov K, Bax B, Ralph A, Lindley P. The X-ray structure of human serum ceruloplasmin at 3.1 Angstroms: nature of the copper centres. JBIC. 1996;1:15–23. [Google Scholar]
- 13.Lollar P, Esmon CT. Involvement of thrombin anion-binding exosites 1 and 2 in the activation of human factor VIII. J Biol Chem. 1996;271:13882–13887. doi: 10.1074/jbc.271.23.13882. [DOI] [PubMed] [Google Scholar]
- 14.Barrow RT, Parker ET, Krishnaswamy S, Lollar P. Inhibition by heparin of the human blood coagulation intrinsic pathway factor X activator. J Biol Chem. 1994;269:26796–26800. [PubMed] [Google Scholar]
- 15.Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. High-level expression of recombinant porcine coagulation factor VIII. J Biol Chem. 2002;277:38345–38349. doi: 10.1074/jbc.M206959200. [DOI] [PubMed] [Google Scholar]
- 16.Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. Identification of porcine coagulation factor VIII domains responsible for high level expression via enhanced secretion. J Biol Chem. 2004;279:6546–6552. doi: 10.1074/jbc.M312451200. [DOI] [PubMed] [Google Scholar]
- 17.Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene splicing by overlap extension. Methods Enzymol. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- 18.Healey JF, Lubin IM, Nakai H, Saenko EL, Hoyer LW, Scandella D, Lollar P. Residues 484-508 contain a major determinant of the inhibitory epitope in the A2 domain of human factor VIII. J Biol Chem. 1995;270:14505–14509. doi: 10.1074/jbc.270.24.14505. [DOI] [PubMed] [Google Scholar]
- 19.Healey JF, Barrow RT, Tamim HM, Lubin IM, Shima M, Scandella D, Lollar P. Residues Glu2181-Val2243 contain a major determinant of the inhibitory epitope in the C2 domain of human factor VIII. Blood. 1998;92:3701–3709. [PubMed] [Google Scholar]
- 20.Barrow RT, Healey JF, Gailani D, Scandella D, Lollar P. Reduction of the antigenicity of factor VIII toward complex inhibitory plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood. 2000;95:557–561. [PubMed] [Google Scholar]
- 21.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duffy EJ, Lollar P. Intrinsic pathway activation of factor X and its activation peptide-deficient derivative, factor X(Des 143-191) J Biol Chem. 1992;267:7821–7827. [PubMed] [Google Scholar]
- 23.Pemberton S, Lindley P, Zaitsev V, Card G, Tuddenham EGD, Kemball-Cook G. A molecular model for the triplicated A domains of human factor VIII based on the crystal structure of human caeruloplasmin. Blood. 1997;89:2413–2421. [PubMed] [Google Scholar]
- 24.Adams TE, Hockin MF, Mann KG, Everse SJ. The crystal structure of activated protein C-inactivated bovine factor Va: Implications for cofactor function. Proc Natl Acad Sci U S A. 2004;101:8918–8923. doi: 10.1073/pnas.0403072101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Murphy ME, Thompson AR. A domain mutations in 65 haemophilia A families and molecular modelling of dysfunctional factor VIII proteins. Br J Haematol. 1998;103:1051–1060. doi: 10.1046/j.1365-2141.1998.01122.x. [DOI] [PubMed] [Google Scholar]
- 26.Higuchi M, Kazazian HH, Jr, Kasch L, Warren TC, McGinness MJ, Phillips JA, III, Kasper C, Janco R, Antonarakis SE. Molecular characterization of severe hemophilia A suggests that about half of the mutations are not within the coding region and splicing junctions of the factor VIII gene. Proc Natl Acad Sci U S A. 1991;88:7405–7409. doi: 10.1073/pnas.88.16.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGinniss MJ, Kazazian HH, Jr, Hoyer LW, Bi L, Inaba H, Antonarakis SE. Spectrum of mutations in CRM-positive and CRM-reduced hemophilia A. Genomics. 1993;15:392–398. doi: 10.1006/geno.1993.1073. [DOI] [PubMed] [Google Scholar]
- 28.Waseem NH, Bagnall R, Green PM, Giannelli F. Start of UK confidential haemophilia A database: analysis of 142 patients by solid phase fluorescent chemical cleavage of mismatch. Haemophilia Centres. Thromb Haemost. 1999;81:900–905. [PubMed] [Google Scholar]
- 29.Bogdanova N, Markoff A, Pollmann H, Nowak-Gottl U, Eisert R, Wermes C, Todorova A, Eigel A, Dworniczak B, Horst J. Spectrum of molecular defects and mutation detection rate in patients with severe hemophilia A. Human Mutation. 2005;26:249–254. doi: 10.1002/humu.20208. [DOI] [PubMed] [Google Scholar]
- 30.Pipe SW, Saenko EL, Eickhorst AN, Kemball-Cook G, Kaufman RJ. Hemophilia A mutations associated with 1-stage/2-stage activity discrepancy disrupt protein-protein interactions within the triplicated A domains of thrombin-activated factor VIIIa. Blood. 2001;97:685–691. doi: 10.1182/blood.v97.3.685. [DOI] [PubMed] [Google Scholar]
- 31.Hakeos WH, Miao H, Sirachainan N, Kemball-Cook G, Saenko EL, Kaufman RJ, Pipe SW. Hemophilia A mutations within the factor VIII A2-A3 subunit interface destabilize factor VIIIa and cause one-stage/two-stage activity discrepancy. Thromb Haemost. 2002;88:781–787. [PubMed] [Google Scholar]
- 32.Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21:731–738. doi: 10.1161/01.atv.21.5.731. [DOI] [PubMed] [Google Scholar]
- 33.Doering CB, Parker ET, Healey JF, Craddock HN, Barrow RT, Lollar P. Expression and characterization of recombinant murine factor VIII. Thromb Haemost. 2002;88:450–458. [PubMed] [Google Scholar]
- 34.Yang Z. Computational Molecular Evolution. Oxford University Press; Oxford: 2006. Neutral and adaptive protein evolution; pp. 259–292. [Google Scholar]
- 35.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology & Evolution. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 36.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 37.Gough J, Chothia C. The linked conservation of structure and function in a family of high diversity: the monomeric cupredoxins. Structure. 2004;12:917–925. doi: 10.1016/j.str.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 38.Vehar GA, Keyt B, Eaton D, Rodriguez H, O'Brien DP, Rotblat F, Oppermann H, Keck R, Wood WI, Harkins RN, et al. Structure of human factor VIII. Nature. 1984;312:337–342. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]