

Abstract
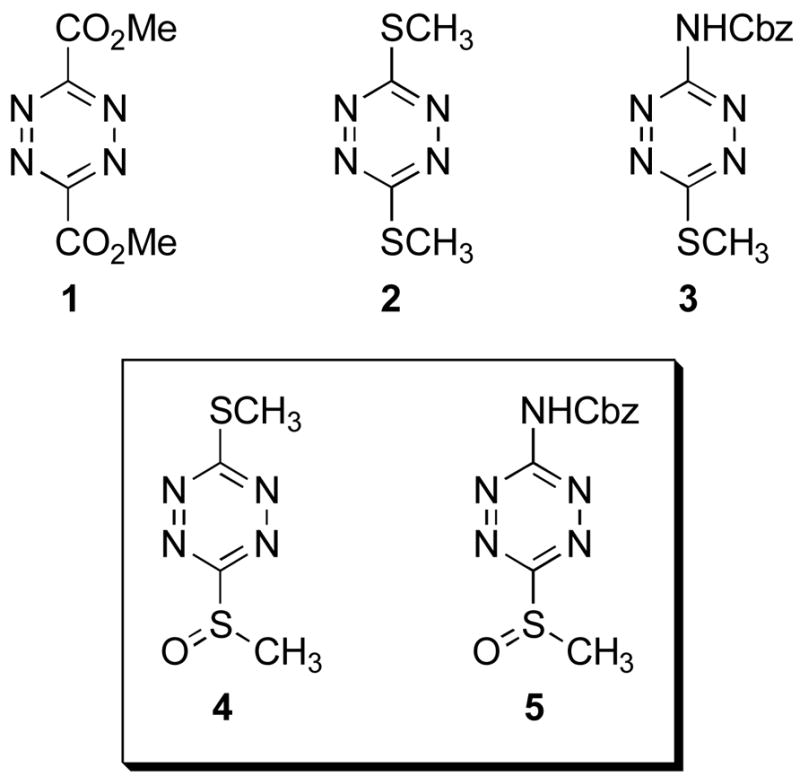
Two new unsymmetrical 1,2,4,5-tetrazines, 3-methylsulfinyl-6-methylthio-1,2,4,5-tetrazine (4) and 3-(benzyloxycarbonyl)amino-6-methylsulfinyl-1,2,4,5-tetrazine (5), were prepared and scope of their participation in intermolecular inverse electron demand Diels–Alder reactions defined. As anticipated, sulfoxides 4 and 5 (4 > 5) display a reactivity that is substantially greater than that of their corresponding sulfides (2 and 3) being derived from their enhanced electron-deficient character and resulting in a wider range of potential dienophile choices or the use of milder reaction conditions. The cycloaddition reactions were expectedly regioselective typically producing a single cycloadduct ensuring their synthetic utility, but both were found to proceed with a regioselectivity opposite what would be anticipated and complementary to that observed with 2 and 3.
Introduction
Electron-deficient heterocyclic azadienes have proven to be useful reagents that often participate in well-defined inverse electron demand Diels–Alder reactions with electron-rich dienophiles providing rapid access to a range of highly substituted heterocyclic systems.1 Of these, the substituted 1,2,4,5-tetrazines are the most reactive and most widely utilized heterocyclic azadienes. Typically, symmetrical 1,2,4,5-tetrazines are employed largely because of their synthetic accessibility, and synthetic studies of their utility have necessarily focused only on their relative reactivities.
In the course of our investigations of such reagents and their applications in complex natural products total synthesis,2–16 we have examined a number of such tetrazines17,18 and introduced several new, useful symmetrical19 or unsymmetrical20,21 1,2,4,5-tetrazines. Of these, dimethyl 1,2,4,5-tetrazine-3,6-dicarboxylate (1)17 and 3,6-bis(thiomethyl)-1,2,4,5-tetrazine (2)18 have been the most widely utilized of the symmetrical tetrazines, and the N-acyl 3-amino-6-methylthio-1,2,4,5-tetrazines (e.g., 3)21 have proven to be the most widely explored of the unsymmetrical tetrazines participating in well-behaved, effective and regioselective [4+2] cycloaddition reactions. Herein, we report the preparation of two new and useful unsymmetrical 1,2,4,5-tetrazines, 3-methylsulfinyl-6-methylthio-1,2,4,5-tetrazine (4) and 3-(benzyloxycarbonyl)amino-6-methysulfinyl-1,2,4,5-tetrazine (5), obtained by S-oxidation of 2 and 3, respectively, and describe studies defining the scope of their Diels–Alder reactions, Figure 1. As anticipated, both 4 and 5 (4 > 5) display a reactivity that is greater than that of either 2 or 3 being derived from their enhanced electron-deficient character resulting in wider range of potential dienophile choices and/or the use of milder reaction conditions for the [4+2] cycloaddition reactions. Moreover, the cycloaddition reactions were expectedly regioselective typically producing a single cycloadduct ensuring their synthetic utility. Remarkably, this regioselectivity proved opposite what one would anticipate based on simple zwitterionic models or more sophisticated FMO analysis of the [4+2] cycloaddition reactions.
Figure 1.
Results and Discussion
Preparation of 1,2,4,5-Tetrazines 4 and 5
To our knowledge, there have been only two reports of the preparation of sulfoxide substituted 1,2,4,5-tetrazines22,23 and only one of these examined their [4+2] cycloaddition reactivity.23 In these latter studies, only their intramolecular [4+2] cycloaddition reaction with tethered unactivated alkynes was examined and no studies of their intermolecular Diels–Alder reactions have been disclosed.23 Both were prepared by oxidation of the corresponding thioether enlisting either the DABCO–Br2 complex24 or oxone.23 The former proved effective for selective oxidation of 2 to provide 4 as a blood red crystalline solid in good yield (0.55 equiv of DABCO, 1.1 equiv of Br2, HOAc–H2O–CH2Cl2, 25 °C, 20 h, 52%) along with small amounts of remaining 2, Scheme 1. Increasing the amount of oxidant increased the conversion without further increasing the yield of 4, likely due to competing over oxidation, and the use of m-CPBA (1.1 equiv) also provided 4, but in lower yield. In either case, the over oxidation led to unidentified water soluble byproducts easily removed in the workup. Tetrazine 4, like 1, is not stable to prolonged exposure to silica gel required of a purification, but can be isolated in pure form by washing the crude reaction product with Et2O/hexane to remove small amounts of unreacted 2 and subsequently recrystallized from EtOAc/hexane (mp 73–74 °C).
Scheme 1.
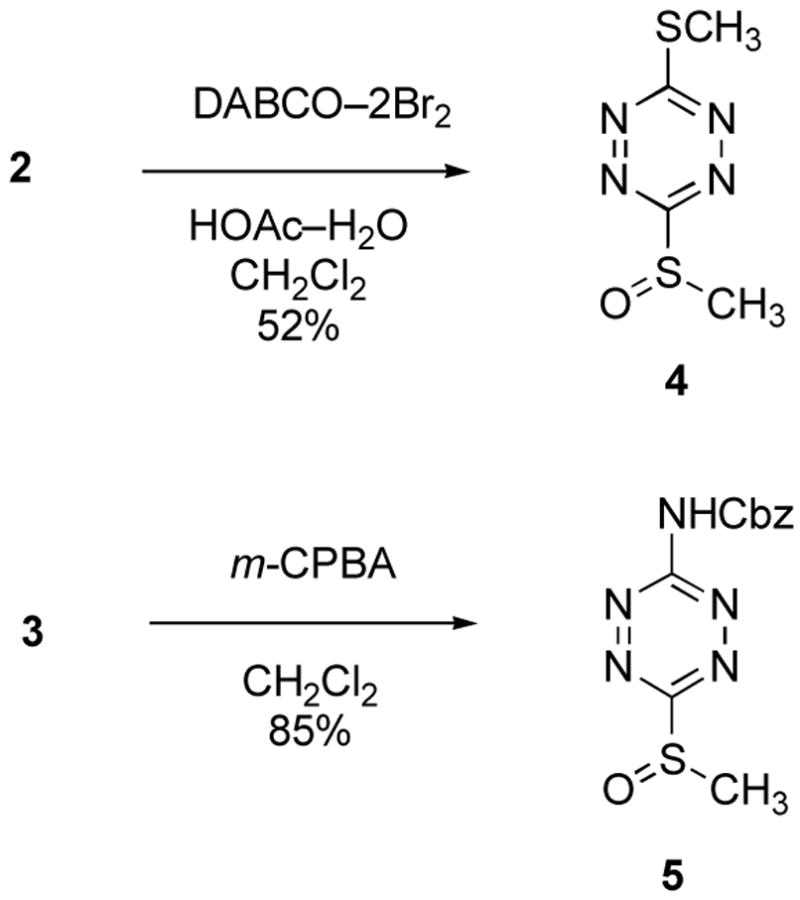
Tetrazine 3 proved essentially unreactive toward DABCO–Br2 under these conditions, but was oxidized to the corresponding sulfoxide with m-CPBA (1.1 equiv, CH2Cl2, 0 °C, 30 min, 85%). Like 1 and 4, 5 was not sufficiently stable to prolonged exposure to silica gel to permit purification by chromatography. However, aqueous workup of the oxidation reaction including an extraction with saturated aqueous NaHCO3 to remove reagent and m-chlorobenzoic acid provided tetrazine 5 as a blood red oil sufficiently pure (>95%) for [4+2] cycloaddition studies.
Diels–Alder Reactions of 4
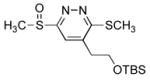
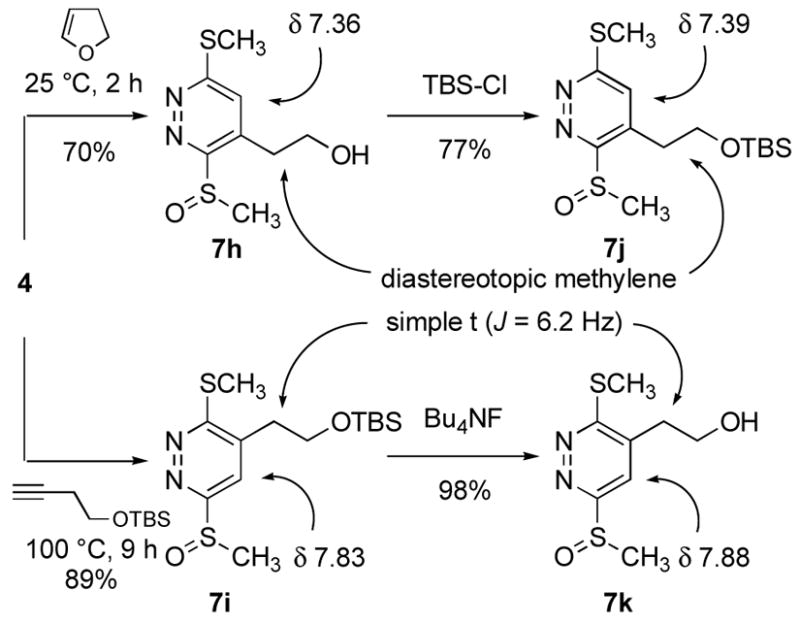
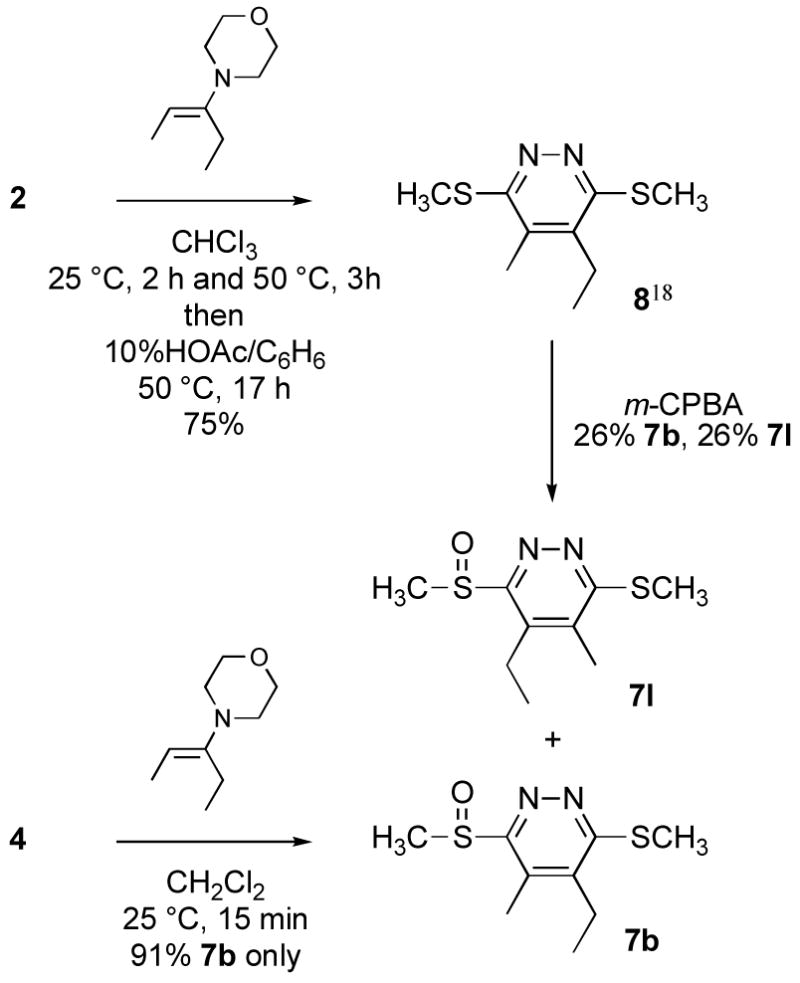
The tetrazine 4 exhibited superb reactivity in prototypical inverse electron demand Diels–Alder reactions and was much more reactive than the corresponding sulfide 2,18 Table 1. The reaction of 4 with enamines was essentially instantaneous at 25 °C and other electron-rich dienophiles including ketene acetals, enol ethers, and enamides readily react smoothly at room temperature to cleanly provide the [4+2] cycloadducts. Notably, no problematic detection of intermediate unaromatized product was observed and the Diels–Alder products were isolated in uniformly high yields. Remarkably, even unactivated dienophiles including phenylacetylene (6h) and alkyne 6k reacted smoothly with 4, albeit slowly at room temperature (ca. 24–48 h), requiring higher reaction temperatures for rapid reaction (100 °C, 1–9 h, 80–90%). As such, tetrazine 4, by virtue of its enhanced electron-deficient character, exhibits a reactivity that accommodates an unusually wide range of potential dienophiles. Moreover, the [4+2] cycloaddition reactions were regioselective typically providing a single detectable product. The only exception to this generalization was dihydrofuran (Table 1, entry 10) where a trace of the second regioisomer (5–11%) was detected. Unexpectedly, this regioselectivity proved to be opposite what one would predict from simple zwitterionic models or FMO analysis of the [4+2] cycloaddition reaction. Although this became apparent in assessing the spectroscopic properties of the cycloaddition products (e.g., 1,2-diazine C4-H vs C5-H chemical shift), single crystal X-ray structures of 7c, 7f and 7g unambiguously established their structures, Figure 2.25 Similarly, dienophiles 6j and 6k provided regioisomeric products clearly distinguishable as the C4-substituted (C5-H δ 7.36) and C5-substituted (C4-H δ 7.88) 1,2-diazines with the latter alkyne regioselectivity being consistent with that observed with phenylacetylene (X-ray), Scheme 2. The structure of cycloadduct 7b, which lacks the distinguishing aryl CH, was confirmed by comparison of its spectroscopic properties with that of the two possible cycloadducts (7b vs 7l) with 7l, but not 7b, exhibiting a characteristic diastereotopic methylene adjacent to the sulfoxide substituent similarly observed with 7h vs 7k, Scheme 3. As such, tetrazine 4 exhibits a very useful Diels–Alder reactivity accommodating an unusually wide range of potential dienophiles, and proceeds with a reaction regioselectivity opposite what one would predict.
Table 1.
[4+2] Cycloaddition Reactions of 4.
| entry | dienophile (equiv) | conditions | product | % yield | ||
|---|---|---|---|---|---|---|
| 1 | 6a |
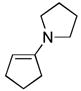 (1.5) (1.5) |
CH2Cl2, 25 °C, 1 min | 7a |
|
96% |
| 2 | 6b26 |
 (2) (2) |
CH2Cl2, 25 °C, 15 min | 7b |
|
91% |
| 3 | 6c |
 (10) (10) |
dioxane, 25 °C, 30 min | 7c |
|
85% |
| 4 | 6d |
|
dioxane, 25 °C, 30 min | 7d |
|
96% |
| 5 | 6e |
 (1.5) (1.5) |
CH2Cl2, 25 °C, 16 h | 7d | 66% | |
| 6 | 6f |
 (10) (10) |
CH2Cl2, 25 °C, 2 h | 7e |
|
91% |
| 7 | 6g |
 (5) (5) |
CH2Cl2, 25 °C, 1 h | 7f |
|
94% |
| 8 | 6h |
|
dioxane, 100 °C, 1 h or dioxane, 25 °C, 24 h or dioxane, 25 °C, 48 h | 7f | 90%
54% 72% |
|
| 9 | 6i27 |
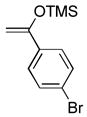 (2) (2) |
CH2Cl2, 25 °C, 2 h | 7g |
|
89% |
| 10 | 6j |
|
CH2Cl2, 25 °C, 2 h | 7h |
|
70% (5–11% regioisomer) |
| 11 | 6k |
|
dioxane, 100 °C, 9 h | 7i |
|
89% |
Figure 2.
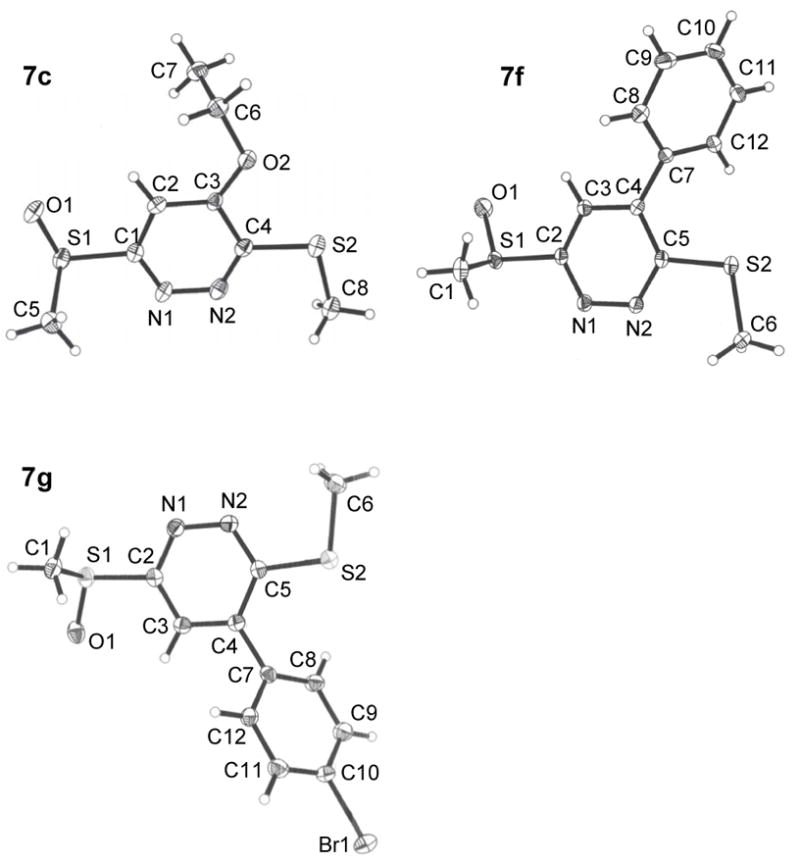
ORTEP drawings of 7c, 7f and 7g.
Scheme 2.
Scheme 3.
Diels–Alder Reactions of 5
Given the useful, but unexpected observations with tetrazine 4, an analogous but less extensive study of the [4+2] cycloaddition reactions of tetrazine 5 was conducted, Table 2. As anticipated, the reactivity of 5, by virtue of its enhanced electron-deficient character, substantially exceeded that of 3. Not only did 5 react with enamines, ketene acetals, and enol ethers rapidly and effectively at room temperature, but even the unactivated dienophile phenylacetylene (6h) provided the [4+2] cycloadduct 9f in excellent conversion (77%) at 25 °C requiring only 24 h for complete reaction. Although tetrazine 5 was slightly less reactive than 4, requiring slightly longer reaction times and providing somewhat lower conversions, it was also found to exhibit a superb [4+2] cycloaddition reactivity. Similarly, the regioselectivity of the [4+2] cycloaddition reactions was often excellent although not always as clean as that observed with 3 or 4 and, like that of 4, was found to be opposite that anticipated. This was first evident upon examination of the spectroscopic properties of the products and confirmed by X-ray for 9c25 or by correlation with the alternative regioisomeric products available through S-oxidation of the analogous cycloadducts derived from tetrazine 3, Scheme 4. These latter studies not only further verified that tetrazines 3 and 5 proceed with opposite regioselectivities in the [4+2] cycloaddition reactions and, more surprisingly, that it is the regioselectivity of 5 that is opposite what one might predict, but they also illustrate that the analogous reactions of 3 require much more vigorous reaction conditions to conduct.
Table 2.
[4+2] Cycloaddition Reactions of 5.
| entry | dienophile (equiv) | conditions | product | % yield | ||
|---|---|---|---|---|---|---|
| 1 | 6a |
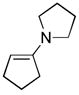 (1.5) (1.5) |
CH2Cl2, 25 °C, 5 min | 9a |
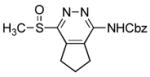
|
77% |
| 2 | 6b26 |
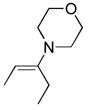 (2) (2) |
CH2Cl2, 25 °C, 15 min | 9b |
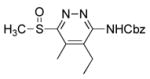
|
29% (10% regioisomer) |
| 3 | 6c |
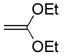 (10) (10) |
dioxane, 25 °C, 30 min | 9c |
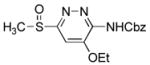
|
47–34% (17% regioisomer) |
| 4 | 6d |
|
dioxane, 25 °C, 1 h | 9d |
|
75% |
| 5 | 6f |
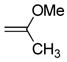 (10) (10) |
CH2Cl2, 25 °C, 6 h | 9e |
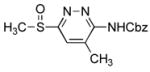
|
58% (7% regioisomer) |
| 6 | 6g |
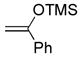 (5) (5) |
CH2Cl2, 25 °C, 2 h | 9f |
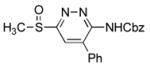
|
83% |
| 7 | 6h |
|
dioxane, 25 °C, 24 h | 9f | 77% | |
| 8 | 6i27 |
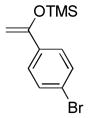 (2) (2) |
dioxane, 25 °C, 6 h | 9g |
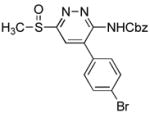
|
69% |
Scheme 4.
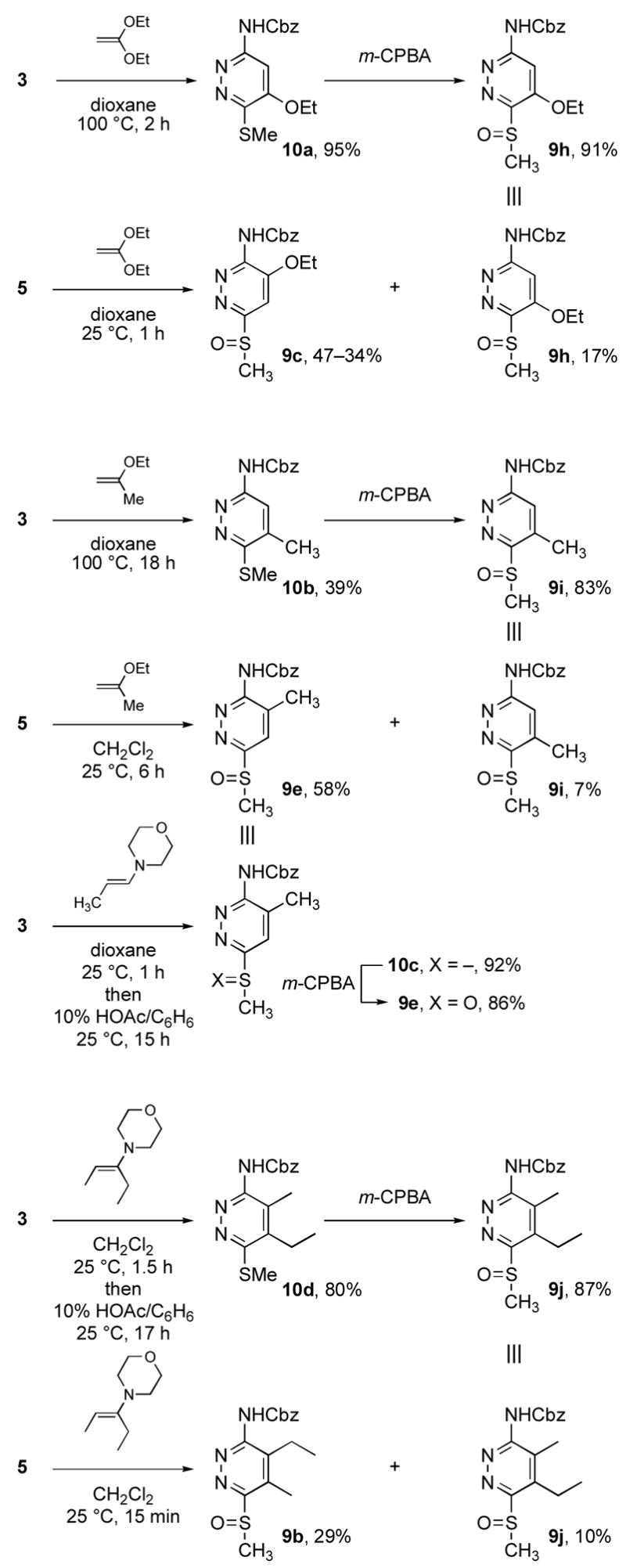
Reactivity and Regioselectivity
The regioselectivity of the cycloadditions is not consistent with the expectation that the methylsulfinyl group would control the reaction orientation by stabilizing a partial negative charge at C3 (Table 3). The dienophile addition does not follow an approach predicted by this stabilization and the complementary ability of the thiomethyl or acylamino group to stabilize a partial positive charge on C6. These intuitive predictions are supported by AM1 and MNDO computational studies where C3 of both 4 and 5 bear a significant partial negative charge while C6 is more electropositive. Moreover, C6 bears the largest LUMO orbital coefficient indicating it should dominate the regioselectivity by preferentially combining with the dienophile C2 center which possesses its largest HOMO orbital coefficient. Thus, both 4 and 5 experimentally display a [4+2] cycloaddition reaction regioselectivity opposite what one would predict based on simple zwitterionic models or FMO analysis of the reactions. Only the LUMO energy levels of the FMO analysis accurately reflect the increased reactivity of 4 and 5 (4 > 5) relative to 2 and 3.
Table 3.
AM1 Computational Results (LUMO) of 1,2,4,5-Tetrazines.
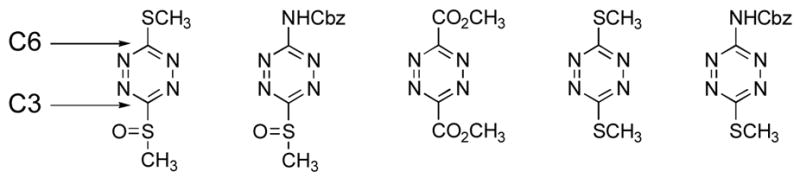 | |||||
|---|---|---|---|---|---|
| 4 | 5a | 1 | 2 | 3a | |
| LUMO (EV) | −1.871 | −1.607 | −2.149 | −1.365 | −1.364 |
| C-3 coefficient | 0.530 | 0.517 | 0.591 | 0.584 | 0.549 |
| C-6 coefficient | 0.617 | 0.625 | 0.591 | 0.584 | 0.596 |
| C-3 net charge | −0.571 | −0.586 | −0.102 | −0.277 | −0.301 |
| C-6 net charge | −0.283 | 0.063 | −0.102 | −0.277 | 0.022 |
Cbz group was replaced with CO2CH3 for the calculations
Consequently, the origin of the reversed regioselectivity is not clear. It is possible that this is related to a destabilizing steric and/or electronic interaction of the dienophile substituents with the larger and more electronegative methylsulfinyl substituent (e.g. destabilizing -NR2/CH3SO- interaction). In part, this may explain the lower regioselectivity typically observed with 5 versus 4 including the relative behavior seen in the reaction of 5 with the terminally substituted dienophile 6b. However, it is also interesting to note that treatment of tetrazine 4 (Et2NH, THF, 0 to 25 °C) or the cycloadducts 7c, 7d and 7e (CH3ONa, CH3OH, 25–70 °C) with nucleophiles only provided products derived from displacement of the methylsulfinyl group and not the methylsulfide. Consequently, it is also possible that the reactions proceed by stepwise addition–cyclization reactions initiated by an analogous nucleophilic addition. Although potentially reasonable for the nucleophilic dienophiles examined, no intercepted simple addition–elimination products (no cyclization) were detected and such a stepwise reaction course is unlikely for the unactivated alkynes examined.
Conclusions
The unsymmetrical 1,2,4,5-tetrazines 4 and 5 participate in well-defined and regioselective inverse electron demand Diels–Alder reactions with a wide range of electron-rich and unactivated dienophiles providing the corresponding 1,2-diazines in excellent yields. As anticipated, their enhanced electron-deficient character relative to 2 and 3 provide dienes that react faster and/or under milder reaction conditions and with a wider range of potential dienophiles. The cycloadditions are regioselective, albeit providing products opposite what is predicted using simple zwitterionic models or FMO analysis of the [4+2] cycloaddition reaction.
Experimental Section
3-Methylsulfinyl-6-methylthio-1,2,4,5-tetrazine (4)
3,6-Bis(methylthio)tetrazine (2, 500 mg, 2.87 mmol) was dissolved in 16 mL of a 5:2:1 mixture of HOAc, H2O and CH2Cl2. DABCO–2Br2 complex (681 mg, 1.58 mmol) was added and the mixture was stirred at room temperature for 20 h. Water was added and the mixture extracted with CH2Cl2, dried (MgSO4) and evaporated. The crude material was washed with Et2O/hexane (1:1, 3×) providing pure tetrazine 4 (286 mg, 52% yield) as a red solid. An analytically pure sample of 4 was obtained by recrystallization from EtOAc/hexane: mp 73–74 °C (EtOAc/hexane); 1H NMR (CDCl3, 500 MHz) δ 3.18 (s, 3H), 2.81 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 178.9, 173.0, 40.1, 13.6; IR (film) νmax 1382, 1242, 1122, 1078, 1049, 961, 892 cm−1; HRMS (MALDI-FTMS) m/z 212.9878 (M+Na+, calculated 212.9875).
6-(Benzyloxycarbonyl)amino-3-(methylsulfinyl)-1,2,4,5-tetrazine (5)
6-(Benzyloxycarbonyl)-amino-3-methylthio-1,2,4,5-tetrazine (3, 100 mg, 0.36 mmol) was dissolved in 4 mL of CH2Cl2 and cooled to 0 °C. m-CPBA (68.5 mg, 0.397 mmol) was added and the mixture was stirred at 0 °C for 30 min. Saturated aqueous NaHCO3 was added and the mixture was extracted with CH2Cl2, dried (MgSO4), and evaporated to give 90 mg (85%) of essentially pure tetrazine 5 (> 95%) as a red foam: 1H NMR (CDCl3, 400 MHz) δ 8.91 (br s, 1H), 7.45–7.35 (m, 5H), 5.34 (s, 2H), 3.16 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 171.9, 160.7, 150.4, 134.6, 128.6, 128.5 (2C), 128.4 (2C), 68.5, 39.6; IR (film) νmax 3196, 3031, 1759, 1557, 1471, 1279, 1211, 1179, 1059, 964, 927, 742 cm−1; HRMS (ESI-TOF) m/z 316.0466 (M+Na+, calculated 316.0475).
General procedure for cycloadditions
Tetrazine 4 or 5 was dissolved in the reaction solvent (Table 1 and 2), the dienophile was added at room temperature and the mixture was stirred at the indicated temperature for the indicated time. After completion of the reaction, the solvent was removed and the crude material was purified by chromatography on silica gel.
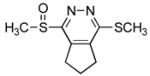
1-(Methylsulfinyl)-4-(methylthio)-6,7-dihydro-5H-cyclopenta[d]pyridazine (7a)
10 mg of 4 yielded 11.5 mg of 7a (96%, white solid) after chromatography (0–5% MeOH/EtOAc): mp 92–93 °C (EtOAc); 1H NMR (CDCl3, 400 MHz) δ 3.44–3.30 (m, 2H), 3.07 (s, 3H), 2.83 (t, J = 7.7 Hz, 2H), 2.77 (s, 3H), 2.27–2.16 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 161.7, 160.4, 145.3, 142.3, 39.3, 30.3, 30.1, 23.3, 12.7; IR (film) νmax 1541, 1427, 1291, 1191, 1058, 964 cm−1; HRMS (MALDI-FTMS) m/z 229.0466 (M+H+, calculated 229.0464).
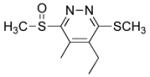
5-Ethyl-4-methyl-3-(methylsulfinyl)-6-(methylthio)pyridazine (7b)
20 mg of 4 yielded 22 mg of 7b (91%, pale yellow viscous oil) after preparative TLC (EtOAc): 1H NMR (CDCl3, 400 MHz) δ 3.11 (s, 3H), 2.74 (q, J = 7.5 Hz, 2H), 2.71 (s, 3H), 2.60 (s, 3H), 1.18 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 164.6, 160.4, 141.3, 134.6, 37.1, 21.8, 13.7, 12.7, 11.2; IR (film) νmax 3460, 2972, 2926, 1715, 1652, 1538, 1057 cm−1; HRMS (ESI-TOF) m/z 231.0620 (M+H+, calculated 231.0620).
5-Ethoxy-3-(methylsulfinyl)-6-(methylthio)pyridazine (7c)
10 mg of 4 yielded 10.4 mg of 7c (85%, white solid) after chromatography (0–2% MeOH/EtOAc gradient elution). A single-crystal X-ray structure determination25 conducted on crystals grown from benzene unambiguously established the structure of 7c: mp 111.5–112.5 °C (benzene); 1H NMR (CDCl3, 400 MHz) δ 7.31 (s, 1H), 4.31 (q, J = 7 Hz, 2H), 2.98 (s, 3H), 2.69 (s, 3H), 1.54 (t, J = 7 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.6, 157.0, 156.2, 99.5, 65.5, 41.8, 14.0, 12.6; IR (film) νmax 1556, 1353, 1193, 1058, 1033 cm−1; HRMS (MALDI-FTMS) m/z 233.0412 (M+H+, calculated 233.0413).
3-(Methylsulfinyl)-6-(methylthio)pyridazine (7d)
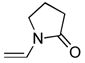
From ethyl vinyl ether (6d): 10 mg of 4 yielded 9.5 mg of 7d (96%, white solid) after chromatography (EtOAc). From vinyl pyrrolidinone (6e): 10 mg of 4 afforded 6.5 mg of 7d (66%) after chromatography (EtOAc): mp 115–117 °C (EtOAc/hexane, lit28 mp 118–119 °C); 1H NMR (CDCl3, 500 MHz) δ 7.91 (d, J = 8.8 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H), 2.98 (s, 3H), 2.76 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.1, 164.6, 127.4, 121.3, 41.7, 13.4; IR (film) νmax 1557, 1390, 1156, 1053, 974, 854 cm−1; HRMS (MALDI-FTMS) m/z 189.0153 (M+H+, calculated 189.0151).
5-Methyl-3-(methylsulfinyl)-6-(methylthio)pyridazine (7e)
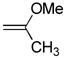
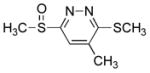
10 mg of 4 yielded 9.6 mg of 7e (91%, white solid) after chromatography (EtOAc): mp 106–107 °C (EtOAc/hexane); 1H NMR (CDCl3, 300 MHz) δ 7.74 (s, 1H), 2.95 (s, 3H), 2.74 (s, 3H), 2.36 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.4, 164.8, 138.5, 121.0, 41.7, 18.5, 13.4; IR (film) νmax 1558, 1347, 1062 cm−1; HRMS (MALDI-FTMS) m/z 203.0308 (M+H+, calculated 203.0307).
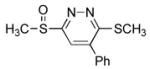
3-(Methylsulfinyl)-6-(methylthio)-5-phenylpyridazine (7f)
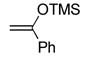
From 1-phenyl-1-(trimethylsilyloxy)ethylene (6g): 10 mg of 4 yielded 13 mg of 7f (94%, white solid) after chromatography (60% EtOAc/hexane). From phenylacetylene (6h): 50 mg of 4 yielded 63 mg of 7f (90%) after chromatography (60% EtOAc/hexane). A single-crystal X-ray structure determination25 conducted on crystals grown from acetone/H2O unambiguously established the structure of 7f: mp 143.2–143.8 °C (acetone/H2O); 1H NMR (CDCl3, 400 MHz) δ 7.81 (s, 1H), 7.50 (s, 5H), 3.02 (s, 3H), 2.70 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.0, 163.5, 141.4, 134.3, 130.1, 128.9 (2C), 128.4 (2C), 120.7, 41.7, 14.3; IR (film) νmax 1344, 1193, 1059 cm−1; HRMS (MALDI-FTMS) m/z 265.0466 (M+H+, calculated 265.0464).
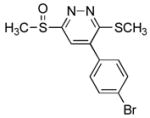
5-(4′-Bromophenyl)-3-(methylsulfinyl)-6-(methylthio)pyridazine (7g)
30 mg of 4 yielded 48 mg of 7g (89%, white solid) after chromatography (50–100% EtOAc/hexane). A single-crystal X-ray structure determination25 conducted on crystals grown from acetone/H2O unambiguously established the structure of 7g: mp 149–150 °C (acetone/H2O); 1H NMR (CDCl3, 500 MHz) δ 7.79 (s, 1H), 7.64 (d, J = 8.8 Hz, 2H), 7.40 (d, J = 8.8 Hz, 2H), 3.02 (s, 3H), 2.70 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.1, 163.1, 140.1, 133.0, 132.2 (2C), 130.0 (2C), 124.6, 120.6, 41.7, 14.2; IR (film) νmax 1487, 1418, 1328, 1142, 1063, 1009, 843, 821, 753 cm−1; HRMS (ESI–TOF) m/z 342.9571 (M+H+, calculated 342.9569).
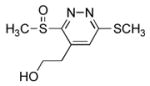
4-(2′-Hydroxyethyl)-3-(methylsulfinyl)-6-(methylthio)pyridazine (7h)
10 mg of 4 yielded 8.5 mg of 7h (70%, colorless oil) after chromatography (0–10% MeOH/EtOAc). The regioisomer (7k) was isolated as a minor product (5–11%): 1H NMR (CDCl3, 500 MHz) δ 7.36 (s, 1H), 4.04–3.98 (m, 1H), 3.87–3.82 (m, 1H), 3.37–3.31 (m, 1H), 3.18 (s, 3H), 3.13–3.08 (m, 1H), 2.74 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.1, 161.7, 139.7, 128.4, 61.7, 38.9, 32.8, 13.3; IR (film) νmax 3383, 1415, 1353, 1193, 1057 cm−1; HRMS (MALDI-FTMS) m/z 233.0412 (M+H+, calculated 233.0413).
5-(2-(tert-Butyldimethylsilyloxy)ethyl)-3-(methylsulfinyl)-6-(methylthio)pyridazine (7i)
10 mg of 4 yielded 16 mg of 7i (89%, colorless oil) after chromatography (40% EtOAc/hexane): 1H NMR (CDCl3, 500 MHz) δ 7.83 (s, 1H), 3.96 (t, J = 6.2 Hz, 2H), 2.95 (s, 3H), 2.90 (t, J = 6.2 Hz, 2H), 2.76 (s, 3H), 0.84 (s, 9H), 0.00 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 165.5, 164.4, 139.6, 121.2, 59.6, 41.7, 35.0, 25.7, 18.1, 13.6, –5.4; IR (film) νmax 2928, 1353, 1256, 1096, 1066, 837, 777 cm−1; HRMS (MALDI-FTMS) m/z 347.1274 (M+H+, calculated 347.1278).
4-(2-(tert-Butyldimethylsilyloxy)ethyl)-3-(methylsulfinyl)-6-(methylthio)pyridazine (7j)
A solution of 7h (7 mg, 0.03 mmol) in DMF (300 μL) was treated with imidazole (3.4 mg, 0.05 mmol) and TBSCl (6.7 mg, 0.045 mmol). The mixture was stirred for 3 h at room temperature before being diluted with EtOAc and washed with water. Preparative TLC (50% EtOAc/hexane) afforded 8 mg (77%) of 7j as a colorless oil: 1H NMR (CDCl3, 500 MHz) δ 7.39 (s, 1H), 3.98–3.90 (m, 2H), 3.27–3.22 (m, 1H), 3.14 (s, 3H), 3.12–3.09 (m, 1H), 2.74 (s, 3H), 0.84 (s, 9H), –0.01 (s, 3H), –0.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 164.7, 161.0, 139.3, 128.5, 61.6, 37.9, 33.0, 25.7 (3C), 18.1, 13.2, –5.5 (2C); IR (film) νmax 2927, 1566, 1360, 1256, 1090, 1065, 836, 777 cm−1; HRMS (MALDI-FTMS) m/z 347.1279 (M+H+, calculated 347.1278).
5-(2′-Hydroxyethyl)-3-(methylsulfinyl)-6-(methylthio)pyridazine (7k)
A solution of 7i (5 mg) in 100 μL of THF was treated with Bu4NF (1.0 M in THF, 30μL, 2 equiv) at room temperature and the mixture was stirred at 25 °C for 1 h. Chromatography (5% MeOH/EtOAc) afforded 3.3 mg of 7k (98%, white solid): mp 110–112 °C (EtOAc); 1H NMR (CDCl3, 400 MHz) δ 7.88 (s, 1H), 4.06 (t, J = 6.2 Hz, 2H), 2.99 (s, 3H), 2.95 (t, J = 6.2 Hz, 2H), 2.77 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 165.4, 164.7, 139.4, 120.8, 59.3, 41.6, 34.5, 13.7; IR (film) νmax 3377, 1567, 1413, 1362, 1193, 1149, 1057, 958 cm−1; HRMS (MALDI-FTMS) m/z 233.0412 (M+H+, calculated 233.0413).
4-Ethyl-5-methyl-3-(methylsulfinyl)-6-(methylthio)pyridazine (7l)
A solution of 8 (46 mg, 0.215 mmol) in CH2Cl2 (1 mL) was treated with m-CPBA (70%, 53 mg, 0.215 mmol, 1 equiv) at 0 °C. The mixture was allowed to warm to room temperature over 1 h before being washed with saturated aqueous NaHCO3. The organic layer was dried over Na2SO4 and preparative TLC (SiO2, EtOAc) afforded 7b (13 mg, 0.056 mmol, 26%, pale yellow viscous oil) and 7l (13 mg, 0.056 mmol, 26%, white solid). For 7l: mp 94–96 °C (EtOAc); 1H NMR (CDCl3, 300 MHz) δ 3.16–2.92 (m, 2H), 3.10 (s, 3H), 2.72 (s, 3H), 2.28 (s, 3H), 1.24 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 165.6, 159.8, 140.5, 135.5, 37.6, 20.7, 14.1, 13.8, 13.6; IR (film) νmax 3475, 2926, 1539, 1294, 1206, 1035, 952 cm−1; HRMS (ESI-TOF) m/z 231.0629 (M+H+, calculated 231.0620).
4-(Benzyloxycarbonyl)amino-1-(methylsulfinyl)-6,7-dihydro-5H-cyclopenta[d]pyridazine (9a)
54 mg of 5 yielded 47 mg of 9a (77%, orange film) after chromatography (0–1% MeOH/EtOAc): 1H NMR (CDCl3, 400 MHz) δ 7.41–7.34 (m, 5H), 5.23 (s, 2H), 3.38 (t, J = 7.6 Hz, 2H), 3.13–3.02 (m, 2H), 3.00 (s, 3H), 2.26–2.08 (m, 2H); 13C NMR (CDCl3, 150 MHz) δ 160.8, 153.4, 153.0, 148.0, 141.5, 135.3, 128.6 (2C), 128.5, 128.2 (2C), 67.8, 39.5, 31.6, 30.4, 24.3; IR (film) νmax 3184, 2960, 1733, 1515, 1232, 1047 cm−1; HRMS (ESI-TOF) m/z 332.1056 (M+H+, calculated 332.1063).
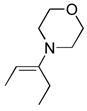
6-(Benzyloxycarbonyl)amino-5-ethyl-4-methyl-3-(methylsulfinyl)pyridazine (9b)
29 mg of 5 yielded 9b (9.6 mg, 29%, colorless oil) after preparative TLC (SiO2, EtOAc). The regioisomer (9j) was isolated as a minor product (3.2 mg, 10%, colorless oil). For 9b: 1H NMR (CDCl3, 400 MHz) δ 7.39–7.36 (m, 5H), 5.23 (s, 2H), 3.10 (s, 3H), 2.76 (q, J = 7.6 Hz, 2H), 2.66 (s, 3H), 1.14 (t, J = 7.6 Hz); 13C NMR (CDCl3, 100 MHz) δ 161.9, 154.2, 153.8, 139.2, 135.3, 128.6 (2C), 128.5, 128.4 (2C), 126.9, 68.0, 37.6, 20.8, 13.3, 12.2; IR (film) νmax 3209, 2976, 1728, 1557, 1498, 1455, 1228, 1051 cm−1; HRMS (ESI-TOF) m/z 334.1221 (M+H+, calculated 334.1220). For minor isomer (9j): 1H NMR (CDCl3, 400 MHz) δ 7.77 (s, 1H), 7.39–7.33 (m, 5H), 5.21 (s, 2H), 3.13–2.95 (m, 2H), 3.06 (s, 3H), 2.30 (s, 3H), 1.26 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 161.3, 155.3, 153.6, 145.1, 135.3, 133.8, 128.65 (2C), 128.56, 128.3 (2C), 68.0, 38.3, 21.1, 13.9, 13.7; IR (film) νmax 3180, 2973, 1732, 1504, 1231, 1043 cm−1; HRMS (ESI-TOF) m/z 334.1215 (M+H+, calculated 334.1220).
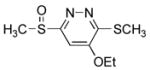
6-(Benzyloxycarbonyl)amino-5-ethoxy-3-(methylsulfinyl)pyridazine (9c)
70 mg of 5 yielded 27 mg of 9c (34%, white solid) after chromatography (0–10% MeOH/CH2Cl2) and preparative TLC (SiO2, 5% MeOH/CH2Cl2). The regioisomer (9h) was isolated as a minor product (14 mg, 17%, white solid). A single-crystal X-ray structure determination25 conducted on crystals grown from EtOAc/CHCl3 unambiguously established the structure of 9c. For 9c: mp 119–121 °C (EtOAc/CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.48–7.38 (m, 6H), 5.30 (s, 2H), 4.29 (q, J = 8.5 Hz, 2H), 2.96 (s, 3H), 1.52 (t, J = 8.5 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 165.9, 151.0, 148.8, 147.2, 135.3, 128.63 (2C), 128.58 (2C), 128.54, 102.6, 67.9, 65.9, 41.9, 14.1; IR (film) νmax 1731, 1572, 1503, 1438, 1221, 1042 cm−1; HRMS (MALDI-FTMS) m/z 336.1016 (M+H+, calculated 336.1012). For minor isomer (9h): mp 144–145 °C (EtOAc); 1H NMR (CDCl3, 400 MHz) δ 8.59 (br s, 1H), 7.89 (s, 1H), 7.42–7.36 (m, 5H), 5.26 (s, 2H), 4.29 (q, J = 7.0 Hz, 2H), 3.00 (s, 3H), 1.53 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.7, 157.3, 152.9, 152.1, 135.1, 128.7 (2C), 128.6, 128.2 (2C), 98.5, 67.8, 65.5, 37.3, 14.0; IR (film) νmax 1731, 1572, 1512, 1228, 1152, 1050, 1029 cm−1; HRMS (MALDI-FTMS) m/z 336.1017 (M+H+, calculated 336.1012).
6-(Benzyloxycarbonyl)amino-3-(methylsulfinyl)pyridazine (9d)
16 mg of 5 yielded 12 mg of 9d (75%, white solid) after chromatography (10% EtOAc/hexane): mp 155.5–155.8 °C (EtOAc/hexane); 1H NMR (CDCl3, 500 MHz) δ 8.55 (d, J = 9.4 Hz, 1H), 8.39 (br s, 1H), 8.16 (d, J = 9.4 Hz, 1H), 7.42–7.39 (m, 5H), 5.28 (s, 2H), 2.94 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.2, 155.6, 152.7, 134.9, 128.78, 128.75 (2C), 128.4 (2C), 124.9, 118.6, 68.0, 41.9; IR (film) νmax 1721, 1573, 1519, 1228, 1056 cm−1; HRMS (MALDI-FTMS) m/z 292.0758 (M+H+, calculated 292.0756).
6-(Benzyloxycarbonyl)amino-5-methyl-3-(methylsulfinyl)pyridazine (9e)
77 mg of 5 yielded 43 mg of 9e (54%, colorless oil) after chromatography (0–10% EtOAc/hexane). The regioisomer (9i) was isolated as a minor product (5.5 mg, 7%, white solid). For 9e: 1H NMR (CDCl3, 400 MHz) δ 8.01 (s, 1H), 7.76 (br s, 1H), 7.42–7.35 (m, 5H), 5.24 (s, 2H), 2.94 (s, 3H), 2.45 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.8, 155.1, 153.5, 136.5, 135.1, 128.68 (2C), 128.64, 128.4 (2C), 125.8, 68.1, 41.8, 18.4; IR (film) νmax 1732, 1506, 1234, 1049 cm−1; HRMS (MALDI-FTMS) m/z 306.0913 (M+H+, calculated 306.0907). For minor isomer (9i): mp 139–141 °C (EtOAc); 1H NMR (CDCl3, 400 MHz) δ 8.22 (s, 1H), 8.15 (br s, 1H), 7.43–7.37 (m, 5H), 5.26 (s, 2H), 3.08 (s, 3H), 2.69 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 160.2, 155.8, 152.7, 141.6, 135.0, 128.7 (2C), 128.3 (2C), 119.1, 67.9, 37.6, 17.7; IR (film) νmax 3182, 2923, 1733, 1558, 1505, 1409, 1224, 1152, 1047, 744 cm−1; HRMS (MALDI-FTMS) m/z 306.0912 (M+H+, calculated 306.0907).
6-(Benzyloxycarbonyl)amino-3-(methylsulfinyl)-5-phenylpyridazine (9f)
From 1-phenyl-1-(trimethylsilyloxy)ethylene (6f): 9.2 mg of 5 yielded 9.5 mg of 9f (83%, white solid) after chromatography (60–100% EtOAc/hexane). From phenylacetylene (6g): 10.4 mg of 5 yielded 10 mg of 9f (77%) after preparative TLC (SiO2, EtOAc): mp 119–121 °C (EtOAc/hexane); 1H NMR (CDCl3, 500 MHz) δ 8.07 (s, 1H), 7.52–7.49 (m, 5H), 7.35–7.33 (m, 3H), 7.28–7.27 (m, 2H), 5.07 (s, 2H), 3.03 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.7, 152.7, 152.0, 135.5, 135.1, 133.8, 129.9, 129.5 (2C), 128.55 (2C), 128.52, 128.4 (2C), 127.5 (2C), 124.5, 67.8, 41.9; IR (film) νmax 1731, 1495, 1213, 1045, 743, 697 cm−1; HRMS (MALDI-FTMS) m/z 368.1064 (M+H+, calculated 368.1063).
6-(Benzyloxycarbonyl)amino-5-(4′-bromophenyl)-3-(methylsulfinyl)pyridazine (9g)
33 mg of 5 yielded 34 mg of 9g (69%, white solid) after chromatography (50–100% EtOAc/hexane): mp 162–163 °C (toluene/CHCl3); 1H NMR (CDCl3, 500 MHz) δ 8.07 (s, 1H), 7.92 (brs, 1H), 7.57–7.55 (m, 2H), 7.40–7.34 (m, 5H), 7.22–7.20 (m, 2H), 5.03 (s, 2H), 3.00 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 167.2, 152.7, 152.2, 135.3, 135.0, 133.4, 132.5 (2C), 128.7 (2C), 128.6, 128.5 (2C), 128.4 (2C), 124.5, 124.2, 67.9, 41.8; IR (film) νmax 3176, 2960, 1733, 1488, 1393, 1250, 1214, 1055, 751 cm−1; HRMS (MALDI-FTMS) m/z 446.0166 (M+H+, calculated 446.0168).
6-(Benzyloxycarbonyl)amino-4-ethoxy-3-(methylthio)pyridazine (10a)
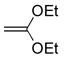
A solution of 3 (20 mg, 0.072 mmol) in dioxane (300μL) was treated with ketene diethyl acetal (6c, 94 μL, 10 equiv). The mixture was heated at 100 °C in a closed vessel for 2 h. Chromatography (30% EtOAc/hexane) afforded 22 mg of 10a (95%, white solid): mp 167–168 °C (EtOAc); 1H NMR (CDCl3, 400 MHz) δ 8.07 (br s, 1H), 7.59 (s, 1H), 7.42–7.34 (m, 5H), 5.23 (s, 2H), 4.21 (q, J = 7.0 Hz, 2H), 2.59 (s, 3H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 155.8, 153.6, 153.3, 150.8, 135.5, 128.6 (2C), 128.4, 128.0 (2C), 96.1, 67.3, 64.7, 14.0, 12.2; IR (film) νmax 2923, 1723, 1589, 1571, 1515, 1373, 1357, 1239, 1156, 1117, 1034, 750 cm−1; HRMS (MALDI-FTMS) m/z 320.1059 (M+H+, calculated 320.1063).
6-(Benzyloxycarbonyl)amino-4-methyl-3-(methylthio)pyridazine (10b)
A solution of 3 (20 mg, 0.072 mmol) in dioxane (300 μL) was treated with 2-methoxypropene (6e, 69 μL, 10 equiv). The mixture was heated in a closed vessel at 100 °C for 18 h. Chromatography (20% EtOAc/hexane) afforded 10b (8 mg, 39%, white solid): mp 151–152 °C (EtOAc); 1H NMR (CDCl3, 300 MHz) δ 7.96 (s, 1H), 7.82 (br s, 1H), 7.43–7.35 (m, 5H), 5.23 (s, 2H), 2.67 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 158.5, 153.0, 152.2, 138.3, 135.4, 128.6 (2C), 128.5, 128.2 (2C), 116.8, 67.5, 18.5, 13.1; IR (film) νmax 1721, 1570, 1502, 1232, 1149, 1110, 1041, 750, 695 cm−1; HRMS (MALDI-FTMS) m/z 290.0956 (M+H+, calculated 290.0958).
6-(Benzyloxycarbonyl)amino-5-methyl-3-(methylthio)pyridazine (10c)
A solution of 3 (25 mg, 0.090 mmol) in dioxane (0.2 mL) was treated with 1-morpholinopropene26 (57 mg, 0.45 mmol, 5 equiv). The mixture was stirred at 25 °C for 1 h before solvent was removed. The residue was dissolved in 10% HOAc/benzene (0.2 ml) and stirred at 25 °C for 15 h. Neutralization with saturated aqueous NaHCO3, extraction (CH2Cl2) and chromatography (40% EtOAc/hexane) affoded 10c (24 mg, 92%, white solid): mp 117–119 °C (EtOAc); 1H NMR (CDCl3, 400 MHz) δ 7.53 (s, 1H), 7.40–7.32 (m, 5H), 7.17 (s, 1H), 5.20 (s, 2H), 2.63 (s, 3H), 2.27 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 145.3, 135.6, 128.6 (2C), 128.3, 128.2 (2C), 67.6, 17.7, 13.2 (4 peaks are undetected); IR (film) νmax 3208, 2926, 1727, 1497, 1239, 1101 cm−1; HRMS (ESI-TOF) m/z 290.0957 (M+H+, calculated 290.0958).
6-(Benzyloxycarbonyl)amino-4-ethyl-5-methyl-3-(methylthio)pyridazine (10d)
A solution of 3 (23 mg, 0.083 mmol) in CH2Cl2 (0.17 mL) was treated with 3-morpholino-2-pentene26 (6b, 26 mg, 0.166 mmol, 2 equiv). The mixture was stirred at 25 °C for 1.5 h before removal of the solvent. The residue was treated with 10% HOAc/benzene (0.2 mL) and the mixture stirred at 25 °C for 17 h. Neutralization with saturated aqueous NaHCO3, extraction (CH2Cl2) and preparative TLC (SiO2, 33% EtOAc/hexane) afforded 10d (21 mg, 80%, pale yellow oil); mp 124–126 °C (CH2Cl2); 1H NMR (CDCl3, 400 MHz) δ 7.41–7.32 (m, 5H), 5.20 (s, 2H), 2.70 (q, J = 7.6 Hz, 2H), 2.64 (s, 3H), 2.23 (s, 3H), 1.17 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 141.6, 135.8, 128.5 (2C), 128.3, 128.2 (2C), 67.6, 22.3, 13.5, 13.3, 11.5 (4 peaks are undetected); IR (film) νmax 3170, 2971, 1731, 1499, 1237, 1071 cm−1; HRMS (ESI-TOF) m/z 318.1279 (M+H+, calculated 318.1271).
General procedure for oxidation of 8 or 10
A solution of 8 or 10 in CH2Cl2 was treated with m-CPBA (1 equiv) at 0 °C. The reaction mixture was allowed to warm to room temperature over 0.5–1 h. Washed with saturated aqueous NaHCO3, dried (Na2SO4) and chromatography afforded 7 or 9.
Supplementary Material
1H NMR spectra of all new compounds and details of the X-ray structure of 7c, 7f, 7g and 9c. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 3.
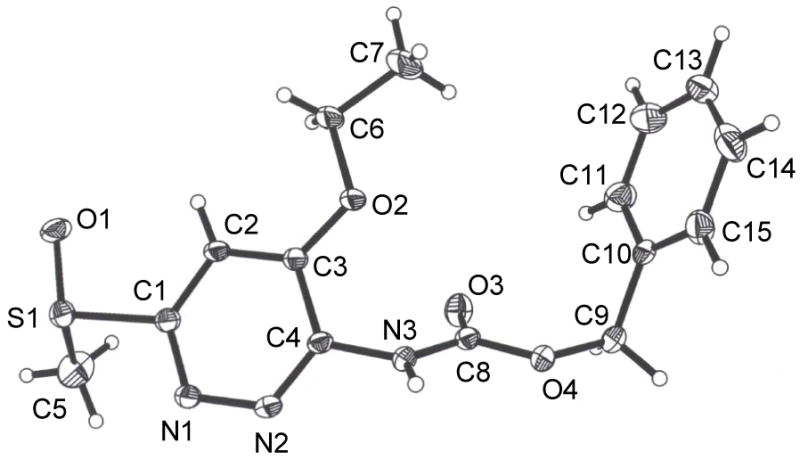
ORTEP drawing of 9c.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA42056) and the Skaggs Institute for Chemical Biology.
References
- 1.Boger DL, Weinreb SM. Hetero Diels–Alder Methodology in Organic Synthesis. Academic; San Diego: 1987. [Google Scholar]
- 2.Reviews: Boger DL. Tetrahedron. 1983;39:2869.Boger DL. Chem Rev. 1986;86:781.Boger DL. Chemtracts: Org Chem. 1996;9:149.
- 3.Streptonigrin: Boger DL, Panek JS, Duff SR. J Am Chem Soc. 1985;107:5745.
- 4.OMP: Boger DL, Coleman RS, Panek JS, Yohannes D. J Org Chem. 1984;49:4405.
- 5.Lavendamycin: Boger DL, Duff SR, Panek JS, Yasuda M. J Org Chem. 1985;50:5790.
- 6.PDE-I and PDE-II: Boger DL, Coleman RS. J Am Chem Soc. 1987;109:2717.
- 7.CC-1065: Boger DL, Coleman RS. J Am Chem Soc. 1988;110:1321, 4796.
- 8.Prodigiosin: Boger DL, Patel M. J Org Chem. 1988;53:1405.
- 9.Trikentrin A: Boger DL, Zhang M. J Am Chem Soc. 1991;113:4230.
- 10.Isochrysohermidin: Boger DL, Baldino CM. J Am Chem Soc. 1993;115:11418.
- 11.Ningalin A, Lamellarin O, Lukianol A, Storniamide A, Boger DL, Boyce CW, Labroli MA, Sehon CA, Jin Q. J Am Chem Soc. 1999;121:54. [Google Scholar]
- 12.Phomazarin: Boger DL, Hong J, Hikota M, Ishida M. J Am Chem Soc. 1999;121:2471.
- 13.Ningalin B: Boger DL, Soenen DR, Boyce CW, Hedrick MP, Jin Q. J Org Chem. 2000;65:2479. doi: 10.1021/jo9916535.
- 14.Anhydrolycorinone and related alkaloids: Boger DL, Wolkenberg SE. J Org Chem. 2000;65:9120. doi: 10.1021/jo0012546.
- 15.Roseophilin: Boger DL, Hong J. J Am Chem Soc. 2001;123:8515. doi: 10.1021/ja011271s.
- 16.Ningalin D: Hamasaki A, Zimpleman JM, Hwang I, Boger DL. J Am Chem Soc. 2005;127:10767. doi: 10.1021/ja0526416.
- 17.Dimethyl 1,2,4,5-tetrazine-3,6-dicarboxylate (1): Sauer J, Mielert A, Lang D, Peter D. Chem Ber. 1965;98:1435.Boger DL, Coleman RS, Panek JS, Huber FX, Sauer J. J Org Chem. 1985;50:5377.Boger DL, Panek JS, Patel M. Org Syn. 1991;70:79.
- 18.3,6-Bis(methylthio)-1,2,4,5-tetrazine (2): Strube RE. Org Syn. IV. Wiley; New York: 1963. p. 967.Sandström J. Acta Chem Scand. 1961;15:1575.Boger DL, Sakya SM. J Org Chem. 1988;53:1415.
- 19.3,6-Bis(3,4-dimethoxybenzoyl)-1,2,4,5-tetrazine: Soenen DR, Zimpleman JM, Boger DL. J Org Chem. 2003;68:3593. doi: 10.1021/jo020713v.
- 20.3-Methoxy-6-methylthio-1,2,4,5-tetrazine: Sakya SM, Groskopf KK, Boger DL. Tetrahedron Lett. 1997;38:3805.
- 21.N-Acyl 6-amino-3-methylthio-1,2,4,5-tetrazine (e.g., 3): Boger DL, Schaum RP, Garbaccio RM. J Org Chem. 1998;63:6329. doi: 10.1021/jo980795g.
- 22.Johnson JL, Whitney B, Werbel LM. J Heterocyclic Chem. 1980;17:501. [Google Scholar]
- 23.Seitz G, Dietrich S, Görge L, Richter J. Tetrahedron Lett. 1986;27:2747. [Google Scholar]
- 24.Blair LK, Baldwin J, Smith WC., Jr J Org Chem. 1977;42:1817. [Google Scholar]
- 25.Atomic coordinates for 7c (CCDC 282318), 7f (CCDC 282319), 7g (CCDC 282321) and 9c (CCDC 282320) have been deposited with the Cambridge Crystallographic Data Centre.
- 26.Stork G, Brizzolara A, Landesman H, Szmuszkovic J, Terrell R. J Am Chem Soc. 1963;85:207. [Google Scholar]
- 27.Cazeau P, Duboudin F, Moulines F, Babot O, Dunogues J. Tetrahedron. 1987;43:2075. [Google Scholar]
- 28.Sega T, Pollak A, Stanovnik B, Tisler M. J Org Chem. 1973;38:3307. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H NMR spectra of all new compounds and details of the X-ray structure of 7c, 7f, 7g and 9c. This material is available free of charge via the Internet at http://pubs.acs.org.