

Abstract
We recently showed that the class Ic ribonucleotide reductase from the human pathogen, Chlamydia trachomatis, uses a MnIV/FeIII cofactor to generate protein and substrate radicals in its catalytic mechanism [Jiang, W., Yun, D., Saleh, L., Barr, E. W., Xing, G., Hoffart, L. M., Maslak, M.-A., Krebs, C., and Bollinger, J. M., Jr. (2007) Science 316, 1188-1191]. Here, we have dissected the mechanism of formation of this novel heterobinuclear redox cofactor from the MnII/FeII cluster and O2. An intermediate with a g = 2 EPR signal that shows hyperfine coupling to both 55Mn and 57Fe accumulates almost quantitatively in a second order reaction between O2 and the reduced R2 complex. The otherwise slow decay of the intermediate to the active MnIV/FeIII-R2 complex is accelerated by the presence of the one-electron reductant, ascorbate, implying that the intermediate is more oxidized than MnIV/FeIII. Mössbauer spectra show that the intermediate contains a high-spin FeIV center. Its chemical and spectroscopic properties establish that the intermediate is a MnIV/FeIV-R2 complex with an S = 1/2 electronic ground state arising from antiferromagnetic coupling between the MnIV (SMn = 3/2) and high-spin FeIV (SFe = 2) sites.
A conventional class I ribonucleotide reductase (RNR1), such as the RNR from Escherichia coli or Homo sapiens, activates O2 at a carboxylate-bridged Fe2II/II cluster in its R2 subunit to oxidize a nearby tyrosine (Y) residue to a stable tyrosyl radical (Y•) (1). The Y• in R2 oxidizes a cysteine residue in the R1 subunit by a long-distance (∼ 35 Å), inter-subunit, proton-coupled electron transfer (PCET), generating a transient cysteine thiyl radical (C•) (2, 3). The C• in R1 initiates reduction of the ribonucleoside diphosphate (NDP) substrate by abstracting the hydrogen atom from C3' (4, 5). After reduction of the substrate 3' radical to the 2'-deoxy (product) 3' radical by two additional cysteine residues in R1 (which become oxidized to a disulfide), the hydrogen originally abstracted from C3' is returned to this position, regenerating the C• and yielding the 2'-deoxyribonucleoside diphosphate (dNDP) product. The C• then re-oxidizes the Y in R2 back to the stable Y•.
When McClarty and co-workers identified the genes encoding the class I RNR subunits from several species of chlamydiae, they noted that the R2 proteins had phenylalanine (F) residues at the position aligning with the Y•-harboring tyrosine residues of the other R2s (6). They found that the Chlamydia trachomatis (Ct) RNR was, nevertheless, catalytically active. Subsequent biochemical and structural characterization of the Ct R2 confirmed the absence of a Y• and location of F at the site normally harboring the Y• (7). Analysis of genome sequences suggested that Y•-less R2 subunits might also be present in other bacteria (including the important human pathogen, Mycobacterium tuberculosis) and archaea, and a new sub-classification of these RNRs as class Ic was proposed. It was further suggested that a Fe2III/IV cluster, formed by reaction of O2 with the Fe2II/II form of R2 and similar to the intermediate, cluster X, which had previously been shown to generate the Y•s in the R2 proteins from E. coli and Mus musculus (8, 9), directly generates the C• in these class Ic RNRs. Two subsequent studies on Ct RNR provided support for this hypothesis by showing that the oxidized diiron cluster could be stabilized for several minutes (10) and even induced to accumulate from Fe2III/III-R2 (11) in a complete RNR reaction solution (containing R1, R2, CDP, ATP, Mg2+, and DTT). However, the relatively meager R2 activity (e.g., < 3% that of E. coli R2) and marked variation thereof (230 U/mg in (10) but only < 75 U/mg in (11)) reported by these authors suggested that another, minor form of the protein, present in varying amounts in different preparations, might be responsible for the observed activity. Indeed, we recently demonstrated that a MnIV/FeIII cluster can be assembled in Ct R2 and that this heterodinuclear form exhibits much greater activity than had been observed in the previous studies (12). Importantly, the reduction of the cofactor to the MnIII/FeIII oxidation state and formation of the well-characterized, nitrogen-centered free radical upon incubation of the enzyme with the radical-trapping mechanism-based inactivator, 2'-azido-2'-deoxyadenosine-5'-diphosphate, established that the MnIV/FeIII cluster is the radical-initiating cofactor of Ct RNR. Additional experiments showed that the active cofactor forms by reaction of the reduced (MnII/FeII) cluster with O2, but the activation process was not studied in detail (12). In this work, we have examined the mechanism of activation of Ct R2 by stopped-flow absorption and freeze-quench EPR and Mössbauer spectroscopies. The results show that the activation mechanism entails (1) the rapid formation of a MnIV/FeIV-R2 intermediate in a bimolecular reaction between the reduced complex and O2 and (2) the slow decay of the intermediate by reduction of the iron site to FeIII. Kinetic characteristics of the decay step suggest that it may be mediated by the protein, perhaps by the same residues required for the intra-subunit radical transfer that initiates turnover (2).
MATERIAL AND METHODS
Expression and Purification of Ct R2
The protein used throughout this study has an additional 20 amino acids (MGS2H6S2GLVPRGSH) appended to the N-terminal methionine residue of Ct R2. The appendage contains a His6 element to permit purification of the protein by metal-ion-affinity chromatography. Preparation of the plasmid that directs over-expression of this protein in E. coli, growth of the over-expression strain, and purification of the metal-depleted form of the R2 protein have been described (12).
Preparation of MnII//FeII-R2 Complex
In an MBraun anoxic chamber, MnII and FeII (natural abundance FeII, which contains 91.8% 56FeII and is hereafter referred to as 56FeII, or ∼95% enriched 57FeII, which is hereafter referred to as 57FeII) were added to the metal-depleted R2 to form the O2-reactive MnII/FeII-R2 complex. It was determined in the course of this study that R2 containing only MnII does not react with O2 (not shown), whereas previous studies had established that the Fe2II/II-R2 complex reacts rapidly (7, 10). The reaction of Fe2II/II-R2 results in development and decay of the absorption spectrum and sharp, isotropic, g = 2.00 EPR signal of the Fe2III/IV cluster (7, 10). To minimize this undesired reaction, stopped-flow absorption and freeze-quench EPR experiments employed a three-fold excess of Mn over Fe. MnII (1.5 equiv relative to R2 monomer) was added to the metal-depleted R2 protein first, and the solution was incubated at ambient temperature (22 °C) to allow for binding. FeII (0.5 equiv) was then added slowly. This solution was loaded into the stopped-flow or freeze-quench apparatus. For preparation of the freeze-quenched sample for Mössbauer characterization of the MnIV/FeIV-R2 intermediate, a two-fold excess of MnII over FeII was employed. The divalent metal ions (1.0 equiv MnII, 0.5 equiv 57FeII) were pre-mixed before being added to the metal-depleted R2. The MnII/FeII-R2 solution was then loaded into the freeze-quench syringe.
Stopped-flow Absorption and Freeze-quench EPR and Mössbauer Experiments
The stopped-flow and freeze-quench apparatus and procedures and the EPR and Mössbauer spectrometers have been described (13). The magnitudes of the static magnetic fields for the low-field Mössbauer spectra were determined using a Digital Tesla meter (model 132D) with a Hall probe LPT 130-20S (Group3 Technologies Inc., Auckland, NZ). Details of reaction and spectroscopy conditions are provided in the appropriate figure legend or in the figure itself.
Simulation of the EPR and Mössbauer Spectra of MnIV/FeIV-R2
The simulations are based on the commonly used spin Hamiltonian formalism (14) and were carried out with respect to the total spin of the electronic ground state, STotal = 1/2. For simulation of the EPR spectra, Equation 1 was used. The first term is the electron Zeeman effect and the second and third terms are the hyperfine couplings between the electron spin and the 55Mn nuclear spin (I = 5/2) and between the electron spin and the 57Fe nuclear spin (I = 1/2), respectively.
| (1) |
The program SimFonia (Bruker, Billerica, MA) was used to simulate EPR spectra by the second-order-perturbation method for powder spectra. The full-matrix diagonalization program, SIM, which was written by Høgni Weihe (University of Copenhagen) (15), was used to simulate the spectra by an independent method to verify the parameters obtained with SimFonia. For simulation of the Mössbauer spectra, the program WMOSS (Web Research, Edina, MN) was used. All simulations were carried out with the assumption that the fluctuation rate of the electron spin is slow compared to the 57Fe Larmor frequency. The first two terms of equation 1 were solved first with the parameters obtained from analysis of the EPR spectra. It was necessary to consider AMn explicitly, because it affects the splitting in the weak-field (B < ∼150 mT) spectra. Coupling between the electron spin of the ground state, S = 1/2, and the 55Mn nuclear spin, I = 5/2, leads to two states with spins F = 2 and F = 3 (with F = S + I). For B > 150 mT, the electron Zeeman effect dominates the S•AMn•I term, the states are “pure” and characterized by the MS [= ± 1/2] and MI [= ± 1/2, ± 3/2, ± 5/2] quantum numbers (Figure S1), and the spin expectation values are at their maxima, |〈S〉| = 0.5. With weaker applied fields, the electron Zeeman interaction and AMn are comparable, leading to mixing of the states. From the solution of the first two terms in eq 1, the spin expectation value, 〈S〉, was calculated. Equation 2, in which all symbols have their usual meaning (14), was then used to compute the Mössbauer spectrum. All tensors were assumed to be collinear.
| (2) |
RESULTS
Freeze-quench EPR evidence for accumulation of an oxidized Mn/Fe intermediate
The EPR spectrum of the O2-reactive MnII/FeII-R2 complex, resolved as the difference of the spectra taken before and after exposure of the reactant solution to O2 (Figure S2), exhibits a broad resonance centered at g ∼ 2 that shows hyperfine coupling characteristic of an I = 5/2 55Mn nucleus. Optimal detection of this signal requires relatively low temperature and high power (e.g., 4.2 K and 20 mW, as in Figure S2). Spectra of samples freeze-quenched during the reaction of this complex with O2 were recorded under less stringent conditions (14 K and 20 μW) to eliminate the contribution from the reactant complex. By subtracting the spectrum of the reactant solution under these conditions from the spectra of the freeze-quenched samples, the contribution of free MnII (resulting from the use of excess MnII in preparation of the reactant complex) was also removed. The time-dependent spectra (Figure 1A) illustrate that an intermediate with a sharp g ∼ 2 EPR signal develops rapidly upon reaction with O2 and then decays slowly. The spectrum of this intermediate has six lines separated by ∼ 80 G that reflect hyperfine coupling to a single 55Mn nucleus. When the intermediate is formed from MnII/FeII-R2 reactant containing 57Fe, the sextet signal also shows hyperfine coupling to this I = 1/2 nucleus (compare the first and third spectra in Figure 1B). Simulation of these spectra (Figure 1B, second and fourth spectra) together with the Mössbauer spectra to extract electronic structural parameters (including the 55Mn and 57Fe hyperfine coupling tensors, AMn and AFe) is presented below.
Figure 1.

X-band EPR spectra at 14.0 (± 0.2) K of the MnIV/FeIV intermediate in activation of Ct R2. A: Spectra of samples freeze-quenched at various reaction times (indicated on the figure) after mixing at 5 °C of an O2-free solution of MnII/FeII-R2 (0.40 mM R2 monomer, 0.5 equiv Fe, 1.5 equiv Mn) with an equal volume of O2-saturated buffer. The spectrum of the recovered product (completion) sample has been scaled to account for the fact that it was manually frozen rather than being freeze-quenched (× 0.6, the “packing factor” typical of freeze-quenched samples). In addition, the appropriately scaled spectrum of the reactant sample (× 0.5 because it wasn't diluted and × 0.6 because it was manually frozen) was subtracted from the experimental spectrum of each sample to generate the spectra shown. B: spectra of samples prepared by manual mixing of an O2-free solution of MnII/FeII-R2 (3.0 mM R2 monomer, 0.75 equiv of each metal ion) at ambient temperature (22 ± 2 °C) with 9 equivalent volumes of O2-saturated buffer and freezing after 20 ± 2 s. The first and third traces are the experimental spectra of the samples prepared with 56Fe and 57Fe, respectively. Spectrometer conditions were: microwave frequency, 9.45 GHz; microwave power, 20 μW; modulation frequency, 100 kHz; modulation amplitude, 10 G; scan time, 167 s; time constant, 167 ms. The second and fourth traces are simulations of the experimental spectra generated as described in Material and Methods with the g, AMn, and AFe tensors given in Table 1.
Kinetics of the reaction by stopped-flow absorption measurements
Accumulation of the Mn/Fe intermediate complex is also apparent in the time-dependent absorption spectra from the reaction (Figure 2A). An intense feature at ∼ 390 nm develops rapidly (black, red and blue traces) and then decays slowly, leaving the spectrum of the MnIV/FeIII-R2 product (green) (16). An overlay of the scaled intensities of the EPR spectra from the experiment of Figure 1A (Figure 1B, filled circles) with the absorbance-versus-time trace from the stopped-flow experiment with the same reaction conditions (Figure 1B, diamonds) illustrates that the g ∼ 2 EPR signal and 390-nm absorption feature arise from the same intermediate. The kinetics of the intermediate were defined and the effects of variation of [O2] and inclusion of a reductant (ascorbate) were interrogated by additional stopped-flow experiments. Formation of the intermediate is kinetically first-order in [O2] (Figure 2B). The re-plot of the apparent first-order rate constant, obtained by fitting the equation for two parallel first-order reactions to the data,2 versus [O2] gives a second-order rate constant (slope) of 13 (± 3) mM−1s−1 (Figure 2B, inset).
Figure 2.
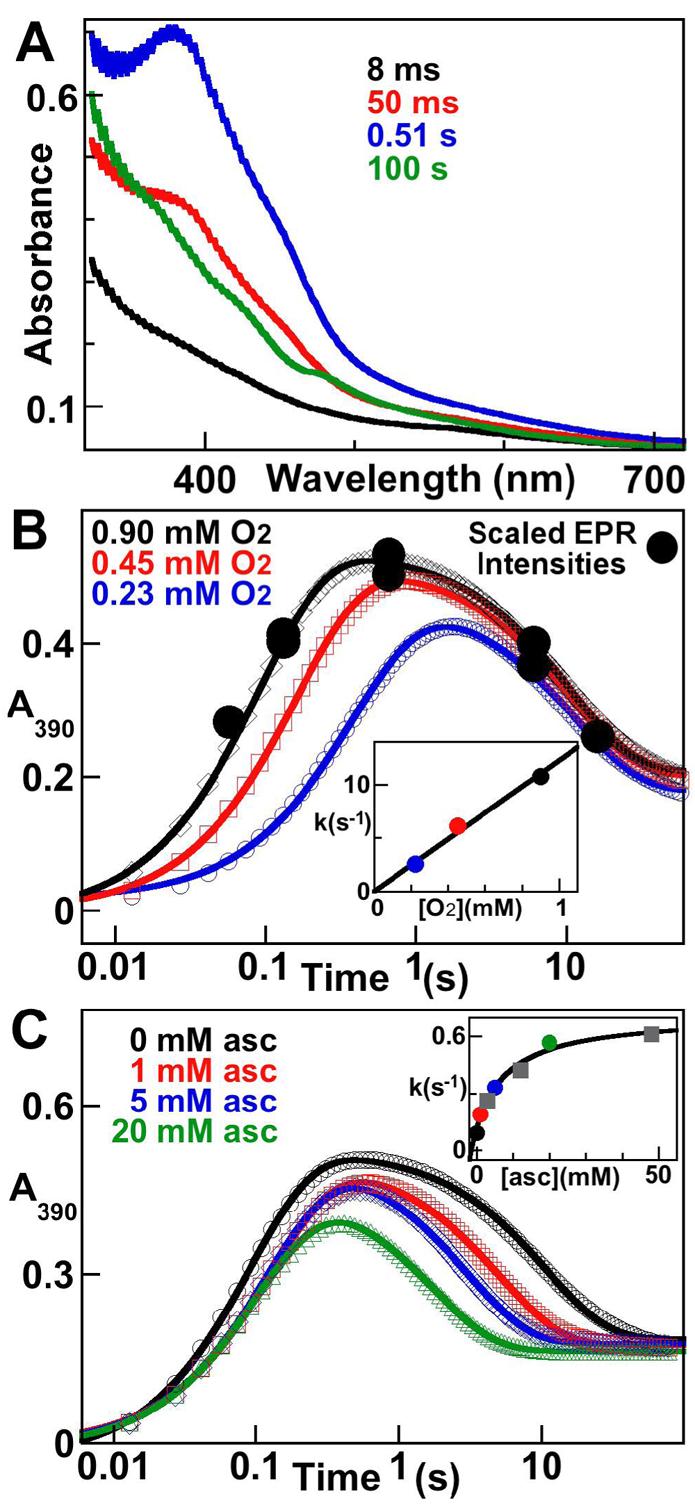
Kinetics of the activation of Ct R2 by stopped-flow absorption spectroscopy. A: spectra acquired at the indicated reaction times after mixing at 5 °C of an O2-free solution of MnII/FeII-R2 (0.40 mM R2 monomer, 0.5 equiv Fe, 1.5 equiv Mn) with an equal volume of O2-saturated buffer. B: dependence of the kinetics of the reaction on [O2]. An equivalent MnII/FeII-R2 solution was mixed with 100% (black), 50% (red), or 25% (blue) O2-saturated buffer. The black circles are the EPR signal intensities from the experiment of Figure 1A (which had identical reaction conditions) scaled for direct comparison to the absorbance changes. The inset shows the apparent first-order rate constant for the formation phase of the reaction (obtained by fitting a “double-exponential” equation to the data2) versus [O2], which gives a second order rate-constant (slope) of 12 (± 3) mM−1s−1. C: Dependence of the kinetics on the concentration of ascorbate. The otherwise-equivalent MnII/FeII-R2 reactant solution, which contained ascorbate at a concentration sufficient to give the indicated [ascorbate] after mixing, was mixed with 100% O2-saturated buffer. The inset shows the apparent first-order rate constant for the decay phase versus [ascorbate], which gives a limiting reduction rate constant (asymptote of hyperbolic fit) of 0.7 (± 0.1) s−1.
Formation of the Y• and Fe2III/III cluster of a conventional class I RNR requires transfer of an “extra” electron to the buried cofactor during its reaction with O2 (17-19). It has been shown that ascorbate can donate this electron (17, 19). Similarly, the MnIV/FeIII cofactor of active Ct R2 is three units more oxidized than the MnII/FeII-R2 complex, which reacts with the four-electron oxidant, O2, to produce the active state (12, 16). Thus, an extra electron is also required in activation of Ct R2. The ability of ascorbate to donate this electron and the timing and mechanism of donation were evaluated. Indeed, ascorbate accelerates decay of the intermediate in concentration-dependent fashion without affecting the kinetics of its formation (Figure 2C). The acceleration of its decay to the MnIV/FeIII state by a reductant is consistent with its assignment from the spectroscopic data as a MnIV/FeIV complex (vide infra). A plot of the observed first-order rate constant for decay versus [ascorbate] is hyperbolic (Figure 2C, inset), suggesting that a unimolecular step is rate-limiting for reduction of the MnIV/FeIV complex at high [ascorbate]. The nature of this step is discussed below.
Characterization of the intermediate by EPR and Mössbauer spectroscopy
The electronic structure of the intermediate was probed further by EPR and Mössbauer spectroscopies. Analysis of the data demonstrates that the intermediate has an S = 1/2 ground state as a consequence of antiferromagnetic coupling between the MnIV (SMn = 3/2) and high-spin FeIV (SFe = 2) ions.
As noted, the EPR spectrum of the intermediate prepared with 56Fe exhibits six “packets” of intensity, due to hyperfine coupling with one 55Mn. The second packet (at ∼ 3210 G) is isotropic (i.e., all transitions are observed at the same magnetic field). The other five packets either are somewhat broadened or exhibit resolved features due to anisotropy of the g- and AMn-tensors. The resolution of the features within the packets, especially for the fifth and sixth packets, permits determination of the g- and AMn-tensors directly from simulation analysis (Table 1). AMn is nearly isotropic [(247, 216, 243) MHz], similar to AMn for the MnIV site of Mn2III/IV catalase3 [(235, 224, 252) MHz] (20) and as expected for a MnIV site (20, 21). In simulating the EPR spectrum of the intermediate prepared with 57Fe, the g- and AMn-tensors determined from the spectrum of the intermediate containing 56Fe were assumed and hyperfine coupling to the 57Fe was then imposed. An isotropic AFe-tensor was assumed first, but it became obvious that the quality of the simulation could be improved considerably with an anisotropic AFe. By considering the EPR spectra together with the field-dependent Mössbauer spectra (vide infra), AFe was determined (Table 1).
Table 1.
Spin-Hamiltonian parameters of the MnIV/FeIV-R2 intermediate.
| Parameter | MnIV/FeIVCt-R2 | |
|---|---|---|
| g | 2.017, 2.030, 2.027 | |
| FeIV site | MnIV site | |
| A (MHz) | (−55.9, −59.3, −40.5)a | (247, 216, 243) |
| δ(mm/s) | 0.17 (6) | - |
| ΔEQ (mm/s) | −0.75 | - |
| η | −10 | - |
Sign determined from Mössbauer spectroscopy
The 4.2-K/53-mT Mössbauer spectra (Figure S4) of the MnII/FeII-R2 complex (top spectrum) and samples prepared by reacting this complex at 5 °C with O2 for 0.090 s (second spectrum from top), 2.0 s (near the time of maximal accumulation of the intermediate; third spectrum from top) or 10 min (completion; bottom two spectra) before freezing illustrate the accumulation of the intermediate to a high level and its subsequent decay to the previously characterized MnIV/FeIII-R2 product (16). Importantly, comparison of the spectra of the 0.09-s and 2-s samples shows that the dominant features in the latter spectrum develop with the same kinetics as for the g ∼ 2 EPR signal and 390-nm absorption feature and are therefore associated with the same intermediate. Specifically, analysis of the spectrum of the 0.09-s sample suggests that ∼40% of the intensity of the spectrum is attributable to the intermediate.
The 4.2-K/variable-field Mössbauer spectra of the 2-s sample are dominated (∼70% of the absorption area) by features of the intermediate (Figure 3). Ideally, the spectral contributions of the minor species would be removed by subtraction of appropriate reference spectra in order to resolve the spectrum of the intermediate for detailed simulation analysis. However, in this case, even though the accumulation of the intermediate compares favorably with the best cases that we have encountered in previous studies, the multiplicity and unknown identities of the minor species make removal of their contributions impossible. The major “contaminant” is a high-spin FeII species of unknown identity.4 Its presence is most clearly revealed in the weak-field (B < 53 mT) spectra by peaks at −0.2 mm/s and +2.8 mm/s (∼17% intensity; Figure 3, middle spectrum, blue line). The magnetic field dependence of the FeII-associated spectral component is unknown, precluding its removal. Fortunately, it is clear that, as expected for high-spin (S = 2) FeII, the Mössbauer features become broader and contribute little to the overall line-shape of the experimental spectrum of the 2-s sample with increasing field strengths. The remaining contaminants are predictable from the previously characterization of the product of the reaction (16), which showed that it contains ∼80% MnIV/FeIII-R2 and ∼20% of the homodinuclear Fe2III/III product. Thus, the iron in the 2-s sample not associated with the MnIV/FeIV intermediate and FeII contaminant should be distributed among the MnIV/FeIII and Fe2III/III products and the Fe2III/IV precursor (X) to the latter product. Given the slow decay of both the MnIV/FeIV intermediate and X, only a small fraction should be in the form of the products at a reaction time of 2 s. Indeed, whereas the experimental spectra can accommodate a small contribution from the product species (≤ 9% total), their contribution could be much less or even negligible. The spectra do reveal the presence of a small quantity of the Fe2III/IV complex.5 In particular, two features of X are nearly fully resolved in the 13-mT spectrum and can be used to estimate the contribution from the complex (∼11%; Figure 3, bottom spectrum, red line). In addition, the highest-energy lines of the sub-spectra of the FeIV and FeIII sites are coincident at 3.7 mm/s in the 8-T spectrum, resulting in a more intense line that reveals the presence of the complex (Figure 3, top spectrum, red line).
Figure 3.
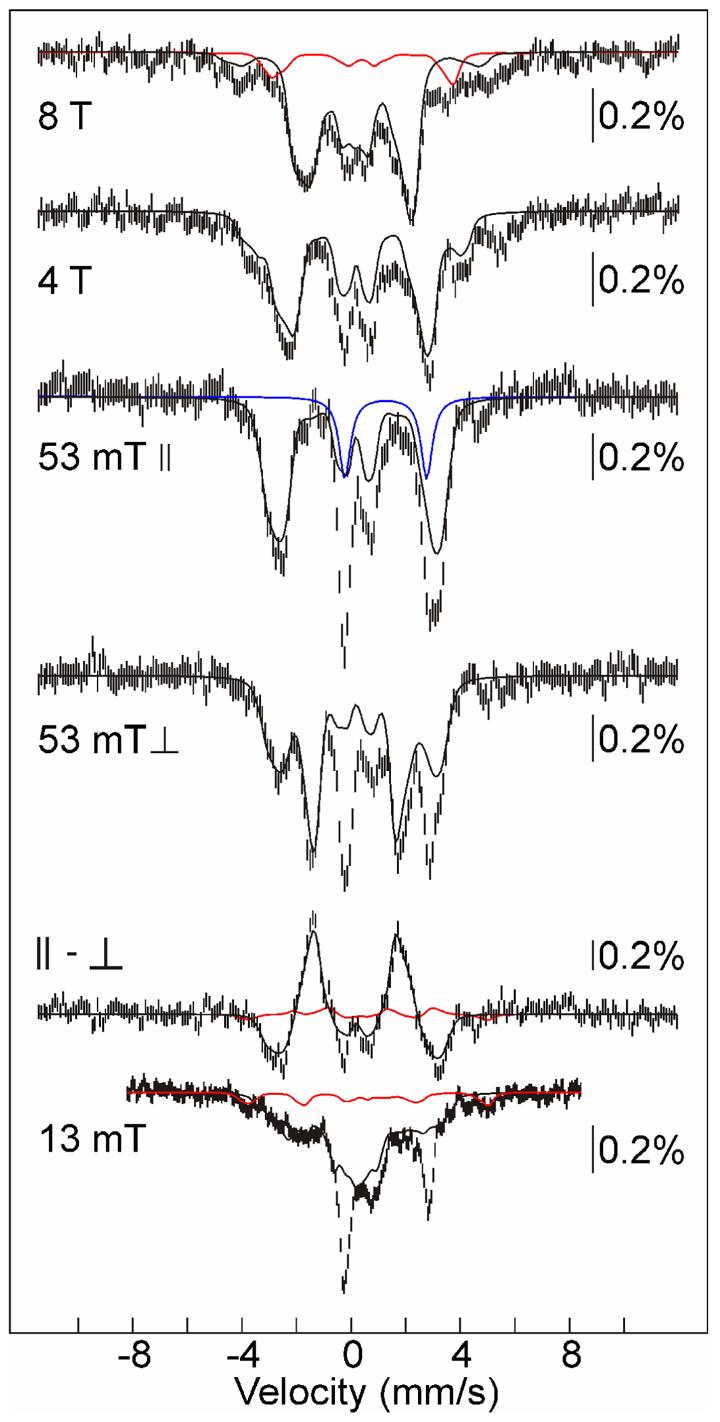
4.2-K Mössbauer spectra in varying magnetic fields (as indicated on the spectra) of a sample containing primarily the MnIV/FeIV intermediate. The sample was prepared by mixing an O2-free solution of MnII/FeII-R2 (3.0 mM R2 monomer, 0.5 equiv Fe and 1.0 equiv Mn) at 5 °C with an equal volume of O2-saturated buffer and freeze-quenching at a reaction time of 2 s. Unless otherwise noted, the field was parallel to the γ-beam. The solid black lines plotted over the spectra are simulations of the spectra of the MnIV/FeIV-R2 intermediate as described in Materials and Methods with the parameters given in Table 1. They are scaled to account for 70% of the total intensity. The red lines are a simulation of the spectrum of the Fe2III/IV complex (11% of total intensity) with the published parameters (26), and the blue line is a quadrupole doublet with δ = 1.3 mm/s and ΔEQ = 3.0 mm/s to illustrate the contribution from the FeII component of the sample (17% of total intensity).
Despite this heterogeneity, the predominance of the MnIV/FeIV intermediate makes the positions and shapes of its features sufficiently clear for simulations to be used to extract spectroscopic parameters (Table 1 and Figure 3, solid black lines). In particular, the contributions of species with integer-electron-spin ground states (FeII species and the MnIV/FeIII and Fe2III/III products) are canceled in field orientation dependent spectra (53 mT ∥ – 53 mT ⊥) and the contributions of the species with half-integer-spin (the MnIV/FeIV and Fe2III/IV intermediates) are resolved (14). The contribution from the Fe2III/IV intermediate (red line) is small compared to that of the MnIV/FeIV intermediate (black line). This difference spectrum provides constraints on the parameters of the MnIV/FeIV intermediate, in particular the isomer shift (δ). Because δ must be determined from magnetically split spectra,6 the uncertainty in this crucial parameter is fairly large (0.06 mm/s). Nevertheless, even with the large uncertainty, the value of δ (0.17 ± 0.06 mm/s) indicates that the intermediate has an FeIV site. Indeed, the center of the range is essentially identical with δ of the Fe2IV/IV complex, Q, in the reaction of soluble methane monooxygenase (22, 23).
The hyperfine tensor for the FeIV site with respect to the total spin of the ground state (S = 1/2), AFe, determines the splitting in the spectra and is given by the product of the intrinsic hyperfine tensor, aFe, and the spin projection factor, cFe (Equation 3) (24).
| (3) |
The components of AFe are negative, as revealed by the decrease of the overall splitting with increasing applied magnetic field (e.g., compare the 53-mT, 4-T, and 8-T spectra in Figure 3) (25). The components of the intrinsic hyperfine tensor for iron, aFe, are negative. Thus, cFe must be positive. A positive value of cFe requires that SFe > SMn (3/2). The FeIV site must therefore be in the high-spin configuration (SFe = 2). For this spin system, cFe = 2 for the S = 1/2 ground state, giving aFe = (−28.0, −29.7, − 20.3) MHz. These values are almost identical to those of the high-spin FeIV site of cluster X in E. coli R2 [aFe = (−27.6, −27.6, − 20.6) MHz] (26), consistent with the assignment of the Fe site of the Ct R2 intermediate as high-spin FeIV.
The g-tensor of the S = 1/2 ground state, given by Equation 4 (24), is nearly isotropic as a result of relatively small anisotropy of gFe and gMn, as was observed before for MnIV and high-spin FeIV species (20, 26).
| (4) |
Comparison of the low-field spectra clearly illustrates the perturbation associated with the hyperfine coupling to the I = 5/2 55Mn nucleus, which is comparable in magnitude to the electron Zeeman term in weak fields. For example, at a field of 13 mT (Figure 3, bottom spectrum), the absolute magnitude of the internal magnetic field [given by – 〈S〉 •(A/gNβN)Fe] is smaller for some of the states as a consequence of sub-saturating values of 〈S〉 (see Figure S1 for plots of the field-dependence of the spin expectation values), resulting in reduced splitting and greater intensity in the center of the spectrum. As the field is increased in the 0 – 150 mT regime, 〈S〉 and the magnetic splitting increase (compare to the 53-mT ∥ spectrum; Figure 3, middle). With much greater fields (B > 150 mT), splitting decreases again (compare 53-mT ∥ spectrum to 8-T spectrum) because for the ground state the applied field opposes the already saturated internal field from the electron spin.
DISCUSSION
The stopped-flow absorption and freeze-quench EPR and Mössbauer data thus establish that the active MnIV/FeIII cofactor of Ct RNR forms via a MnIV/FeIV intermediate that decays by reduction of the FeIV site (Scheme 1, top). The MnIV (SMn = 3/2) and high-spin FeIV (SFe = 2) sites of the intermediate couple antiferromagnetically to yield an S = 1/2 ground state. Whereas a MnIV/FeIII complex has been reported (27), the Ct R2 intermediate is, to our knowledge, the first example of a MnIV/FeIV complex. In view of the X-ray crystal structure of the (presumptively) Fe2III/III form of the Ct R2 protein by Högbom, et al. (7), which suggested a bis-(μ-hydroxo)-dimetal core, and previous studies suggesting formation of a (μ-O)2-Fe2IV/IV complex, Q, in the catalytic cycle of soluble methane monooxygenase (28), we consider it very likely that the MnIV/FeIV intermediate also has this [M2O2(H)n](4+n)+ “diamond core” structure. Its half-integer (S = 1/2) electron-spin ground state, which contrasts with the S = 0 ground state of Q, and heterodinuclear rather than homodinuclear nature should afford unique opportunities to test this hypothesis and probe details of the core structure by electron-nuclear double resonance (ENDOR) and X-ray absorption experiments.
Scheme 1.

Mechanisms of the R2 activation reactions in Ct R2 (top) and E. coli R2 (bottom).
Q and the Y•-generating Fe2III/IV intermediate, X, form from the corresponding μ-peroxo-Fe2III/III intermediates in methane monooxygenase (29) and conventional RNR-R2 proteins (29, 30), respectively. However, no Fe2IV/IV complex has ever been detected in an R2 protein, either because O-O cleavage occurs reductively or because the Fe2IV/IV complex is reduced too rapidly to accumulate. In the best-studied R2 reaction, in E. coli R2, tryptophan (W) 48 near the protein's surface is the proximal electron source for this step, and the resultant W48 cation radical is readily reduced by a variety of compounds (FeIIaq, ascorbate, thiols) (Scheme 1, bottom) (31). A radical of the corresponding W residue in Ct R2, W51, has been detected during O2 activation by the Fe2II/II forms of variants of Ct R2 (W. Jiang, L. Saleh, J. M. Bollinger, Jr., unpublished observations), proving that this residue can function equivalently in the class Ic R2 and could, in principle, rapidly reduce the MnIV/FeIV cluster to limit its accumulation. Apparently, changes accompanying replacement of one Fe by Mn (e.g., of the mechanistic pathway or reduction potentials of constituent complexes), structural adjustments to the cluster site (e.g., the presence of E89 in Ct R2 in place of the D84 found in E. coli R2), or both allow the IV/IV state to build up uniquely in the Ct R2 protein. Nevertheless, the “saturation” of the observed rate constant in Figure 2C suggests that reduction of the MnIV/FeIV complex by ascorbate might also take place by a two-step mechanism, with the first step being the oxidation of W51 (or perhaps another residue). A rate-constant of 0.7 ± 0.1 s−1, the asymptotic value of kobs for decay of the intermediate, for the first step in this hypothetical electron-shuttling mechanism would rationalize the saturation of the decay rate constant at this value. This speculation should be testable by use of alternative reductants and variant R2 proteins.
It remains to be seen whether the MnIV/FeIV intermediate, like Q and X, forms from a (μ-peroxo)-M2III/III intermediate. The stopped-flow and freeze-quench EPR data provide no evidence for the accumulation of such a complex. Thus, as in E. coli R2, it might prove necessary to perturb the reaction kinetics (e.g., by replacement of a ligand, as in the D84E substitution in E. coli R2 that was shown to stabilize the peroxide intermediate (32)) to permit accumulation of a peroxide precursor to the MnIV/FeIV intermediate.
Supplementary Material
Footnotes
This work was supported by the National Institutes of Health (GM-55365 to JMB), the Beckman Foundation (Young Investigator Award to CK), and the Dreyfus Foundation (Teacher Scholar Award to CK).
Abbreviations: Ct, Chlamydia trachomatis; EPR, electron paramagnetic resonance; PCET, proton-coupled electron transfer; RNR, ribonucleotide reductase; NDP, ribonucleoside diphosphate ; dNDP,: 2′-deoxyribonucleoside 5′-diphosphate.
It is generally considered appropriate to invoke the pseudo-first-order approximation implicit in this fitting analysis only when one reactant is in 10–20-fold excess over the other. In these experiments, O2 is in excess over the theoretical concentration of reactive MnII/FeII-R2 complex by a minimum of 2.3-fold and a maximum of 9.0-fold. However, within this range, the apparent first-order rate constant still behaves as a nearly linear function of the concentration of the excess reactant (see Figure S3) and the error introduced into the second-order rate constant by the approximation is small (∼ 10%) in comparison with other sources (e.g., ∼ 25% in the values of [O2]).
The MnIV sites of the Mn2III/IV cluster of catalase and the MnIV/FeIV intermediate in Ct R2 have the same spin projection factors, and therefore the magnitudes of the AMn-tensors with respect to the total spin of the S = 1/2 ground state can be directly compared.
Candidates for the FeII complex(es) are aqueous FeII, complexes in which the divalent metal is bound to R2 either in mononuclear fashion or in a homodinuclear or heterodinuclear cluster, or some combination of these. The FeII-associated features remaining in the spectrum of the 2-s sample are different from the spectrum of the reactant complex and thus cannot simply be removed by subtraction of this spectrum.
The 4.2-K/53-mT Mössbauer features of the Fe2III/IV cluster, X, of Ct R2, which accumulates to a large amount in the reaction of the Fe2-form of Ct R2 are almost identical to those of X from E. coli R2 (unpublished results). Therefore, we used the published parameters of E. coli X (26) to simulate X of Ct R2.
The standard tactic of raising the temperature to make the electronic fluctuation rapid with respect to the nuclear precession frequency and thereby collapse the magnetic spectrum into a quadrupole doublet for more accurate determination of δ and ΔEQ failed. The 120 K/zero-field spectrum is very broad and featureless, implying that the fluctuation rate of the electronic states is comparable to the 57Fe Larmor frequency (the intermediate relaxation regime) at this temperature.
Supporting Information Available. Calculated energies and spin expectation values of the 55Mn hyperfine interaction in low magnetic fields, EPR spectrum of the O2-reactive MnII/FeII-R2 complex, analysis proving the applicability of the pseudo-first-order approximation in analysis of the [O2]-dependent stopped-flow data, and a comparison of the 4.2-K/53-mT Mössbauer spectra of the MnII/FeII-R2 reactant complex and samples frozen at various times after reacting this complex with O2. This material is available free of charge at the journal website http://pubs.acs.org.
REFERENCES
- 1.Stubbe J. Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 2.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the Class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 3.Nordlund P, Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 4.Licht S, Gerfen GJ, Stubbe J. Thiyl radicals in ribonucleotide reductases. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 5.Mao SS, Yu GX, Chalfoun D, Stubbe J. Characterization of C439SR1, a mutant of Escherichia coli ribonucleotide diphosphate reductase: evidence that C439 is a residue essential for nucleotide reduction and C439SR1 is a protein possessing novel thioredoxin-like activity. Biochemistry. 1992;31:9752–9759. doi: 10.1021/bi00155a031. [DOI] [PubMed] [Google Scholar]
- 6.Roshick C, Iliffe-Lee ER, McClarty G. Cloning and Characterization of Ribonucleotide Reductase from Chlamydia trachomatis. J. Biol. Chem. 2000;275:38111–38119. doi: 10.1074/jbc.M006367200. [DOI] [PubMed] [Google Scholar]
- 7.Högbom M, Stenmark P, Voevodskaya N, McClarty G, Gräslund A, Nordlund P. The radical site in Chlamydial ribonucleotide reductase defines a new R2 subclass. Science. 2004;305:245–248. doi: 10.1126/science.1098419. [DOI] [PubMed] [Google Scholar]
- 8.Bollinger JM, Jr., Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 9.Yun D, Krebs C, Gupta GP, Iwig DF, Huynh BH, Bollinger JM., Jr. Facile electron transfer during formation of cluster X and kinetic competence of X for tyrosyl radical production in protein R2 of ribonucleotide reductase from mouse. Biochemistry. 2002;41:981–990. doi: 10.1021/bi011797p. [DOI] [PubMed] [Google Scholar]
- 10.Voevodskaya N, Lendzian F, Gräslund A. A stable FeIII-FeIV replacement of tyrosyl radical in a class I ribonucleotide reductase. Biochem. Biophys. Res. Commun. 2005;330:1213–1216. doi: 10.1016/j.bbrc.2005.03.104. [DOI] [PubMed] [Google Scholar]
- 11.Voevodskaya N, Narvaez AJ, Domkin V, Torrents E, Thelander L, Gräslund A. Chlamydial ribonucleotide reductase: tyrosyl radical function in catalysis replaced by the FeIII-FeIV cluster. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9850–9854. doi: 10.1073/pnas.0600603103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak M-A, Krebs C, Bollinger JM., Jr. A Manganese(IV)/Iron(III) Cofactor in Chlamydia trachomatis Ribonucleotide Reductase. Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 13.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr., Krebs C. The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 14.Münck E. In: Physical Methods in Bioinorganic Chemistry. Que L Jr., editor. University Science Books; Sausalito, CA: 2000. pp. 287–319. [Google Scholar]
- 15.Glerup J, Weihe H. Magnetic Susceptibility and EPR Spectra of (μ-Hydroxo)bis[pentaamminechromium(III)] Chloride Monohydrate. Inorg. Chem. 1997;36:2816–2819. doi: 10.1021/ic970029c. [DOI] [PubMed] [Google Scholar]
- 16.Jiang W, Bollinger JM, Jr., Krebs C. The active form of Chlamydia trachomatis ribonucleotide reductase R2 protein contains a heterodinuclear Mn(IV)/Fe(III) cluster with S = 1 ground state. J. Am. Chem. Soc. 2007 doi: 10.1021/ja072528a. published on the web on May 27, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochiai E, Mann GJ, Gräslund A, Thelander L. Tyrosyl free radical formation in the small subunit of mouse ribonucleotide reductase. J. Biol. Chem. 1990;265:15758–15761. [PubMed] [Google Scholar]
- 18.Elgren TE, Lynch JB, Juarez-Garcia C, Münck E, Sjöberg BM, Que L., Jr. Electron transfer associated with oxygen activation in the B2 protein of ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 1991;266:19265–19268. [PubMed] [Google Scholar]
- 19.Bollinger JM, Jr., Tong WH, Ravi N, Huynh BH, Edmonson DE, Stubbe J. Mechanism of Assembly of the Tyrosyl Radical-Diiron(III) Cofactor of E. coli Ribonucleotide Reductase. 2. Kinetics of The Excess Fe2+ Reaction by Optical, EPR, and Moessbauer Spectroscopies. J. Am. Chem. Soc. 1994;116:8015–8023. [Google Scholar]
- 20.Zheng M, Khangulov SV, Dismukes GC, Barynin VV. Electronic structure of dimanganese(II,III) and dimanganese(III,IV) complexes and dimanganese catalase enzyme: a general EPR spectral simulation approach. Inorg. Chem. 1994;33:382–387. [Google Scholar]
- 21.Sinnecker S, Neese F, Lubitz W. Dimanganese catalase-spectroscopic parameters from broken-symmetry density functional theory of the superoxidized MnIII/MnIV state. J. Biol. Inorg. Chem. 2005;10:231–238. doi: 10.1007/s00775-005-0633-9. [DOI] [PubMed] [Google Scholar]
- 22.Lee S-K, Fox BG, Froland WA, Lipscomb JD, Münck E. A transient intermediate of the methane monooxygenase catalytic cycle containing an FeIVFeIV cluster. J. Am. Chem. Soc. 1993;115:6450–6451. [Google Scholar]
- 23.Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. Spectroscopic detection of intermediates in the reaction of dioxygen with the reduced methane monooxygenase/hydroxylase from Methylococcus capsulatus (Bath) J. Am. Chem. Soc. 1994;116:7465–7466. [Google Scholar]
- 24.Bencini A, Gatteschi D. EPR of exchange coupled systems. Springer; Berlin, Germany: 1990. [Google Scholar]
- 25.Krebs C, Price JC, Baldwin J, Saleh L, Green MT, Bollinger JM., Jr. Rapid Freeze-Quench 57Fe Mössbauer Spectroscopy: Monitoring Changes of an Iron-Containing Active Site during a Biochemical Reaction. Inorg. Chem. 2005;44:742–757. doi: 10.1021/ic048523l. [DOI] [PubMed] [Google Scholar]
- 26.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. Reconsideration of X, the diiron intermediate formed during cofactor assembly in E. coli ribonucleotide reductase. J. Am. Chem. Soc. 1996;118:7551–7557. [Google Scholar]
- 27.Hotzelmann R, Wieghardt K, Flörke U, Haupt HJ, Weatherburn DC, Bonvoisin J, Blondin G, Girerd JJ. Spin exchange coupling in asymmetric heterodinuclear complexes containing the μ-oxo-bis(μ-acetato)dimetal core. J. Am. Chem. Soc. 1992;114:1681–1696. [Google Scholar]
- 28.Shu L, Nesheim JC, Kauffmann KE, Münck E, Lipscomb JD, Que L., Jr. An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 29.Tong WH, Chen S, Lloyd SG, Edmondson DE, Huynh BH, Stubbe J. Mechanism of assembly of the diferric cluster-tyrosyl radical cofactor of Escherichia coli ribonucleotide reductase from the diferrous form of the R2 subunit. J. Am. Chem. Soc. 1996;118:2107–2108. [Google Scholar]
- 30.Yun D, Garcia-Serres R, Chicalese BM, An YH, Huynh BH, Bollinger JM., Jr. (μ-1,2-Peroxo)diiron(III/III) Complex as a Precursor to the Diiron(III/IV) Intermediate X in the Assembly of the Iron-Radical Cofactor of Ribonucleotide Reductase from Mouse. Biochemistry. 2007;46:1925–1932. doi: 10.1021/bi061717n. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM., Jr. Mechanism of Rapid Electron Transfer during Oxygen Activation in the R2 Subunit of Escherichia coli Ribonucleotide Reductase. 1. Evidence for a Transient Tryptophan Radical. J. Am. Chem. Soc. 2000;122:12195–12206. [Google Scholar]
- 32.Bollinger JM, Jr., Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. Engineering the diiron site of Escherichia coli ribonucleotide reductase protein R2 to accumulate an intermediate similar to Hperoxo, the putative peroxodiiron(III) complex from the methane monooxygenase catalytic cycle. J. Am. Chem. Soc. 1998;120:1094–1095. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.