

Abstract
Human exposure to dithiocarbamates results from their uses as pesticides, in manufacturing and as pharmaceutical agents. Neurotoxicity is an established hazard of dithiocarbamate exposure and has been observed in both humans and experimental animals. Previous studies have shown that the neurotoxicity of certain dithiocarbamates, including N,N-diethyldithiocarbamate (DEDC), disulfiram, and pyrrolidine dithiocarbamate can manifest as a primary myelinopathy of peripheral nerve. Because increased levels of copper in peripheral nerve and elevated levels of lipid peroxidation products accompany DEDC induced lesions it has been suggested that disruption of copper homeostasis and increased oxidative stress may contribute to myelin injury. To further assess the biological impact of DEDC-mediated lipid peroxidation in nerve the changes in protein expression levels resulting from DEDC exposure were determined. In addition, protein carbonyl content in peripheral nerve was also determined as an initial assessment of protein oxidative damage in DEDC neuropathy. Rats were exposed to DEDC by intraabdominal osmotic pumps for 8 weeks and proteins extracted from sciatic nerves of DEDC exposed animals and from non-exposed controls. The comparison of protein expression levels using two dimensional difference gel electrophoresis demonstrated significant changes in 56 spots of which 46 were identified by MALDI-TOF/MS. Among the proteins showing increased expression were three isoforms of glutathione transferase, important for detoxification of reactive αβ,-unsaturated aldehydes generated from lipid peroxidation. The increased expression of one isoform, glutathione transferase pi, was localized to the cytoplasm of Schwann cells using immunohistochemistry. Immunoassay for nerve protein carbonyls demonstrated a significant increase of approximately 2 fold for the proteins isolated from the DEDC exposed rats. These data support the ability of DEDC to promote protein oxidative damage in peripheral nerve and to produce sufficient lipid peroxidation in either myelin or another component of the Schwann cell to elicit a protective cellular response to oxidative stress.
Keywords: oxidative stress, diethyldithiocarbamate, protein carbonyls, copper, sciatic nerve, demyelination, proteomic analysis, myelinopathy
Introduction
Dithiocarbamates are a non-systemic group of pesticides widely used in agriculture to control insects and fungi, and also have multiple applications in industry including the manufacture of sugar, rubber, paper, and water treatment. The use of disulfiram for alcohol aversion therapy is probably the best recognized pharmaceutical application of a dithiocarbamate but additional new therapeutic applications of dithiocarbamates are currently being investigated for cancer (1), AIDS (2), inflammatory diseases, and atherosclerosis (3, 4). In addition, dithiocarbamates have been proposed as biological tools to modulate gene transcription (5, 6) and as antiviral agents (7-9). Delineating the hazards and mechanisms of dithiocarbamates is important for determining the risks associated with environmental exposures to dithiocarbamates and for developing safer and more efficacious dithiocarbamates for therapeutic purposes.
Previous studies have demonstrated the ability of dithiocarbamates to produce neurotoxicity in both humans and in experimental animals (10-14). Depending on the route of exposure and the chemical structure of the dithiocarbamate, two types of neuropathies have been observed. Oral administration of an acid labile dithiocarbamate, e.g., N,N-diethyldithiocarbamate (DEDC)1, can produce a CS2-mediated axonopathy (15); whereas oral administration of an acid stable dithiocarbamate, e.g., pyrrolidine dithiocarbamate or disufiram (16, 17), as well as parenteral administration of DEDC (18) has been shown to result in a primary myelinopathy characterized by intramyelinic edema, thin myelin and demyelination. In addition to the myelin structural lesions observed in peripheral nerve, increases in copper levels and F2 isoprostanes result from parenteral administration of DEDC or pyrrolidine dithiocarbamate (19, 20). Similarly, increases in brain copper levels and thiobarbituric acid reactive substances have been reported following administration of disulfiram or DEDC (21-24). Interestingly, the ability of dithiocarbamates to alter metal levels within the nervous system appears to be limited to copper with no significant DEDC-mediated changes in other metals observed by inductively coupled plasma-atomic emission spectroscopy in previous studies (17, 20). As a result, copper accumulation and lipid peroxidation have been proposed as contributing processes to the observed myelin injury in peripheral nerve produced by dithiocarbamates.
Although previous studies have demonstrated increases in nervous system copper levels and markers of lipid peroxidation they have not localized the sites of elevated copper or lipid peroxidation and have not determined whether the excess copper exists in a redox active form or if the level of lipid peroxidation is biologically significant. Since lipid peroxidation appears to be an ongoing process within the nervous system, as evidenced by detectable levels of thiobarbituric acid reactive substances and F2 isoprostanes in controls (20, 21), this latter point appears particularly relevant to assessing whether oxidative stress contributes to DEDC-mediated myelinopathy. To further characterize the biological impact of oxidative stress in DEDC myelinopathy, changes in protein expression and protein oxidative damage were assessed in peripheral nerve using an established model for DEDC-mediated myelinopathy in the rat. Subchronic intraabdominal administration of DEDC was performed using osmotic pumps after which proteins were isolated from the sciatic nerves of exposed animals and non-exposed controls. The relative expression levels of nerve proteins were determined using two dimensional difference gel electrophoresis (2D-DIGE) and matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF/MS). As an initial assessment of protein oxidative damage, protein carbonyls were quantified by immunoassay after their derivatization with fluoresceineamine. Additionally, the localization of structural changes and expression of glutathione transferase pi (GST-pi) were performed using light microscopy and immunohistochemistry.
Experimental Procedures
Materials
2ML4 Alzet® osmotic pumps were obtained from Braintree Scientific (Braintree, MA). Sodium N,N-diethyldithiocarbamate (DEDC) was obtained from Alfa Aesar (Ward Hill, MA). Fluoresceinamine and sodium cyanoborohydride (NaCNBH3) were purchased from VWR Scientific (Atlanta, GA). N-hydroxy succinimide ester Cy2, Cy3 and Cy5 dyes were purchased from Amersham Biosciences (Piscataway NJ); Sypro Ruby gel stain was purchased from Molecular Probes (Eugene, OR). Protease and phosphatase inhibitors were purchased from Amersham Biosciences (Piscataway, NJ) and Sigma-Aldrich (Saint Louis, MO).
Animals
All treatments and procedures using animals were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Vanderbilt University. Adult male Sprague-Dawley rats obtained from Harlan Bioproducts (Indianapolis, IN) were caged at Vanderbilt University Animal facilities in a temperature (21-22 °C) controlled room with a 12 h dark-light cycle, supplied with Purina Lab Diet 2001 and water ad libitum. Body weights were determined prior to surgery and then twice weekly during the course of the experiment. Twelve rats (351.7 g ± 3.5) were administrated DEDC at 0.3 mmol/Kg/day for 4 weeks using surgically implanted intraabdominal Alzet® osmotic pumps. After 4 weeks, the pumps were replaced using the same procedure to extend the exposure period to 8 weeks. DEDC was delivered as an aqueous solution, the concentration of which was determined from the UV absorbance at 282 nm (ε= 13,000 M-1cm-1) prior to filling the pumps. DEDC was sterilized prior to filling the pumps by filtering through a 0.22 μm-syringe filter. Hind limb grip strengths were measured using a digital force gauge to assess neuromuscular function at the end of the exposure (25).
Morphology and Immunohistochemistry
For morphologic evaluations, four DEDC exposed and four non-exposed control animals were perfused through the left ventricle of the heart under deep anesthesia (100 mg/kg pentobarbital ip) with a solution of 0.8% NaCl, 0.025% KCl, 0.05% NaHCO3, in 0.01 M phosphate buffer followed by 4% glutaraldehyde in 0.1M phosphate buffer, pH 7.4. After perfusion, sciatic nerves were dissected and immersed in 4% glutaraldehyde in 0.1M phosphate buffer overnight and then transferred to 0.1 M phosphate buffer (pH 7.4). Sciatic nerve sections were post-fixed with osmium and embedded in Epon; thick sections (1 μm) were cut and stained with toluidine blue. The thick sections of peripheral nerve were evaluated by light microscopy on an Olympus BX41 microscope equipped with an Optronics Microfire digital camera. One cross section of sciatic nerve was examined per animal and the total number of lesions counted by one observer (WMV). The lesions quantified were: degenerated axons, axons with thin myelin (axon/axon with myelin diameter ratio (g ratio) greater than 0.7, intramyelinic edema, and demyelinated axons. Thin (70 nm) sections were prepared from sciatic nerves and evaluated using a Phillips CM-12 electron microscope, 120 keV with a high resolution CCD camera system.
For immunohistochemistry, four DEDC exposed animals and four controls were perfused and the nerves prepared similarly to the samples for morphologic assessments except that 4% paraformaldehyde was used as the protein fixative instead of glutaraldehyde; and the nerves were embedded in paraffin without prior osmium fixation. Sections of rat nerve on + slides were then deparaffinized by placing in 3 xylene baths, 10 minutes each, rehydrated through a series of graded alcohols (twice in 100%, once in 95% and once in 80%) 5 minutes each, and finally placed in TBS buffer. The slides were then heated in a 0.01 M citrate buffer for 25 minutes in a preheated steamer. Slides were allowed to cool at room temperature for an additional 15 minutes and then placed in a buffer of TBS-0.1% Tween-20. Endogenous peroxide was blocked for 5 minutes using a peroxidase blocking reagent (Dako, Catalog #S2001) for 5 min followed by buffer rinse. The predetermined optimal dilution of primary antibody (1:500 of Rabbit anti-rat glutathione transferase pi (GST-pi) from Alpha Diagnostic International, Catalog #GSTP11-S) was applied for 1 hour at room temperature (diluent used: Dako Diluent, Catalog #S0809). Slide staining was completed using the Dako Polyclonal EnVision + kit (Catalog #K4010) polymer system following the manufacturer’s directions exactly.
Protein Extraction and Quantification
Four DEDC-exposed and four control rats were deeply anesthetized and exsanguinated by cardiac puncture. The sciatic nerves were then removed and immediately frozen in liquid nitrogen and stored at -80 °C. One frozen sciatic nerve of ≥ 15mm length was thawed and homogenized using a mini-bead beater for 3 min at 4,800 rpm in chilled TNE buffer (2mM EDTA, 150 mM NaCl, 50mM Tris-base, 2 mM DTT, NP-40 (1%, v/v)), containing phosphatase and protease inhibitors, and then sonicated at 0 °C for 1 min. The homogenate was centrifuged at 10,000g for 20 min at 4°C and the supernatant collected. The extraction step was repeated using the remaining pellet, and the supernatant collected and combined with the previous extract and stored at -80 °C. Protein concentration in the supernatant of control and exposed nerve samples was measured by a modified Bradford method (26) using bovine serum albumin as standard.
RP-HPLC Analysis of Globins
Globin isolation and analyses to quantify cysteine carbamylation was performed as previously described (16, 27). Blood was collected from anesthetized animals via cardiac puncture and 0.5-1 mL used to isolate globin for analysis by HPLC. Blood was separated into plasma and hemolysate as described (27) and 100 μL of 1 N ascorbic acid added to each 1 mL hemolysate. The resulting solution was added dropwise to 10 mL of 2.5% oxalic acid in acetone. Globin was allowed to precipitate on ice for 30 min, then centrifuged at 10,000g for 10 min at 4°C and washed in 5 mL ice-cold acetone. The pellet, containing crude globin, was dried under a stream of N2 and stored at -80°C. Dried globin was solubilized with 0.1% trifluoroacetic acid (TFA) to produce a solution for HPLC analysis.
Globin chains were separated by RP-HPLC on a Phenomenex Jupiter 5μm column (150 5 460 mm) using a Waters 2690 liquid chromatograph after adjusting sample concentration to a UV absorption of 1.0 ± 0.2 at 280 nm. Globins were separated using a linear gradient from 56% solvent A, 44% solvent B to 30% solvent A, 70% solvent B over 30 min followed by a linear gradient to 100% solvent B over 10 min. Solvent A was 20:80:0.1 acetonitrile:water:TFA and solvent B was 60:40:0.08 acetonitrile:water:TFA. The elution of globin peaks was monitored by their UV absorption at 220 nm using a Waters 996 photodiode array detector.
Carbonyl determination
Protein carbonyl content of nerve samples and standards were determined by the fluorescein amine-cyanoborohydride method using immunochemical detection as previously described (28). Briefly, 25 μg aliquots of protein were treated with fluoresceinamine (12 μL of 0.25 M) and sodium cyanoborohydride (10 μL of 0.4 M) for 1 hour at 37°C. Next, protein was precipitated at room temperature with methanol:water:chloroform (4:3:1, v/v). Precipitates were washed 5 times with acidified ethanol:ethyl acetate (1:1) for 5 min at 37°C followed by centrifugation (13,000 rpm, 9 min) and then solubilized in 250 μL sodium hydroxide (0.1 N) for 15 min at 37°C. Treated proteins (controls or DEDC exposed) were bound to Immobilon-P membranes (Millipore, MA) using the Bio-Dot® Blot apparatus (BioRad, CA). Four replicates per sample containing approximately 0.4 μg of sciatic nerve protein per well were loaded. Protein carbonyls were detected using the CDP-Star Universal Alkaline Phophatase kit (Sigma-Aldrich Co., MO) following the manufacturer’s instructions. Kodak X-Omat Blue XB-1 film was exposed to the treated membrane at room temperature for 20 min. For quantification, films were scanned with a GS-700 densitometer and analyzed using Quantity One® 1-D Analysis Program version 4.1 (Bio-Rad, CA). The protein carbonyl content of samples was determined from a standard curve generated using oxidized bovine serum albumin (BSA) for which carbonyl content was determined spectrophotometrically (ε=86,000 M-1cm-1 at 490 nm) using a Shimadzu UV-2401 PC. Oxidized BSA standards were prepared by incubating 10 mg of BSA dissolved in 1 ml of 20 mM TrisHCl (pH 7.4) with 100 mM Fe2+ and 100 mM hydrogen peroxide, at room temperature for 1 hour. Reduced BSA was prepared by mixing oxidized-BSA (10 mg/mL) with 5 mg of sodium borohydride for 30 min at 37°C. The quantity of nerve protein bound to the PVDF membrane was determined using the MemCode™Reversible Protein Stain Kit for PVDF membranes (Pierce, IL) or the Colloidal Gold Protein stain (Bio-Rad, CA) using BSA as standards.
Cy-dye Labeling
The mixed internal standard methodology, i.e. standard samples comprising equal amounts of each experimental control and DEDC-exposed sciatic nerve protein (N=3 for each) was used in this study (29). Aliquots (250 μg) of each sciatic nerve protein extract from DEDC and control were individually precipitated with methanol:water:chloroform (4:3:1, v/v) at room temperature (30) and re-suspended in 30 μL labeling buffer (7 M urea, 2 M thiourea, 4% w/v CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 30 mM Tris, 5 mM magnesium acetate). One-third of each sample (83.3 μg; 10 μL) was combined in a new tube for bulk-labeling with Cy2 (pooled-sample internal standard), and the remaining two-thirds (166.7 μg; 20 μL) were individually labeled with 200 pmol of Cy3 or Cy5 per the manufacturer’s recommended protocols according to the following:
| Cy3 | Cy5 | Cy2 | |
|---|---|---|---|
| Gel 1 | con1 | DEDC1 | std |
| Gel 2 | DEDC2 | con2 | std |
| Gel 3 | con3 | DEDC3 | std |
The pooled-sample internal standard was similarly labeled in bulk with 600 pmol Cy2. Cy3- and Cy5-labeled samples were then mixed with an equal aliquot of the Cy2-labeled internal standard and co-resolved across the three DIGE gels as indicated above. Mixed samples were reduced by the addition of equal volume of 2x sample buffer (7 M urea, 2 M thiourea, 4% w/v CHAPS, 4 mg/mL DTT), and brought up to a final volume of 450 μL with 1x sample buffer (same as 2x except for 2 mg/mL DTT) and IPG buffer (3-10NL) was added to 0.5% (v/v).
2-D DIGE Analysis
Tripartite-labeled samples were separated by standard 2D gel electrophoresis using an IPGphor first dimension isoelectric focusing unit and 24-cm pH 3-10 NL immobilized pH gradient (IPG) strips (Amersham Biosciences, Uppsala, Sweden). Prior to the second-dimensional SDS-PAGE, the focused strips were equilibrated in buffer (30% glycerol, 2% SDS, 6 M urea, 50 mM Tris pH 8.8, trace bromophenol blue) supplemented with 1% DTT for 20 min at room temperature, followed by 2.5% iodoacetamide in fresh equilibration buffer for an additional 20 min room temperature incubation. After equilibration, proteins were separated on 12 % SDS-PAGE gels using an Ettan-DALT 12 unit (Amersham Biosciences) according to the manufacturer’s protocols.
The Cy2 (standard sample), Cy3 (sample-x), and Cy5 (sample-y) components of each gel were individually imaged using mutually exclusive excitation/emission wavelengths of 480/530 nm for Cy2, 520/590 nm for Cy3, and 620/680 nm for Cy5 using a 2D 2920 Master Imager (Amersham Biosciences). After 2D-DIGE and image analysis, the protein spot pattern was visualized by Sypro Ruby stain using standard protocols (Molecular Probes), and images acquired on the same imager using 400/633nm wavelengths. DeCyder software (Amersham Biosciences) was used for simultaneous comparison of abundance changes across all control/DEDC-exposed sample pairs with statistical confidence and without interference from gel-to-gel variation (29, 31). Control:standard and DEDC-exposed:standard volume ratios for each resolved protein were calculated within each gel (where no gel-to-gel variation exists), and then these Cy3:Cy2 and Cy5:Cy2 abundance ratios were normalized across the three DIGE gels using the Cy2 signal for each protein separately. Spots with abundance changes of ≥1.3-fold increase or decrease and p values less than 0.05 (Student’s t-test, N=3) were identified and analyzed by Matrix-assisted laser desorption ionization, time-of-flight mass spectrometry (MALDI-TOF MS).
Protein Identification by MS and Database Analysis
Proteins of interest were robotically excised and processed using an Ettan Spot Picker and Digester workstation (Amersham Bioscences). Gel plugs were excised and digested in-gel with 10 μL porcine modified trypsin protease (Promega, Madison, WI) in 20 mM ammonium bicarbonate for 3 h at 37°C. Peptide extracts were used for protein identification using MALDI-TOF MS and tandem TOF/TOF mass spectrometry on a Voyager 4700 (Applied Biosystems, Foster City, CA). MALDI-TOF peptide mass maps and accompanying tandem mass spectra were used to interrogate rat sequences deposited in the SWISS-PROT and National Center for Biotechnology Information (NCBInr) databases using the MASCOT (www.matrixscience.com) and ProFound (prowl.rockefeller.edu) search algorithms, respectively. MALDI-TOF peptide mass maps were internally calibrated to within 20 ppm mass accuracy using trypsin autolytic peptides (m/z = 842.51 and 2211.10). Searches were performed without constraining protein molecular weight or isoelectric point, and allowed for carbamidomethylation of cysteine, partial oxidation of methionine residues, and one missed trypsin cleavage.
Networks, Pathways and Functional Mapping
Data were analyzed through the use of Ingenuity Pathways Analysis (Ingenuity® Systems, www.ingenuity.com) as previously described (32, 33). A data set containing protein identifiers and corresponding expression values was uploaded in the application. Each protein identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. A fold change cutoff of ≥ 1.3 (p < 0.05) was set to identify genes whose expression was significantly differentially regulated. These genes, called focus genes, were overlaid onto a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. Networks of these focus genes were then algorithmically generated based on their connectivity. The Functional Analysis of a network identified the biological functions and/or diseases that were most significant to the genes in the network. The network genes associated with biological functions and/or diseases in the Ingenuity Pathways Knowledge Base were considered for the analysis. Fischer’s exact test was used to calculate a p-value determining the probability that each biological function and/or disease assigned to that network is due to chance alone.
Statistical Analysis
The one-tailed unpaired t-test, one-sided Fisher’s exact test and square root transformations were performed using Prism 4.0 (GraphPad Software, Inc.) Statistical significance was p< 0.05 unless otherwise noted.
Results
2D-DIGE
A representative image from the 3 gel 2D-DIGE experiment is shown in Figure 1. Fifty-six protein features with an increase or decrease change larger than 1.3-fold (p<0.05) were picked from the gel and excised for trypsin digestion. The resulting peptide mixtures were then subjected to MALDI-TOF MS and TOF/TOF tandem MS followed by database interrogation to produce statistically significant candidate protein identifications. Forty-six features corresponding to 33 non-redundant proteins were identified in this manner, of which 18 were up regulated and 16 down regulated after DEDC-exposure (Table 1 and 2). Among these was one protein, dihydropyrimidase-like 2 protein, that produced a spot that was up regulated as well as several spots that were down regulated suggesting a shift in the population of post translationally modified isoforms.
Figure 1.

Fluorescent 2D DIGE image showing protein expression differences in TNE buffer extracts of rat sciatic nerve from control and DEDC exposed rats; pH 3-10 2D gel. Sciatic nerve proteins were labeled with either Cy3 (control), Cy5 (DEDC) and subjected to 2D gel electrophoresis with a Cy2-labeled pooled standard using pH 3-10 IPG strips. Differential expression of proteins was determined as detailed in the Material and Methods section. Approximate isoelectric points are shown along the horizontal axis and approximate molecular weights are shown along the vertical axis. Numbered spots represent proteins that underwent a significant increase or decrease in expression (≥ 1.3 fold change p<0.05) in DEDC exposed rats relative to controls. The gel shown is representative of results obtained for 3 different 2D DIGE comparisons. Enlargements for spot 51 (glutathione transferase alpha 3) are shown to the right with the corresponding contour plot for the Cy2, Cy3, and Cy5 images.
Table 1.
Identified proteins up-regulated in the DEDC-exposure group vs. control group
| Average ratio DEDC Level vs. controls | Spot No. | t-test | Accession No. | Protein |
|---|---|---|---|---|
| 1.68 | 26 | 0.002 | 70912366 | ARP3 actin related protein 3 homolog (yeast) |
| 2.43 | 25 | 0.04 | 62511242 | aldehyde dehydrogenase-9A1 (ALDH 9, E3 isoenzyme) |
| 2.34 - 2.79 | 44, 45 | 0.03 | 9845234 | annexin A2 |
| 2.51 | 55 | 0.001 | 6680924 | cofilin-1 (non-muscle) |
| 1.82 - 1.89 | 13, 14 | 0.01 | 1351260 | dihydropyrimidase- like 2 |
| 1.43 | 15 | 0.01 | 25742598 | translocase of inner mitochondrial membrane 10 homolog |
| 1.91 | 19 | 0.02 | 29789106 | fibrinogen beta chain |
| 1.47 | 2 | 0.03 | 51854226 | gelsolin (amyloidosis, Finnish type) |
| 2.57 | 51 | 0.006 | 31981724 | glutathione S-transferase A3 |
| 1.31 | 53 | 0.01 | 6680121 | glutathione S-transferase M1 |
| 2.08 | 54 | 0.006 | 1170099 | glutathione S-transferase pi |
| 6.3 | 27 | 0.03 | 121043 | immunoglobulin heavy chain 1a (serum IgG2a) |
| 1.47 - 2.75 | 8 | 0.0001 | 758263 | kinnogen-1 |
| 4.57 - 6.84 | 28, 29, 30, 31 | 0.02 | 62901522 | immunoglobulin heavy chain (IgG) |
| 2.75 | 9, 10, 11, 12 | 0.001 | 205308 | alpha-1 major acute phase protein prepetide |
| 2.92 | 21 | 0.05 | 6981416 | peripherin |
| 1.31 | 17 | 0.05 | 16757994 | pyruvate kinase, muscle |
| 1.6 | 16 | 0.02 | 2506796 | pyruvate kinase, isozyme M2 |
Table 2.
Identified proteins down-regulated in the DEDC-exposure group vs. control group
| Average ratio DEDC Level vs. controls | Spot No. | t-test | Accession No. | Protein |
|---|---|---|---|---|
| (-2.16) - (-2.23) | 20, 22 | 0.01 | 8393538 | 3-hydroxy-3-methylglutaryl-coenzyme A synthase 1 |
| -1.58 | 42 | 0.006 | 33086552 | acetyl coenzyme A acetyltransferase-2 (acetoacetyl coenzyme A thiolase) |
| (-1.55) - (-1.64) | 3,4 | 0.003 | 40538860 | acotinase 2, mitochondrial |
| -1.97 | 37 | 0.003 | 52851386 | aminocylase-1 |
| -1.53 | 43 | 0.02 | 13242314 | aspartoacylase (Canavan disease) |
| -2.93 | 18 | 0.04 | 1351260 | dihydropyrimidase- like 2 |
| -1.55 | 34 | 0.01 | 46195798 | dolichyl-diphosphooligosaccharide - protein glycosyltransferase |
| -2.96 | 39 | 0.01 | 68186 | fructose-biphosphate aldolase |
| (-1.25) - (-1.39) | 48,49 | 0.01 | 2981437 | annexin A5 |
| -1.58 | 47 | 0.03 | 19923092 | monoglyceride lipase |
| -1.37 | 50 | 0.02 | 5803225 | monooxyegenase activaion protein, epsilon polypeptide |
| -2.84 | 36 | 0.002 | 6753304 | serine (or cysteine) protease inhibitor |
| (-1.39) - (-1.67) | 5,6 | 0.01 | 6175089 | transferrin |
| -2.14 | 38 | 0.01 | 14389299 | vimentin |
| -1.36 | 1 | 0.01 | 109501157 | vinculin |
| -1.41 | 52 | 0.01 | 464457 | proteosome (prosome, macropain) subunit, alpha type, 5 |
Dose, weight gain and neuromuscular function
The level of DEDC exposure was monitored by analysis of globin preparations using RP-HPLC (16, 27). At the end of 8 weeks, the level of modified ß3-globin (from the formation of S-(diethylaminocarbonyl)-cysteine adducts on Cys-125) expressed as a percent of total ß-globin was 27.6 ± 0.8 in DEDC exposed animals and was below the limit of quantification in controls. No significant difference was observed in mean body weights measured prior to DEDC exposure (352.7 ± 3.5g SEM) relative to those obtained post exposure (348.7 ± 4.2g SEM)) whereas a significant decrease in hind limb grip strength resulted from DEDC administration (971 ± 31g SEM prior to exposure compared to 804 ± 48g SEM post exposure).
Protein carbonyl content
Dot-blot and subsequent immunochemical detection of protein carbonyls demonstrated increased carbonyl content by approximately 2 fold in sciatic nerve proteins isolated from rats exposed to DEDC relative to control rats (Figure 2).
Figure 2.

Total protein carbonyl level in nerve proteins isolated from DEDC exposed rats and control rats determined by slot blot analysis. Proteins were isolated from sciatic nerves, derivatized with fluoresceinamine, slot blotted to PVDF and the amount of protein and protein carbonyls determined by protein staining with densitometry and immunodetection with chemiluminescence, respectively (see Materials and Methods for details). The total carbonyl level was significantly increased (p < 0.01, eight replications per animal, n = 4 for each group) in the sciatic nerve of the DEDC exposed group when compared to the control group. Bars represent mean ± SEM.
Morphology and Immunohistochemistry
Representative sections of sciatic nerve obtained from controls and animals exposed to DEDC are shown at the light microscope level in Figure 3 (A and B) and at the electron microscope level in Figure 4. Peripheral nerve lesions were observed in animals exposed to DEDC that were not present in control animals. Since the relative variability of lesion counts was high among individuals within the same treatment group the lesion counts were square root transformed to equalize the variance for statistical comparisons. A significant increase in the number of axons exhibiting thin myelin, demyelination and intramyelinic edema was observed in the DEDC group relative to controls (Table 3).
Figure 3.

Morphology and immunostaining of sciatic nerve sections stained with toluidine blue or probed with polyclonal rat anti-GST-pi. (A) Cross section obtained from a control rat showing the presence of large and small myelinated axons. The axons (a) are surrounded by normal compact myelin. One Schwann cell has been sectioned at the middle of the internode through its cytoplasm (*) and nucleus. (B) Cross section of a nerve obtained from a DEDC exposed rat showing thinly myelinated and demyelinated axons (light blue arrows) and intramyelinic edema (green arrowheads). A Schwann cell sectioned through its cytoplasm (*) and nucleus is present. (C) Cross section of a nerve obtained from a control animal that has been probed with anti-GST-pi antibody and counterstained with hematoxylin demonstrating binding of antibody within the axoplasm (a) but not within the Schwann cell cytoplasm (green arrows). (D) Cross section from a DEDC exposed rat probed with anti-GST-pi demonstrating positive immunostaining within the Schwann cell cytoplasm (blue arrows) in addition to the axons. (E) Longitudinal section from a DEDC exposed rat showing localization of positive anti-GST-pi immunostaining to a Schwann cell cytoplasm (blue arrow) and the presence of a nonimmunopositive Schwann cell (green arrow). (F) Cross section from a control processed identical to C, D and E but without anti-GST-pi primary antibody. (Black bars in A-F represent 20 μm)
Figure 4.
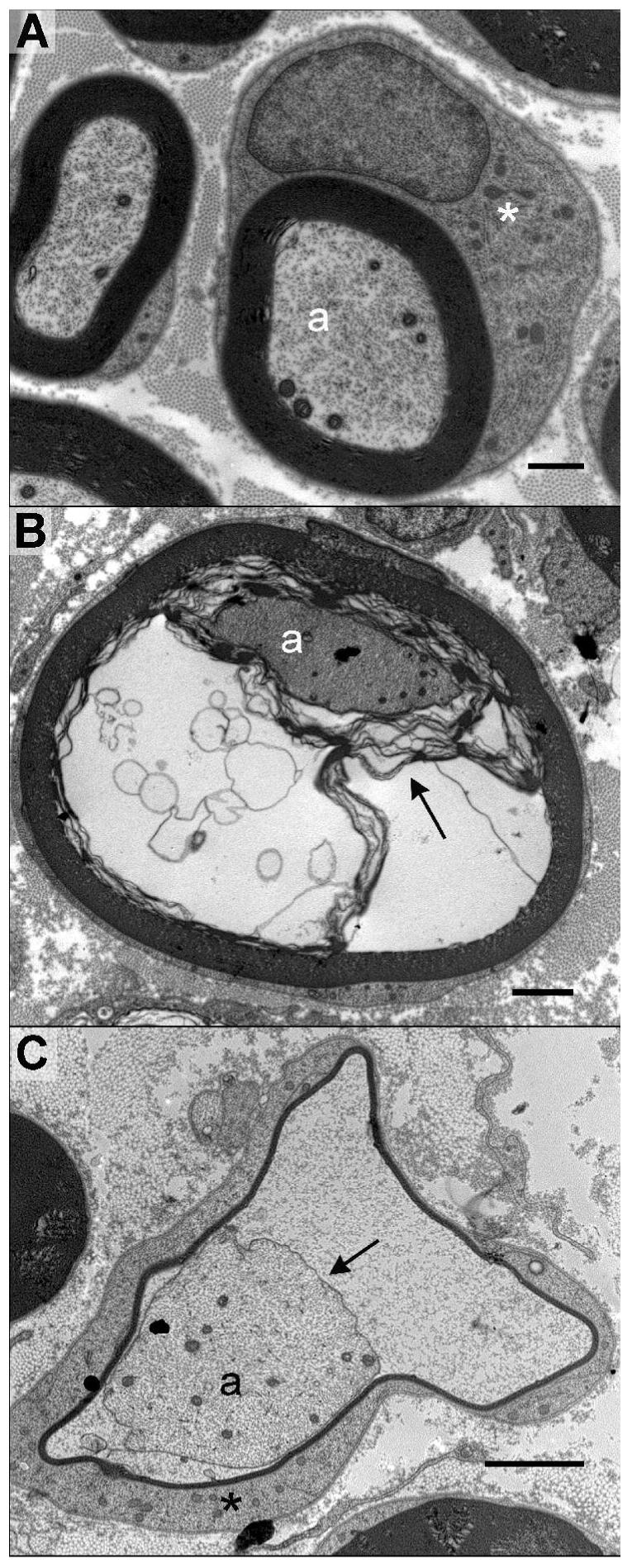
Electron micrographs of sciatic nerve cross sections from control and DEDC treated rats. (A) Large myelinated axons in a control. The myelinated axons (a) are surrounded by compact myelin, and Schwann cell cytoplasm (*) that has been sectioned through the nucleus. (B and C) Sections from a rat exposed to DEDC at 0.3 mmol/Kg/day for 8 weeks using intraabdominal osmotic pumps. (B) A myelinated axon (a) showing separation of the inner lamellae of myelin (arrow) creating fluid filled spaces within the myelin. (C) A myelinated axon (a) with its axolemma (arrow) separated from the myelin sheath. The myelin is very thin and surrounded by Schwann cell cytoplasm (*). (Black bars represent 1 μm in A and 2 μm in B and C)
TABLE 3. Incidence and Severity of Sciatic Nerve Lesionsa.
| Degenerated axons | Thin myelinb | Demyelination | Intramyelinic edema | |||||
|---|---|---|---|---|---|---|---|---|
| Treatment group | Incidencec | Severityd | Incidencec | Severityd | Incidencec | Severityd | Incidencec | Severityd |
| Controls | 4/4 | 2.16 (0.16) | 0/4 | 0 (0) | 0/4 | 0 (0) | 0/4 | 0 (0) |
| Rangee | (4 - 5) | |||||||
| DEDC Exposed | 4/4 | 2.38 (0.32) | 3/4 | 3.5 (1.2)f | 4/4g | 4.1 (0.9)f | 4/4g | 5.6 (1.5)f |
| Rangee | (3 - 10) | (0 - 35) | (9 - 48) | (13 - 99) | ||||
Epon cross sections (1μm) of peripheral nerve stained with toluidine blue were evaluated by light microscopy on an Olympus BX41 microscope using the 40X and 60X objectives. One cross section of sciatic nerve was examined per animal (4 animals per group) and the total number of lesions per section were counted by one observer (WMV). The lesions quantified were: degenerated axons, axons with thin myelin, axons with intramyelinic edema, and demyelinated axons.
Defined as having an axon/axon with myelin diameter ratio (g ratio) greater than 0.7.
Number of animals with positive observations/number of animals in treatment group.
Mean square root value of axons with lesion (SEM).
The least to greatest number of lesions observed for individual nerve sections within a treatment group.
p < 0.05 by one-tailed unpaired t test as relative to Controls.
p < 0.05 by one-sided Fisher’s exact test relative to Controls
Sciatic nerve sections obtained from the DEDC and control groups that were probed with anti-rat GST-pi antibody are shown in Figure 3 (C-E). Positive staining was observed within the axons of both treatment groups. In addition to the positive staining within the axoplasm, a significant number of Schwann cells exhibited positive staining within their cytoplasm in the nerves obtained from the DEDC exposed rats (Figure 3 (D and E) and Figure 5). No positive staining was observed in nerve sections obtained from either treatment group that were processed identically except for the omission of anti-rat GST-pi primary antibody (Figure 3 (F)).
Figure 5.

Percentages of Schwann cells showing GST-pi immunoreactivity in sciatic nerve. The Schwann cells within an entire cross-section of sciatic nerve from each control and DEDC exposed rat (n=4 per group) were counted and evaluated for positive immunostaining. The number of positively staining cells as a percent of total and the SEM were calculated (total Schwann cell count per section ranged from 24-56). Immunostaining of Schwann cell cytoplasm was increased significantly in the DEDC exposed rats (* p < 0.01 unpaired Student’s t-test).
Interaction Networks and Functional Mapping of the Nerve Proteomic Changes
Functional analyses were carried out on the data set using the Ingenuity Pathways Analysis as described in the Experimental Procedures. Protein-protein interactions were analyzed to determine the most significant networks and the network and global functions represented by the proteins undergoing change. The networks were scored (negative log of the p value) according to the number of focus proteins and the size of the network to approximate the significance of the network.
From the 33 sciatic nerve proteins that showed a significant change in expression from DEDC exposure 28 were eligible for network analysis based upon the presence of the gene in the Ingenuity Pathway Knowledge Base. Two high scoring networks (≥ 20) and two low scoring networks (both with a score of 2) were identified. A score of 31 was obtained for the network shown in Figure 6 (A) that involved 15 focus proteins. Six of the focus proteins were down regulated: annexin 5 (ANXA5), aconitase 2, mitochondrial (ACO2), acetyl-Coenzyme A acetyltransferase 2 (ACAT2), 3-hydroxy-3-methylglutaryl-Coenzyme A synthase (HMGCS1), vimentin (VIM), 5-monoxygenase activation protein (YWHAE); and 9 focus proteins were up regulated: serum IgG2a or immunoglobulin heavy chain 1a (IGH1A), ARP3 actin-related protein 3 homolog (ACTR3), kinnogen 1 (KNG1), pyruvate kinase (PKM2), UDP-glucose pyrophosphorylase 2 (UGP2), dihydropyrimidase-like 2 (DPYSL2), glutathione S-transferase A3 (GSTA3), glutathione S-transferase pi (GSTP1), glutathione S-transferase M1 (GSTM1). The most significant functions and diseases associated with this network were cancer (p = 6.00 × 10-8 involving 18 genes), cellular movement (p = 6.00 × 10-8 involving 16 genes) and cell to cell signaling and interaction (p = 8.42 × 10-8 involving 16 genes).
Figure 6.
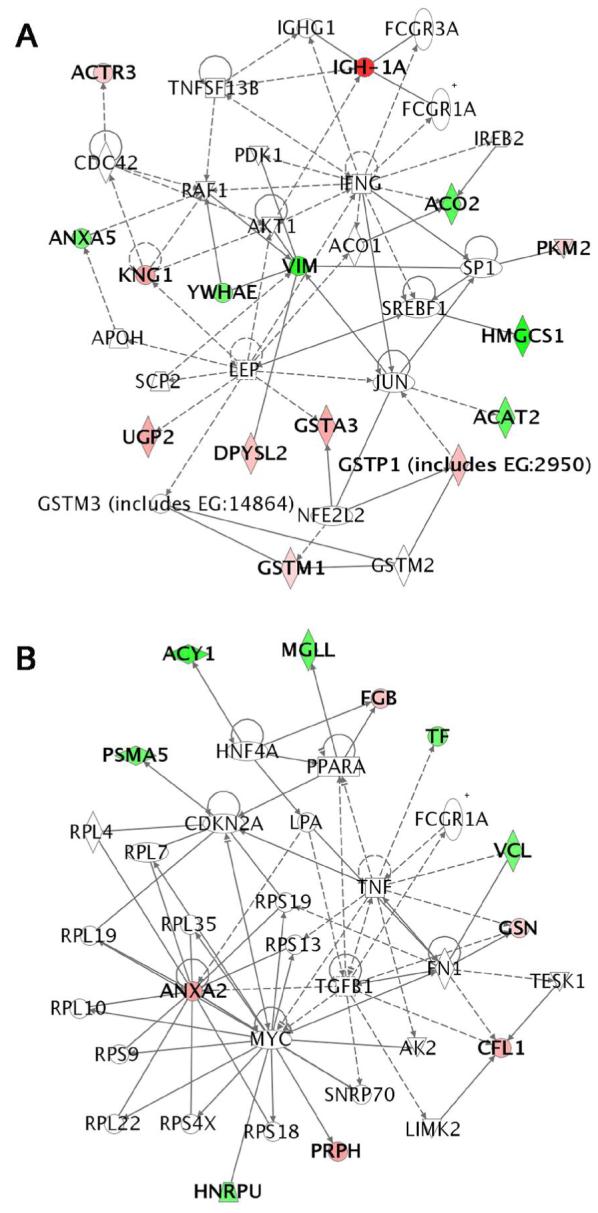
Graphical representation of the molecular relationships between genes/gene products of the two most significant networks determined by Ingenuity Pathways Analysis. Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as a line. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation. (A) The highest scoring network scored 30 and contained 15 focus proteins. Significant diseases and functions associated with this network included cancer, cellular movement, and cell to cell signaling and interaction. (B) The second highest scoring network scored 20 and contained 11 focus proteins. Significant functions and diseases associated with this network included protein synthesis, cellular movement, and connective tissue development and function.
The second highest scoring network is shown in Figure 6 (B) with a score of 20 and 11 focus proteins. There were 6 down regulated proteins: heterogeneous nuclear ribonucleoprotein U (HNRPU), proteosome subunit alpha type 5 (PSMA5), aminoacylase 1 (ACY1), monoglyceride lipase (MGLL), transferrin (TF), vinculin (VCL); and 5 up regulated proteins: annexin A2 (ANXA2), fibrinogen beta chain (FGB), gelsolin (GSN), cofilin (CFL1), and peripherin (PRPH). The most significant functions associated with this network were protein synthesis (p = 1.06 × 10-12 involving 13 genes), cellular movement (p = 2.97 × 10-8 involving 14 genes) and connective tissue development and function (p = 2.97 × 10-8 involving 8 genes).
The most significant global functions associated with the entire data set were organismal injury and abnormalities (p= 8.93 × 10-5 involving 4 genes) and cellular assembly and organization (p = 9.87 × 10-5 involving 12 genes). The most significant (p < 0.01) global canonical pathways were synthesis and degradation of ketone bodies (involving ACAT2 and HMGCS1) and glutathione metabolism (involving GSTA3, GSTM1 and GSTP1).
Discussion
In the current investigation protein oxidative damage and protein expression in peripheral nerve were examined to help delineate the role of oxidative stress in DEDC-mediated peripheral neuropathy. Protein oxidative damage was assayed using the fluoresceineamine-cyanoborohydride assay. Because sodium cyanoborohydride does not reduce the Schiff base resulting from flurosceinamine and ketones, this assay is thought to be relatively selective for protein aldehydes (28). Previous investigations suggest that at least two processes may have contributed to the generation of protein carbonyls by DEDC and that glutamic semialdehyde and aminoadipic semialdehydes are the major protein oxidative modifications (34, 35). Metal catalyzed protein oxidation due to excess copper is one possibility. Copper generated hydroxyl radicals could react with prolyl and arginyl residues or lysyl residues to generate glutamic semialdehydes or aminoadipic aldehydes, respectfully (34). Secondly, the increase in protein aldehydes may also be due in part to the reaction of lipid peroxidation products such as 4-hydroxynonenal and malondialdehyde with nucleophilic sites on proteins to form Michael adducts and N-propenal adducts as well. Regardless of the underlying mechanisms the observed elevation of protein carbonyls is consistent with increased oxidative protein damage (35).
Studies examining the disposition of 4-hydroxynonenal (HNE) as a representative αβ-unsaturated aldehyde produced by lipid peroxidation have identified three major metabolic pathways for HNE: conjugation with glutathione by glutathione transferases, reduction of the conjugated HNE by aldol reductase and oxidation of free or conjugated HNE by aldehyde dehydrogenases (36). A previous comparison of the expression levels of several antioxidant enzymes following exposure to elevated levels of HNE in vitro detected significant increases in glutathione transferase (GST) levels (37). This suggests that members of the GST class are an important component of the cellular response to elevations in lipid peroxidation and may serve as a marker for this injury. Interestingly, significant elevations in three isoforms of GST occurred in the present study. Although HNE can react with glutathione nonenzymatically the reaction is enhanced up to 500-600 fold in the presence of GSTs (37). All isoforms of GST have activity toward HNE but the alpha class demonstrates the greatest substrate preference for HNE and was the isoform showing the greatest increase in expression in the present study. Although their substrate preference for HNE is lower than the alpha forms, the mu and pi GSTs are present at higher intracellular concentrations than alpha GST and thus probably play a role in response to oxidative stress involving lipid peroxidation. In addition to their conjugating activity, GSTs also exhibit peroxidase activity important for the reduction of phospholipid peroxides within biological membranes (38). Thus the increased expression of the GSTs in the present study is consistent with a protective cellular response to oxidative stress.
In contrast, the increased expression of aldehyde dehydrogenase E3 is more difficult to interpret. Certain isoenzymes of aldehyde dehydrogenase, e.g., the mitochondrial E2 form, are commonly thought to play a role in metabolism of lipid peroxidation products (39). But aldehyde dehydrogenase E3 is a member of this class that shows high activity toward gamma-aminobutyraldehyde, a metabolite of the neurotransmitter gamma-aminobutyric acid, and betaine aldehyde a precursor of betaine, an important osmolyte (40) making its role in DEDC neuropathy less clear. Additionally, considering the well-recognized property of DEDC and disulfiram to inhibit members of the aldehyde dehydrogenase family and produce elevated levels of acetaldehyde after ethanol consumption, it is not clear what contribution aldehyde dehydrogenases play in response to DEDC neurotoxicity (41). Indeed, inhibition of these enzymes, in particular the E2 form, could conceivably contribute to the development of lesions through compromising the protective cellular response to lipid peroxidation.
Tellurium produces a primary demyelination in peripheral nerve of developing rats through its ability to decrease cholesterol synthesis from inhibition of squalene expoxidase. Previous studies using tellurium to examine changes in gene expression of peripheral nerve in demyelinating injury have supported a concerted down-regulation of several genes involved in myelin synthesis including 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA), the rate limiting enzyme for cholesterol biosynthesis (42, 43). The decreased expression of HMG-CoA observed for DEDC parallels the results obtained for the demyelinating injury produced by tellurium and is consistent with the dedifferentiation of Schwann cells to a nonmyelinating mitotically active phenotype that facilitates clearing of myelin debris and axon regeneration following injury. The decreased levels of HMG-CoA also suggest that the injury is still progressing in severity as an increase in HMG-CoA levels have been associated with the remyelinating phase after recovery from tellurium myelin injury. Two other proteins exhibiting decreased expression, transferrin and aspartoacylase, may also reflect a down regulation of myelin synthesis. Transferrin has been localized to oligodendrocytes and Schwann cells and experiments modulating levels of transferrin have supported its ability to enhance myelin synthesis either in normal animals or after demyelinating injury (44, 45). Aspartoacylase is recognized to be important for the generation of acetyl groups from N-acetylaspartate for utilization in the synthesis of myelin lipids and a loss of function mutation for aspartoacylase results in Canavan’s disease, a central nervous system myelinopathy that is typically fatal by 10 years of age (46-48). Thus the down-regulation of these two proteins also appears consistent with decreased myelin synthesis by Schwann cells.
The greatest increases in protein levels were observed for members of the IgG class. Immune-mediated demyelinating injury is thought to play a part in several neurological diseases in humans including multiple sclerosis and Guillain-Barré syndrome (49, 50). Experimental allergic encephalomyelitis and experimental allergic neuritis are also characterized by demyelinating injury and can be initiated through inoculation of experimental animals with various protein components isolated from the central or peripheral nervous systems (51, 52). In certain cases the sera isolated from these animals can produce demyelination in tissue culture that is complement dependent and associated with the IgG fraction of serum (53). This raises the possibility that antibodies directed at either native or modified proteins in the peripheral nervous system may be contributing to the observed myelinopathy. Generation of autoantibodies could result from either a decrease in the selectivity of the blood nerve barrier or from generation of oxidatively modified proteins within nerve being recognized as foreign. The increased expression observed for kinnogen-1 and alpha-1 major acute phase protein prepeptide is also suggestive of an inflammatory component.
Both the morphology and immunostaining results suggest a component of the Schwann cell as a site of oxidative stress. The structural changes observed were characteristic of a primary myelinopathy and consistent with those reported previously following parenteral administration of DEDC and oral administration of pyrrolidine dithiocarbamate or disulfiram (16-18). Significant increases in thin myelin, demyelination and intramyelinic edema resulted from DEDC administration with no accompanying significant changes occurring in the incidence or severity of axonal degeneration. The observation that the earliest and most pronounced structural changes occurred in myelin suggests that either a component of myelin or another component of the Schwann cell may be a principal target for DEDC. Because myelin contains a high concentration of polyunsaturated fatty acids and has a high ratio of membrane surface area relative to cytoplasmic volume it presents a likely target for metal catalyzed oxidation and a potential source for the elevated lipid peroxidation products reported in previous studies. Similarly, the immunostaining provided evidence that the increased expression of GST-pi was localized to the Schwann cell cytoplasm again supporting either myelin or another component of the Schwann cell as a potential site of lipid peroxidation.
The results obtained for protein expression and protein oxidation are consonant with enhanced oxidative stress during DEDC-induced myelinopathy. Additionally, the localization of GST-pi increased expression support the Schwann cell as a site for lipid peroxidation. Collectively these data are consistent with dithiocarbamate-mediated oxidative stress in nerve resulting from an elevation of redox active copper. But because these indices were examined at a relatively advanced stage of injury, whether or not oxidative stress is an important contributing process to the observed structural changes is not clear. It is possible that the observed changes may be due in part or whole from inflammatory changes since activation of resident and recruited circulating macrophages is common to peripheral nerve injury (54). Additional studies to determine the temporal relationship of oxidative stress to the onset of structural changes will help to address this question. Additionally, studies directed at determining the chemical species and subcellular localization of copper will help define the role of elevated copper in the observed oxidative stress.
Acknowledgments
This work was supported by NIEHS Grants ES06387 and P30 ES00267. We thank Corbin W. Whitwell for assistance with DIGE/MS, and the Vanderbilt Venture Capitol Fund for support of Proteomics.
Footnotes
- DEDC
- N,N-diethyldithiocarbamate
- 2-D DIGE
- two dimensional difference gel electrophoresis
- MALDI-TOF/MS
- matrix assisted laser desorption ionization time of flight mass spectrometry
- BSA
- bovine serum albumin
- TBS
- tris buffered saline
- GST
- glutathione transferase
- GST-pi
- glutathione transferase pi
- HNE
- 4-hydroxynonenal
References
- (1).Gandara DR, Perez EA, Weibe V, De Gregorio MW. Cisplatin chemoprotection and rescue: pharmacologic modulation of toxicity. Semin. Oncol. 1991;18:49–55. [PubMed] [Google Scholar]
- (2).Lang JM, Touraine JL, Trepo C, Choutet P, Kirstetter M, Falkenrodt A, Herviou L, Livrozet JM, Retornaz G, Touraine F. Randomised, double-blind, placebo-controlled trial of ditiocarb sodium (‘Imuthiol’) in human immunodeficiency virus infection. Lancet. 1988;2:702–706. doi: 10.1016/s0140-6736(88)90184-5. [DOI] [PubMed] [Google Scholar]
- (3).Somers PK, Medford RM, Saxena U. Dithiocarbamates: effects on lipid hydroperoxides and vascular inflammatory gene expression. Free Radic. Biol. Med. 2000;28:1532–1537. doi: 10.1016/s0891-5849(00)00257-4. [DOI] [PubMed] [Google Scholar]
- (4).Tsai JC, Jain M, Hsieh CM, Lee WS, Yoshizumi M, Patterson C, Perrella MA, Cooke C, Wang H, Haber E, Schlegel R, Lee ME. Induction of apoptosis by pyrrolidinedithiocarbamate and N-acetylcysteine in vascular smooth muscle cells. J. Biol. Chem. 1996;271:3667–3670. [PubMed] [Google Scholar]
- (5).Hartsfield CL, Alam J, Choi AM. Transcriptional regulation of the heme oxygenase 1 gene by pyrrolidine dithiocarbamate. FASEB J. 1998;12:1675–1682. doi: 10.1096/fasebj.12.15.1675. [DOI] [PubMed] [Google Scholar]
- (6).Munoz C, Pascual-Salcedo D, Castellanos MC, Alfranca A, Aragones J, Vara A, Redondo JM, de Landazuri MO. Pyrrolidine dithiocarbamate inhibits the production of interleukin-6, interleukin-8, and granulocyte-macrophage colony-stimulating factor by human endothelial cells in response to inflammatory mediators: modulation of NF- kappa B and AP-1 transcription factors activity. Blood. 1996;88:3482–3490. [PubMed] [Google Scholar]
- (7).Krenn BM, Holzer B, Gaudernak E, Triendl A, van Kuppeveld FJ, Seipelt J. Inhibition of polyprotein processing and RNA replication of human rhinovirus by pyrrolidine dithiocarbamate involves metal ions. J. Virol. 2005;79:13892–13899. doi: 10.1128/JVI.79.22.13892-13899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Uchide N, Ohyama K. Antiviral function of pyrrolidine dithiocarbamate against influenza virus: the inhibition of viral gene replication and transcription. J. Antimicrob. Chemother. 2003;52:8–10. doi: 10.1093/jac/dkg282. [DOI] [PubMed] [Google Scholar]
- (9).Sohn RH, Deming CB, Johns DC, Champion HC, Bian C, Gardner K, Rade JJ. Regulation of endothelial thrombomodulin expression by inflammatory cytokines is mediated by activation of nuclear factor-kappa B. Blood. 2005;105:3910–3917. doi: 10.1182/blood-2004-03-0928. [DOI] [PubMed] [Google Scholar]
- (10).Bouldin TW, Hall CD, Krigman MR. Pathology of disulfiram neuropathy. Neuropath. Appl. Neurobiol. 1980;6:155–160. doi: 10.1111/j.1365-2990.1980.tb00286.x. [DOI] [PubMed] [Google Scholar]
- (11).Edington N, Howell JM. The neurotoxicity of sodium diethyldithiocarbamate in the rabbit. Acta Neuropath. 1969;12:339–347. doi: 10.1007/BF00809130. [DOI] [PubMed] [Google Scholar]
- (12).Frisoni GB, di Monda V. Disulfiram neuropathy: a review and report of a case. Alcohol Alcohol. 1989;24:429–437. [PubMed] [Google Scholar]
- (13).Howell JM, Edington N. The neurotoxcity of sodium diethyldithiocarbamate in the hen. J. Neuropath. Exp. Neurol. 1968;27:464–472. [Google Scholar]
- (14).Howell JM, Ishmael J, Ewbank R, Blakemore WF. Changes in the central nervous system of lambs following the administration of sodium diethydithiocarbamate. Acta Neuropath. 1970;15:197–207. doi: 10.1007/BF00686766. [DOI] [PubMed] [Google Scholar]
- (15).Johnson DJ, Graham DG, Amarnath V, Amarnath K, Valentine WM. Release of carbon disulfide is a contributing mechanism in the axonopathy produced by N,N- diethyldithiocarbamate. Toxicol. Appl. Pharmacol. 1998;148:288–296. doi: 10.1006/taap.1997.8344. [DOI] [PubMed] [Google Scholar]
- (16).Tonkin EG, Erve JCL, Amarnath K, Valentine WM. Disulfiram produces a non-carbon disulfide-dependent schwannopathy in the rat. J. Neuropathol. Exp. Neurol. 2000;59:786–797. doi: 10.1093/jnen/59.9.786. [DOI] [PubMed] [Google Scholar]
- (17).Valentine HL, Amarnath K, Amarnath V, Valentine WM. Dietary copper enhances the peripheral myelinopathy produced by oral pyrrolidine dithiocarbamate. Toxicol. Sci. 2006;89:485–494. doi: 10.1093/toxsci/kfj047. [DOI] [PubMed] [Google Scholar]
- (18).Tonkin EG, Valentine HL, Zimmerman LJ, Valentine WM. Parenteral N,N-diethyldithiocarbamate produces segmental demelination in the rat that is not dependent on cysteine carbamylation. Toxicol. Appl. Pharmacol. 2003;189:139–150. doi: 10.1016/s0041-008x(03)00093-0. [DOI] [PubMed] [Google Scholar]
- (19).Calviello G, Filippi GM, Toesca A, Palozza P, Maggiano N, Nicuolo FD, Serini S, Azzena GB, Galeotti T. Repeated exposure to pyrrolidine- dithiocarbamate induces peripheral nerve alterations in rats. Toxicol. Lett. 2005;158:61–71. doi: 10.1016/j.toxlet.2005.02.008. [DOI] [PubMed] [Google Scholar]
- (20).Tonkin EG, Valentine HL, Milatovic DM, Valentine WM. N,N- Diethyldithiocarbamate produces copper accumulation, lipid peroxidation, and myelin injury in rat peripheral nerve. Toxicol. Sci. 2004;81:160–171. doi: 10.1093/toxsci/kfh190. [DOI] [PubMed] [Google Scholar]
- (21).Delmaestro E, Trombetta LD. The effects of disulfiram on the hippocampus and cerebellum of the rat brain; A study of oxidative stress. Toxicol. Lett. 1995;75:235–243. doi: 10.1016/0378-4274(94)03187-c. [DOI] [PubMed] [Google Scholar]
- (22).Iwata H, Watanabe K, Miichi H, Matsui Y. Accumulation of copper in the central nervous system on prolonged administration of sodium diethyldithiocarbamate to rats. Pharmacol. Res. Commun. 1970;2:213–220. [Google Scholar]
- (23).Koutensky J, Eybl V, Koutenska M, Sykora J, Mertl F. Influence of sodium diethyldithiocarbamate on the toxicity and distribution of copper in mice. Eur. J. Pharmacol. 1971;14:389–392. doi: 10.1016/0014-2999(71)90194-4. [DOI] [PubMed] [Google Scholar]
- (24).Allain P, Krari N. Diethyldithiocarbamate and brain copper. Res. Commun. Chem. Pathol. Pharmacol. 1993;80:105–112. [PubMed] [Google Scholar]
- (25).Meyer OA, Tilson HA, Byrd WC, Riley MT. A method for the routine assessment of fore- and hindlimb grip strength of rats and mice. Neurobehav. Toxicol. 1979;1:233–236. [PubMed] [Google Scholar]
- (26).Ramagli LS. Quantifying protein in 2-D PAGE solubilization buffers. Methods Mol. Biol. 1999;112:99–103. doi: 10.1385/1-59259-584-7:99. [DOI] [PubMed] [Google Scholar]
- (27).Erve JCL, Jensen ON, Valentine HS, Amarnath V, Valentine WM. Disulfiram generates a stable N,N-diethylcarbamoyladduct on Cys-125 of rat hemoglobin β-chains in vivo. Chem. Res. Toxicol. 2000;13:237–244. doi: 10.1021/tx990191n. [DOI] [PubMed] [Google Scholar]
- (28).Levine RL, Garlans D, Oliver CN, Amici A, Climent I, Lenz A-G, Ahn B-W, Shaltiel S, Stadtmen ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- (29).Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, Currie I. A novel experimental design for comparative two-dimensional gel analysis: two- dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3:36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- (30).Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- (31).Friedman DB, Hill S, Keller JW, Merchant NB, Levy SE, Coffey RJ, Caprioli RM. Proteome analysis of human colon cancer by two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics. 2004;4:793–811. doi: 10.1002/pmic.200300635. [DOI] [PubMed] [Google Scholar]
- (32).Meneses-Lorente G, Watt A, Salim K, Gaskell SJ, Muniappa N, Lawrence J, Guest PC. Identification of early proteomic markers for hepatic steatosis. Chem. Res. Toxicol. 2006;19:986–998. doi: 10.1021/tx060007f. [DOI] [PubMed] [Google Scholar]
- (33).Siripurapu V, Meth J, Kobayashi N, Hamaguchi M. DBC2 significantly influences cell-cycle, apoptosis, cytoskeleton and membrane-trafficking pathways. J. Mol. Biol. 2005;346:83–89. doi: 10.1016/j.jmb.2004.11.043. [DOI] [PubMed] [Google Scholar]
- (34).Requena JR, Chao CC, Levine RL, Stadtman ER. Glutamic and aminoadipic semialdehydes are the main carbonyl products of metal-catalyzed oxidation of proteins. Proc. Nat. Acad. Sci. 2001;98:69–74. doi: 10.1073/pnas.011526698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem. Res. Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- (36).Choudhary S, Xiao T, Srivastava S, Zhang W, Chan LL, Vergara LA, Van Kuijk FJ, Ansari NH. Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cells. Toxicol. Appl. Pharmacol. 2005;204:122–134. doi: 10.1016/j.taap.2004.08.023. [DOI] [PubMed] [Google Scholar]
- (37).Awasthi YC, Ansari GA, Awasthi S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzymol. 2005;401:379–407. doi: 10.1016/S0076-6879(05)01024-4. [DOI] [PubMed] [Google Scholar]
- (38).Yang Y, Sharma R, Zimniak P, Awasthi YC. Role of alpha class glutathione S-transferases as antioxidant enzymes in rodent tissues. Toxicol. Appl. Pharmacol. 2002;182:105–115. doi: 10.1006/taap.2002.9450. [DOI] [PubMed] [Google Scholar]
- (39).Ohsawa I, Nishimaki K, Yasuda C, Kamino K, Ohta S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J. Neurochem. 2003;84:1110–1117. doi: 10.1046/j.1471-4159.2003.01619.x. [DOI] [PubMed] [Google Scholar]
- (40).Lin SW, Chen JC, Hsu LC, Hsieh CL, Yoshida A. Human gamma- aminobutyraldehyde dehydrogenase (ALDH9): cDNA sequence, genomic organization, polymorphism, chromosomal localization, and tissue expression. Genomics. 1996;34:376–380. doi: 10.1006/geno.1996.0300. [DOI] [PubMed] [Google Scholar]
- (41).Hart BW, Faiman MD. In vitro and in vivo inhibition of rat liver aldehyde dehydrogenase by S-methyl N,N-diethylthiolcarbamate sulfoxide, a new metabolite of disulfiram. Biochem. Pharmacol. 1992;43:403–406. doi: 10.1016/0006-2952(92)90555-w. [DOI] [PubMed] [Google Scholar]
- (42).Toews AD, Goodrum JF, Lee SY, Eckermann C, Morell P. Tellurium-induced alterations in 3-hydroxy-3-methylglutaryl-CoA reductase gene expression and enzyme activity: differential effects in sciatic nerve and liver suggest tissue-specific regulation of cholesterol synthesis. J. Neurochem. 1991;57:1902–1906. doi: 10.1111/j.1471-4159.1991.tb06401.x. [DOI] [PubMed] [Google Scholar]
- (43).Toews AD, Roe EB, Goodrum JF, Bouldin TW, Weaver J, Goines ND, Morell P. Tellurium causes dose-dependent coordinate down-regulation of myelin gene expression. Brain Res. Mol. Brain Res. 1997;49:113–119. doi: 10.1016/s0169-328x(97)00132-0. [DOI] [PubMed] [Google Scholar]
- (44).Adamo AM, Paez PM, Escobar Cabrera OE, Wolfson M, Franco PG, Pasquini JM, Soto EF. Remyelination after cuprizone-induced demyelination in the rat is stimulated by apotransferrin. Exp. Neurol. 2006;198:519–529. doi: 10.1016/j.expneurol.2005.12.027. [DOI] [PubMed] [Google Scholar]
- (45).Lin HH, Snyder BS, Connor JR. Transferrin expression in myelinated and non-myelinated peripheral nerves. Brain Res. 1990;526:217–220. doi: 10.1016/0006-8993(90)91224-5. [DOI] [PubMed] [Google Scholar]
- (46).Chakraborty G, Ledeen R. Fatty acid synthesizing enzymes intrinsic to myelin. Brain Res. Mol. Brain Res. 2003;112:46–52. doi: 10.1016/s0169-328x(03)00033-0. [DOI] [PubMed] [Google Scholar]
- (47).Kirmani BF, Jacobowitz DM, Namboodiri MA. Developmental increase of aspartoacylase in oligodendrocytes parallels CNS myelination. Brain Res. Dev. Brain Res. 2003;140:105–115. doi: 10.1016/s0165-3806(02)00592-8. [DOI] [PubMed] [Google Scholar]
- (48).Namboodiri AM, Peethambaran A, Mathew R, Sambhu PA, Hershfield J, Moffett JR, Madhavarao CN. Canavan disease and the role of N-acetylaspartate in myelin synthesis. Mol. Cell. Endocrinol. 2006;252:216–223. doi: 10.1016/j.mce.2006.03.016. [DOI] [PubMed] [Google Scholar]
- (49).Cross AH, Stark JL. Humoral immunity in multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis. Immunol. Res. 2005;32:85–97. doi: 10.1385/IR:32:1-3:085. [DOI] [PubMed] [Google Scholar]
- (50).Sheikh KA, Zhang G, Gong Y, Schnaar RL, Griffin JW. An anti- ganglioside antibody-secreting hybridoma induces neuropathy in mice. Ann. Neurol. 2004;56:228–239. doi: 10.1002/ana.20173. [DOI] [PubMed] [Google Scholar]
- (51).Kadlubowski M, Hughes RA. Identification of the neuritogen for experimental allergic neuritis. Nature. 1979;277:140–141. doi: 10.1038/277140a0. [DOI] [PubMed] [Google Scholar]
- (52).Seil FJ, Kies MW, Bacon ML. A comparison of demyelinating and myelination-inhibiting factor induction by whole peripheral nerve tissue and P2 protein. Brain Res. 1981;210:441–448. doi: 10.1016/0006-8993(81)90924-0. [DOI] [PubMed] [Google Scholar]
- (53).Appel SH, Bornstein MB. The application of tissue culture to the study of experimental allergic encephalomyelitus. II. Serum factors responsible for demyelination. J. Exp. Med. 1961;119:303–312. doi: 10.1084/jem.119.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Mueller M, Leonhard C, Wacker K, Ringelstein EB, Okabe M, Hickey WF, Kiefer R. Macrophage response to peripheral nerve injury: the quantitative contribution of resident and hematogenous macrophages. Lab. Invest. 2003;83:175–185. doi: 10.1097/01.lab.0000056993.28149.bf. [DOI] [PubMed] [Google Scholar]