

Abstract
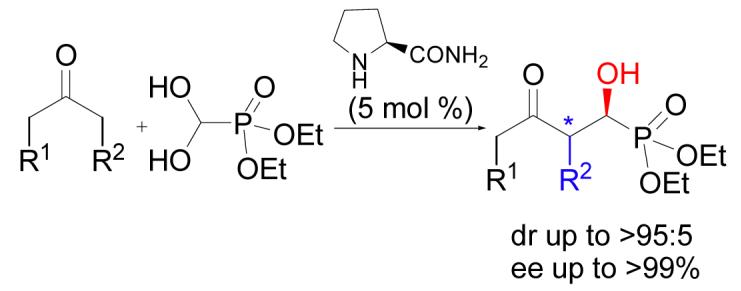
The first organocatalytic cross aldol reaction of ketones and diethyl formylphosphonate hydrate has been realized by using readily available L-prolinamide as the catalyst. Secondary α-hydroxyphosphonates have been synthesized in high enantioselective (up to >99% ee) and good diastereoselectivity.
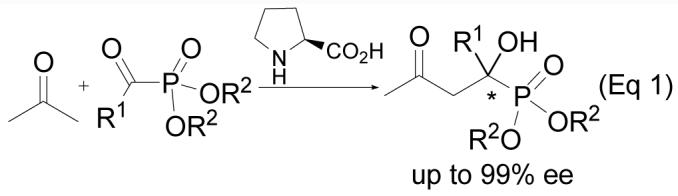
As close analogs of α-amino acids, αhydroxyphosphonate derivatives have been shown to be very important enzyme inhibitors.1 For example, they are inhibitors of renin2 or human immunodeficiency virus (HIV) protease and polymerase.3 They also show anti-virus4 and anti-cancer activities.5 Because of their important biological activities, achieving high enantioselectivity in the synthesis of α-hydroxyphosphonates has been the goal of organic chemists.6 Recently, we reported the first prolinecatalyzed asymmetric aldol reaction of α-ketophosphonates for the highly enantioselective synthesis of tertiary α-hydroxyphosphonates (Eq 1).7 However, from a biological point of view, secondary α-hydroxyphosphonates seem more significant, as all chiral natural amino acids are secondary amines. The optically enriched forms of secondary α-hydroxyphosphonates are mainly obtained through enzymatic methods,6a such as kinetic resolution of racemic mixture by bacteria, fungi or
 |
lipases8 or through asymmetric reduction of α-ketophosphonate with baker’s yeast or fungi.6a,9 Only a few chemical methods are available,6b,c, which include the asymmetric reduction of α-ketophosphonates,10 asymmetric oxidation of benzylphosphonates11 and diastereoselective addition of dialkyl phosphites to aldehydes (phosphoaldol reaction).12a,b These methods are either not catalytic, use special reagents that are difficult to handle, or have very limited substrate scope. A catalytic method based on the phosphoaldol reaction was also reported,12c-g but the enantioselectivities obtained were dependent on the substrates. Most recently, Chen and co-workers reoported a vanadium-catalyzed oxidative kinetic resolution for the high enantioselective synthesis of secondary α-hydroxyhosphonates;13 nonetheless, the disadvantage of this method is the sacrifice of 50% of the starting material.13 Thus, the development of a catalytic highly enantioselective method for the synthesis of secondary α-hydroxyphosphonates is warranted. Herein, we wish to report our preliminary results of a highly enantioselective synthesis of secondary α-hydroxyphosphonates via a prolinamide-catalyzed asymmetric aldol reaction.14
Synthesizing secondary α-hydroxyphosphonates by using our reported protocol7 would require formylphosphonate as the starting material (Eq 1, R1 = H). However, although diethyl formylphosphonate is a known compound,15 it is unstable, and all our attempts to react this substance with acetone failed. Then we turned our attentions to its hydrate (4), because it was reported to be more stable and in equilibrium with its formyl form.16
By using acetone (5a) as the model compound, we screened some readily available L-proline-derivatives (Figure 1) as the catalyst for the cross aldol reaction of diethyl formylphosphonate hydrate (4). The results are summarized in Table 1.
Figure 1.
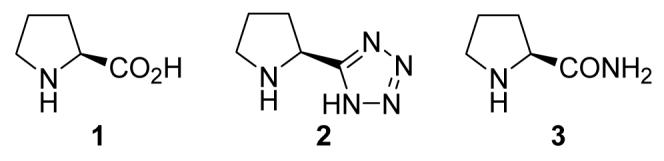
Catalysts screened for the cross aldol reaction.
Table 1.
Catalyst Screening and Reaction Condition Optimizationsa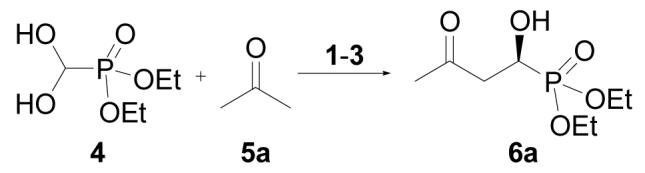
| entry | catalyst | solvent | temp (°C) |
time (h) |
yield (%)b |
ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 1 | acetone | rt | 24 | — | — |
| 2 | 2 | acetone | rt | 24 | 46 | 72 |
| 3 | 3 | acetone | rt | 3 | 93 | 84 |
| 4 | 3 | CH2Cl2 | rt | 10 | 76 | 84 |
| 5 | 3 | DMSO | rt | 10 | 45 | 79 |
| 6 | 3 | DMF | rt | 10 | 55 | 60 |
| 7 | 3 | acetone | 0 | 10 | 95 | 92 |
| 8d | 3 | acetone | 0 | 24 | 95 | 94 |
| 9d | 3 | acetone | -20 | 24 | 62 | 94 |
| 10 | ent-3 | acetone | 0 | 24 | 91 | 92e |
Unless otherwise indicated, all reactions were performed with freshly prepared diethyl formylphosphonate hydrate (4, 0.5 mmol), acetone (0.2 mL), solvent (0.4 mL) and catalyst (10 mol %) at 0 ° C.
Yield of isolated product after column chromatography.
The enantiomeric excess was determined by chiral GC analysis with a Chiraldex GTA column.
With 5 mol % of catalyst loading.
(R)-6a was obtained as the major enantiomer.
As shown in Table 1, although L-proline is a good catalyst for the cross aldol reaction of α-ketophosphonates,7 it failed to catalyze the aldol reaction of 4 (entry 1), presumably because 4 is incompatible with its acidity. In contrast, less acidic L-proline tetrazole (2) and L-prolinamide (3) proved to be good catalysts for the desired reaction. At 10 mol % catalyst loading and room temperature, the aldol product 6a was obtained in 72% (entry 2) and 84% ee (entry 3), respectively. The reaction conditions were further optimized for 3, as it is more reactive and enantioselective. Catalyst 3 is slightly less reactive in CH2Cl2 (entry 4), but the enantioselectivity maintains at the same level as in acetone. Other common solvents used for aldol reactions, such as DMSO (entry 5) and DMF (entry 6), proved to be less effective than excessive acetone (entry 3). Nevertheless, lowering the reaction temperature to 0 °C resulted in an increase of the enantioselectivity (to 92% ee, entry 7). It is interesting to note that the catalyst loading can be further reduced to 5 mol %, without affecting the enantioselectivity, although the reaction is a little bit slower (entry 8). Further dropping of the reaction temperature to -20 °C did not improve the enantioselectivity, instead has an adverse effect on the reactivity of the catalyst (entry 9). Similar reaction with D-prolinamide (ent-3) as the catalyst produced comparable results as those of L-prolinamide, except that the opposite enantiomer of the product was obtained as the major one (entry 10).
In order to determine the absolute configuration of the product, the carbonyl group in product 6a was reduced to the methylene group in two steps (Eq 2) to give compound 7, for which the absolute configuration is known.17 Compound 7 is dextrorotary, which indicates that the newly formed chiral center is S-configured.17
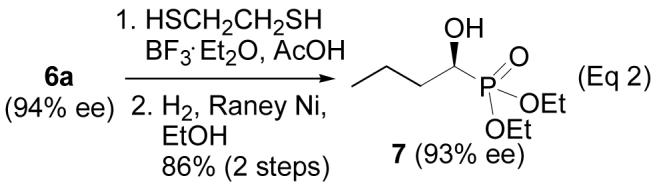 |
Further studies were carried out under the optimized conditions (0 °C, 5 mol % catalyst loading, and excessive ketone as solvent if possible) to understand the scope of this new reaction, and the results are summarized in Table 2.
Table 2.
Enantioselective Cross Aldol Reaction of Diethyl Formylphosphonate Hydrate (4) and Various Ketones.a
| entry | R1 | R2 | product/ yield (%)b |
drc | ee (%)d |
|---|---|---|---|---|---|
| 1 | H | H | 6a/95 | — | 94e |
| 2 | Me | H | 6b/62f | — | 93e |
| H | Me | 6b’ /20f | 95:5g | 99e | |
| 3 | Et | H | 6c/58f | — | 97e |
| H | Et | 6c’/5f | >95:5g | 99e | |
| 4 | Cl | H | 6d/65 | — | 90e |
| 5 | OH | H | 6e/75 | — | 43h |
| 6 | OMe | H | 6f/77 | — | 85 |
| 7 | AcCH2 | H | 6g/60 | — | 86 |
| 8 | —(CH2)2— | 6h/ 94 | 95:5i | >99 | |
| 9 | —(CH2)3— | 6i/82 | 65:35g | 93(86) | |
| 10j | —(CH2)3— | 6i/90k | 65:35g | 96(95) | |
| 11j | —CH2OCH2— | 6j/85k | 70:30g | 97(99) | |
| 12j | —CH2SCH2— | 6k/88k | 70:30g | 93(97)l | |
Unless otherwise indicated, all reactions were performed with freshly prepared diethyl formylphosphonate hydrate (4, 0.5 mmol), ketone (0.6 mL) and L-prolinamide (5 mol %) at 0 °C; the absolute configuration is tentatively assigned for 6b-k on the basis of the reaction mechanism.
Yield of isolated product after column chromatography.
Determined by 1H NMR.
Unless otherwise noted, enantiomeric excess was determined by HPLC analyses with a Chiralpak AD-H column; values in parentheses are for the minor diastereomer.
The enantiomeric excess was determined by chiral GC analysis with a Chiraldex GTA column.
Yields of individual regioisomer as determined by 1H NMR.
anti/syn ratio as determined by NMR.
With a Chiralcel OJ-H column.
syn/anti ratio as determined by NMR.
The reaction was performed with 4 (0.5 mmol), ketone (0.2 mL), CH2Cl2 (0.5 mL) and L-prolinamide (10mol %) at -20 °C for 24 h.
Combined yield of two inseparable diastereomers.
With a Chiralcel OD-H column.
Besides acetone (entry 1), other unsubstituted aliphatic ketones, such as 2-butanone (5b, entry 2) and 2-pentanone (5c, entry 3), also participate in this reaction. The reaction of 5b and 4 gives rise to a mixture of two inseparable regioisomers (6b and 6b’) in a ratio of 3:1. The major regioisomer (6b) is a kinetic product, which was obtained in 93% ee. The thermodynamic product (6b’) was obtained in a 90% de for anti product and the ee value for this diastereomer is 99% (entry 2). Similarly, the reaction 5c and 4 yielded these two products in about 12:1 ratio, with a 97% ee for the major kinetic product (6c), and a single anti diastereomer for the minor product (6c’) in high enantioselectivity (99% ee, entry 3). However, 3-pentanone reacts very slowly and leads to the decomposition of 4 (data not shown).
Substituted ketones may also be used in this reaction. For example, the aldol reaction of α-chloroacetone (5d) produces only the kinetic regioisomer in 65% yield and 90% ee (entry 4). In contrast, acetol (5e) generates very poor enantioselectivity (43% ee) of the product (entry 5), presumably due to the interference of the free hydroxy group. Once the hydroxy group is methylated, compound5f again generates excellent enantioselectivity for the product (6f, 85% ee, entry 6). Similar enantioselectivity (86% ee) was also obtained for the aldol product (6g) of 2,5-hexanedione.
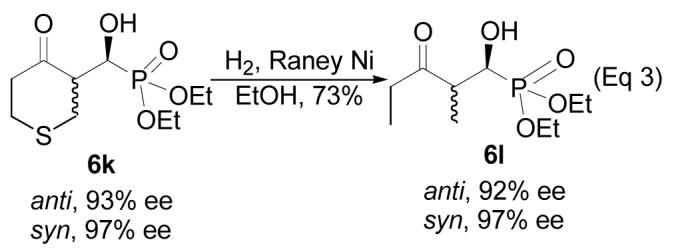
Some cyclic ketones were also studied. Cyclopentanone yielded the aldol product (6h) in excellent yield (94%) and diastereoselectivity (95:5, entry 8). The major diastereomer, which was determined to be syn by NOE, was obtained essentially as a pure enantiomer (>99% ee). The reaction of cyclohexanone generates a 65:35 anti/syn mixture (6i) in excessive cyclohexanone with 93% and 86% ee for these two diastereomers, respectively (entry 9). Similar reaction using CH2Cl2 as solvent with 10 mol % of the catalyst (entry 10) led to improved enantioselectivities of these two diastereomers (96% and 95% ee, respectively). Under these conditions, 4-oxacyclohexanone (5j) and 4-thiacyclohexanone (5k) also perform better. A 70:30 anti/syn mixture was obtained for 6j, with 97% and 99% ee for the major and minor diastereomers, respectively (entry 11). The same anti/syn diastereoselectivity was also obtained for the product of 5k, with 93% and 97% ee, respectively (entry 12). As a potential application of this product, 6k may be readily desulfurized to give product 6l in good yield (Eq 3), with total retention of the stereochemistry. Product 6l can be viewed as the aldol product of 3-pentanone and 4; however, as aforementioned, such a direct aldol reaction is not feasible with the L-prolinamide catalysis at this moment.
 |
To account for the formation of the S-configured α-hydroxyphosphonate as the major enantiomer, we propose a reaction mechanism as shown in Scheme 1, which is similar to other proline derivative-catalyzed cross aldol reactions.18 The re face attack is disfavored due to the unfavorable steric interaction between the large axial phosphonate group and the axial methyl group (Scheme 1, right). Thus, the si face attack is favored, which leads to the observed product (Scheme 1, left).
Scheme 1.
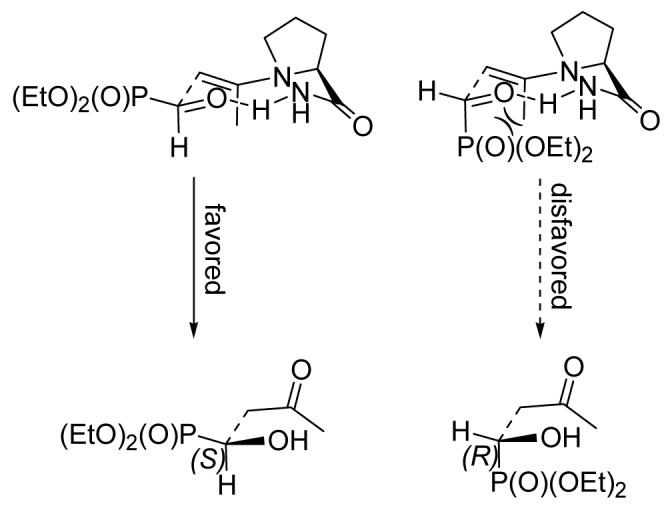
Proposed Transition State Structures
In summary, we have developed the first organocatalytic cross aldol reaction of ketones and diethyl formylphosphonate hydrate by using readily available L-prolinamide as the catalyst. Secondary α-hydroxyphosphonates have been synthesized in high enantioselectivity (up to >99% ee) and good diastereoselectivity.
Supplementary Material
Acknowledgment
The generous financial support of this project from the Welch Foundation (Grant No. AX-1593) and the NIH-MBRS program (Grant No. S0608194) is gratefully acknowledged.
Footnotes
Affectionately dedicated to Professor Waldemar Adam on the occasion of his 69th birthday.
Supporting Information Available Experimental procedures, NMR spectra for new compounds, and HPLC analysis data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For review, see:Kolodiazhnyi OI. Tetrahedron: Asymmetry. 2005;16:3295–3340.
- (2)(a).Dellaria JF, Jr., Maki RG, Stein HH, Cohen J, Whittern D, Marsh K, Hoffman DJ, Plattner JJ, Perun TJ. J. Med. Chem. 1990;33:534–542. doi: 10.1021/jm00164a011. [DOI] [PubMed] [Google Scholar]; (b) Tao M, Bihovsky R, Wells GJ, Mallamo JP. J. Med. Chem. 1998;41:3912–3916. doi: 10.1021/jm980325e. [DOI] [PubMed] [Google Scholar]
- (3).Stowasser B, Budt K-H, Li J-Q, Peyman A, Ruppert D. Tetrahedron Lett. 1992;33:6625–6628. [Google Scholar]
- (4).Snoeck R, Holy A, Dewolf-Peeters C, Van Den Oord J, De Clercq E, Andrei G. Antimicrob. Agents Chemother. 2002;46:3356–3361. doi: 10.1128/AAC.46.11.3356-3361.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5)(a).Peters ML, Leonard M, Licata AA. Clev. Clin. J. Med. 2001;68:945–951. doi: 10.3949/ccjm.68.11.945. [DOI] [PubMed] [Google Scholar]; (b) Leder BZ, Kronenberg HM. Gastroenterology. 2000;119:866–869. doi: 10.1053/gast.2000.17841. [DOI] [PubMed] [Google Scholar]
- (6) (a).For reviews, see:Kafarski P, Lejczak B. J. Mol. Cat. B: Enzym. 2004;29:99–104.Gröger H, Hammer B. Chem. Eur. J. 2000;6:943–948.Wiemer DF. Tetrahedron. 1997;53:16609–16644.
- (7).Samanta S, Zhao C-G. J. Am. Chem. Soc. 2006;128:7442–7443. doi: 10.1021/ja062091r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8) (a).Li Y-F. Tetrahedron: Asymmetry. 1993;4:109–120. [Google Scholar]; (b) Drescher, Li Y-F, Hammerschmidt F. Tetrahedron. 1995;51:4933–4946. [Google Scholar]; (c) Drescher M, Hammerschmidt F, Kahling H. Synthesis. 1995:1267–1272. [Google Scholar]; (d) Wuggenig F, Hammerschmidt F. Monatsh. Chem. 1998;129:423–436. [Google Scholar]; (e) Khushi T, O’Toole KJ, Sime JT. Tetrahedron Lett. 1993;34:2375–2378. [Google Scholar]
- (9) (a).Brzezinska-Rodak M, Zymanczyk-Duda E, Kafarski P, Lejczak B. Biotechnol. Prog. 2002;18:1287–1291. doi: 10.1021/bp0200090. [DOI] [PubMed] [Google Scholar]; (b) Maly A, Lejczak B, Kafarski P. Tetrahedron: Asymmetry. 2003;14:1019–1024. [Google Scholar]
- (10)(a).Meier C, Laux WHG. Tetrahedron: Asymmetry. 1996;7:89–94. [Google Scholar]; (b) Meier C, Laux WHG. Tetrahedron: Asymmetry. 1995;6:1089–1092. [Google Scholar]; (c) Meier C, Laux WHG. Tetrahedron. 1996;52:589–598. [Google Scholar]; (d) Gajda T. Tetrahedron: Asymmetry. 1994;5:1965–1972. [Google Scholar]; (e) Nesterov V, Kolodyazhnyi OI. Russ. J. Gen. Chem. 2005;75:1161–1162. [Google Scholar]; (f) Nesterov VV, Kolodiazhnyi OI. Tetrahedron: Asymmetry. 2006;17:1023–1026. [Google Scholar]
- (11)(a).Pogatchnik DM, Wiemer DF. Tetrahedron Lett. 1997;38:3495–3498. [Google Scholar]; (b) Cermak DM, Du Y, Wiemer DF. J. Org. Chem. 1999;64:388–393. [Google Scholar]; (c) Skropeta D, Schmidt RR. Tetrahedron: Asymmetry. 2003;14:265–273. [Google Scholar]
- (12) (a).For examples, see:Wroblewski AE, Balcerzak KB. Tetrahedron: Asymmetry. 2001;12:427–431.Yokomatsu T, Yamagishi T, Shibuya S. Tetrahedron: Asymmetry. 1993;4:1401–1404.Rowe BJ, Spilling CD. Tetrahedron: Asymmetry. 2001;12:1701–1708.Arai T, Bougauchi M, Sasai H, Shibasaki M. J. Org. Chem. 1996;61:2926–2927. doi: 10.1021/jo960180o.Sasai H, Bougauchi M, Arai T, Shibasaki M. Tetrahedron Lett. 1997;38:2717–2720.Saito B, Katsuki T. Angew. Chem., Int. Ed. 2005;44:4600–4602. doi: 10.1002/anie.200501008.Groaning MD, Rowe BJ, Spilling CD. Tetrahedron Lett. 1998;39:5485–5488.
- (13).Pawar VD, Bettigeri S, Weng S-S, Kao J-Q, Chen C-T. J. Am. Chem. Soc. 2006;128:6308–6309. doi: 10.1021/ja060639o. [DOI] [PubMed] [Google Scholar]
- (14) (a).For examples of asymmetric cross aldol reaction of activated carbonyl compounds, see:Enders D, Grondal C. Angew. Chem., Int. Ed. 2005;44:1210–1212. doi: 10.1002/anie.200462428.Luppi G, Cozzi PG, Monari M, Kaptein B, Broxterman QB, Tomasini C. J. Org. Chem. 2005;70:7418–7421. doi: 10.1021/jo050257l.Shen Z, Li B, Wang L, Zhang Y. Tetrahedron Lett. 2005;46:8785–8788.Tokuda O, Kano T, Gao W-G, Ikemoto T, Maruoka K. Org. Lett. 2005;7:5103–5105. doi: 10.1021/ol052164w.Tang Z, Cun L-F, Cui X, Mi A-Q, Jiang Y-Z, Gong L-Z. Org. Lett. 2006;8:1263–1266. doi: 10.1021/ol0529391.Samanta S, Zhao C-G. Tetrahedron Lett. 2006;47:3383–3386.
- (15).Leitzke A, Flyunt R, Theruvathu JA, von Sonntag C. Org. Biomol. Chem. 2003;1:1012–1019. doi: 10.1039/b212194h. [DOI] [PubMed] [Google Scholar]
- (16).Hamilton R, McKervey MA, Rafferty MD, Walker BJ. J. Chem. Soc., Chem. Commun. 1994:37–38. [Google Scholar]
- (17)(a).Pàmies O, Bäckvall JE. J. Org. Chem. 2003;68:4815–4818. doi: 10.1021/jo026888m. [DOI] [PubMed] [Google Scholar]; (b) Skwarczynski M, Lejczak B, Kafarski P. Chirality. 1999;11:109–114. [Google Scholar]
- (18).For theoretical treatment, see,Clemente FR, Houk KN. J. Am. Chem. Soc. 2005;127:11294–11302. doi: 10.1021/ja0507620.for reviews, see:List B.Tetrahedron 2002585573–5590.List B. Acc. Chem. Res. 2004;37:548–557. doi: 10.1021/ar0300571.Notz W, Tanaka F, Barbas CF., III Acc. Chem. Res. 2004;37:580–591. doi: 10.1021/ar0300468.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.