

Abstract
A total synthesis of (+)-aspidospermidine (1) is described. The key reactions used in the synthesis of this pentacyclic Aspidosperma alkaloid were a deracemizing imine alkylation/Robinson annulation sequence, a selective “redox ketalization” and an intramolecular Schmidt reaction. A Fischer indolization step carried out on a tricyclic ketone mirrored the sequence reported by Stork and Dolfini in their classic aspidospermine synthesis.
Introduction
The Aspidosperma alkaloids have inspired considerable interest since the early days of complex organic synthetic chemistry research.1 Over the years, a number of racemic2,3 syntheses as well as enantioselective4 approaches to (+)-aspidospermidine and (−)-aspidospermine, the prototypical members of the group, have been reported.
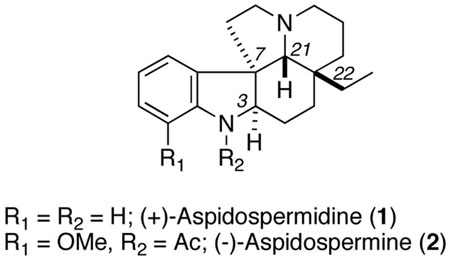
The first successful foray into this class was the total synthesis of (±)-aspidospermine 2 by Stork and Dolfini.2 One highlight of this synthesis was the use of a Fischer indole condensation to generate the core structure of the natural product (Scheme 1). This reaction proceeded through iminium ion 4, so the configurations of C-7 and C-21 were thermodynamically controlled relative to the quaternary center C-22. The final stereocenter (C-3) was set through a stereoselective reduction of imine 5. Thus, a stereoselective route to aspidospermine or aspidospermidine could arise, in principle, from any diastereomer of the Stork ketone 3. Accordingly, an asymmetric synthesis of 3 presents a worthwhile challenge.5 Herein we disclose a total synthesis of (+)-aspidospermidine (1) that uses the Stork approach in the context of modern enantioselective synthesis and also provided an opportunity to carefully examine the late-stage Fischer indole step.
Scheme 1.
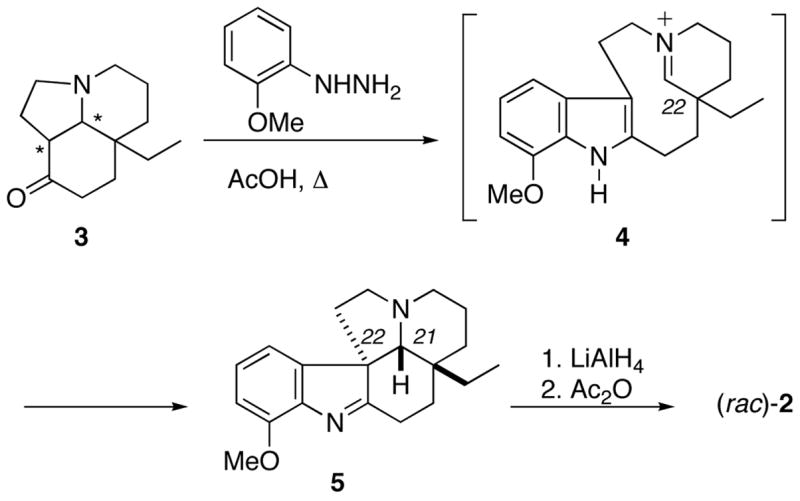
Our plan for the construction of enantiomerically pure 6 involved an intramolecular Schmidt reaction6 of 7 (Scheme 2), which could be derived from bicyclic enone 8. To be successful, this step would require the selective Schmidt reaction of the azide with the cyclopentanone embedded in 7 (path a) to the exclusion of the six-membered cyclic ketone (path b). Although such selectivity had not been demonstrated prior to this work, model studies showed that compounds with four carbons separating the two reactive moieties underwent ring expansion chemistry much more easily than those with only three, which would support the selective formation of the desired product.6 We envisioned an asymmetric synthesis of a precursor enone 8 or 9 from 2-ethylcyclopentanone 12 using the asymmetric variant of the Robinson annulation invented by d’Angelo’s group,7 thus establishing the critical quaternary stereocenter in the desired configuration.
Scheme 2.
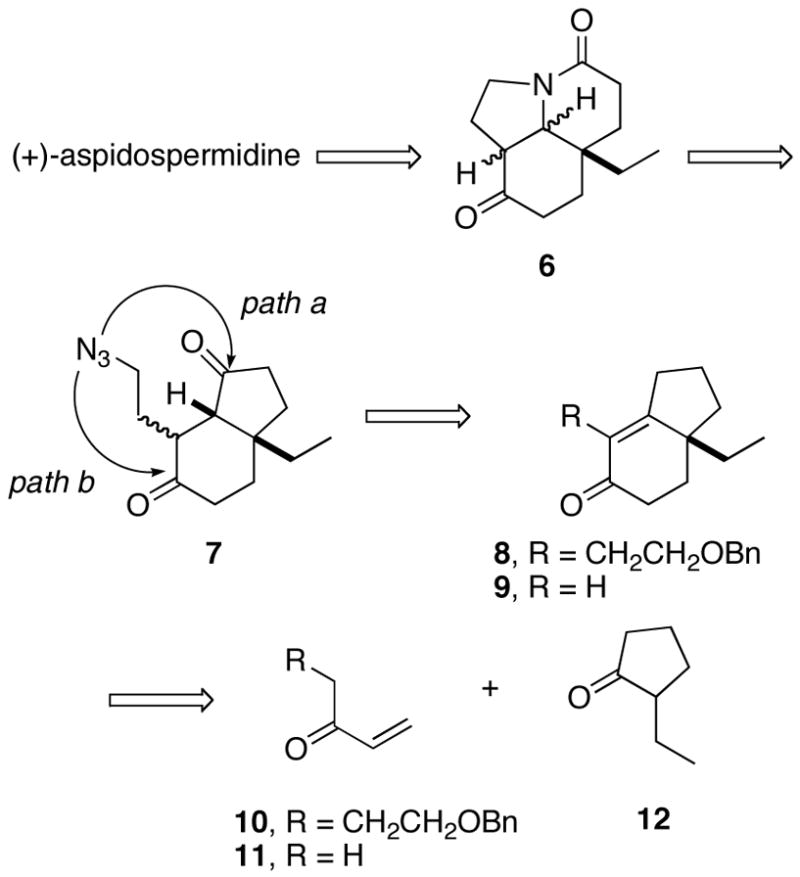
Herein, we describe the successful implementation of this strategy. The discussion is arranged along three major themes: (1) the development of a synthesis of dione 7, (2) investigation of the key intramolecular Schmidt reaction, and (3) the conversion of enantiomerically pure Stork intermediate 6 into (+)-aspidospermidine.8 Finally, a ketal-mediated rearrangement reaction, discovered during the course of this work, was the first time a bridged ring system was observed to result from an intramolecular Schmidt reaction.
Results and Discussion
Synthesis of the key bicyclic diketo azide: Early results
Our first route to the bicyclic ring system revolved around the intermediacy of enone 9 (Scheme 3). The plan was to synthesize compound 13 from 2-ethyl-1,3-pentanedione using the well-documented Hajos-Parrish reaction,9 followed by reduction of the saturated ketone carbonyl group. Once in hand, we planned to remove the extraneous alcohol in 13 using a deoxygenation or elimination reaction.10 Although 13 was readily synthesized as planned, all attempts at converting 13 to 9 or 14 were unsuccessful. In contrast, the application of the d’Angelo annulation protocol to racemic 2-ethylcyclohexanone was successful and provided enone 9 in modest yield. The enone function provided a convenient handle for installing a carbonyl in the γ position. Thus, enone 9 was allowed to reflux briefly with isopropenyl acetate and p-TsOH to effect its quantitative transformation to the corresponding dienol acetate depicted. Oxidation of this material with Oxone (which gave an allylic alcohol; not shown) followed by PCC afforded enedione 15 in excellent overall yields for the three-step protocol.
Scheme 3.
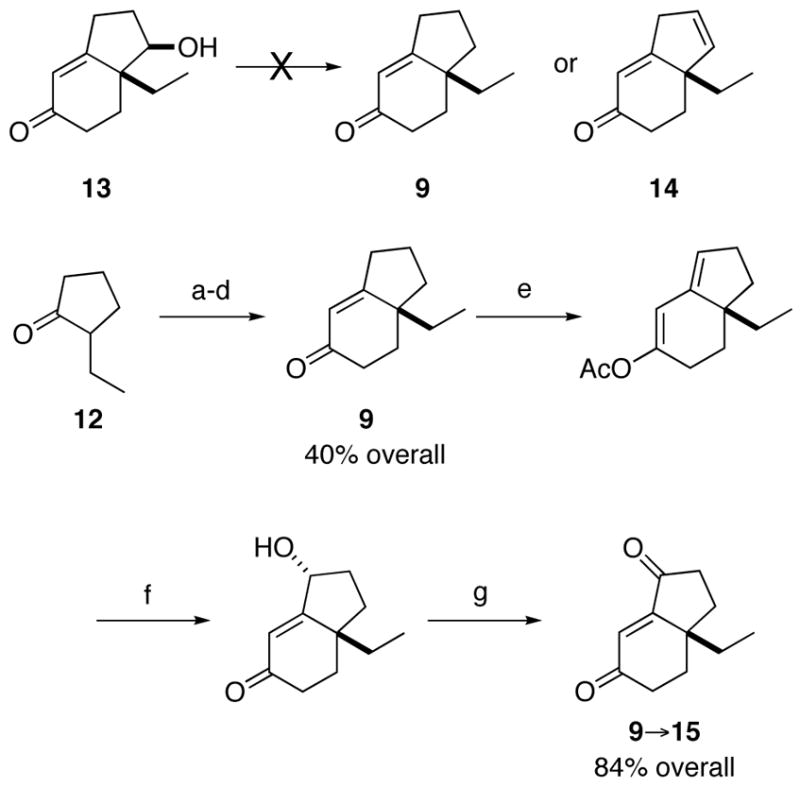
aReagents and conditions: (a) (S)-α-methylbenzylamine; (b) 11, hydroquinone, 60 °C; (c) 10% aq AcOH; (d) NaOMe, MeOH, reflux; (e) isopropenyl acetate; (f) Oxone, acetone; (g) PCC, CH2Cl2.
The direct installation of an alkyl side chain by vinyl cuprate 1,4-addition to enedione 15 was unsuccessful, affording only reduction products. A variety of other reactions, including allylsilane additions to 15 or various reduction products of same, were unsuccessful, non-selective, or inefficient. Accordingly, we redirected our efforts to obtain enone 8, already bearing the necessary side chain, in a more convergent fashion (Scheme 4). To this end, the α,β-unsaturated ketone 10 was synthesized from 1,4-butanediol. Turning again to the d’Angelo deracemization protocol, cyclopentanone 12 was condensed with (S)-α-methylbenzylamine and the resulting enamine was condensed with vinyl ketone 10. Alkene polymerization was moderated by the addition of a small amount of 1,4-hydroquinone (HQ). The multiday reaction was followed by acidic hydrolysis (10% aqueous CH3COOH) and intramolecular aldol reaction under basic conditions (NaOMe, MeOH) to afford the bicyclic enone 8 in 49% overall yield and 66% ee (HPLC, Chiralcel AS, 20% isopropanol/hexanes, 254 nm). The relatively low enantioselectivity was not surprising because the literature indicates that additional steric bulk in the enamine intermediate may result in poor face selectivity.7 Ultimately, the selectivity was improved by applying Lewis acid activation11 (fused ZnCl2, refluxing ether, 3 d), followed by the usual cyclization methods, to generate 84–86% ee material in similar yield as before. This material was brought to enantiomeric purity at a later step.
Scheme 4.
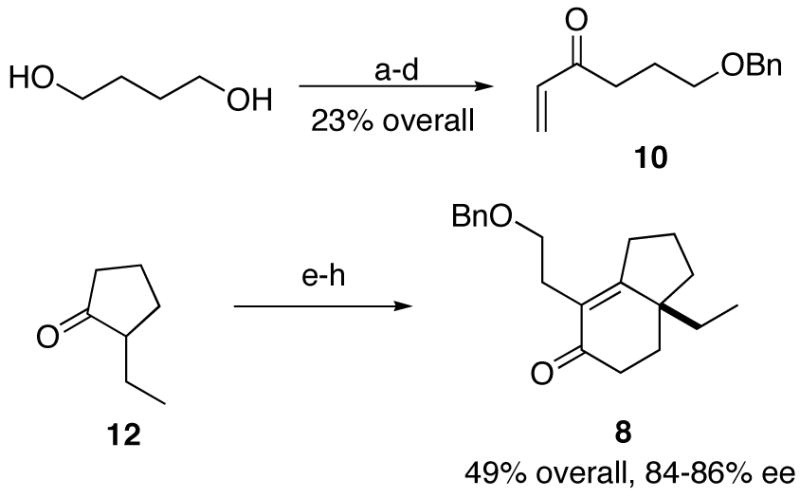
aReagents and conditions: (a) NaH, BnBr, DMF; (b) PCC, CH2Cl2; (c) CH2=CH2 MgBr, THF; (d) CrO3/H2SO4, acetone; (e) (S)-α-methylbenzylamine; (f) 10, hydroquinone, fused ZnCl2, Et2O, reflux; (g) 10% aq AcOH; (h) NaOMe, MeOH, reflux.
With an appropriately substituted [4.3.0]bicyclononenone in hand, it remained to install the azide nucleophile and an additional ketone function to arrive at the required Schmidt reaction substrate. It soon became apparent that precise choice and ordering of the steps was crucial. Initially, we envisioned utilizing a Mitsunobu reaction to transform the primary alcohol derived from 8 into the requisite azide. However, catalytic debenzylation of 8 performed under a variety of conditions also led to unwanted reduction of the enone function. Selective deprotection of the side chain could be achieved by brief exposure of the substrate to either boron tribromide or boron trichloride to directly yield the corresponding alkyl bromide 16a or chloride 16b, respectively (Scheme 5).12 The unexpected halogenation of the terminal alcohol was considered a bonus as alkyl halides easily succumb to nucleophilic azide displacement. The chloride (prepared in higher yield and more stable than 16a) was therefore transformed to the corresponding enedione 17 as in Scheme 5.
Scheme 5.
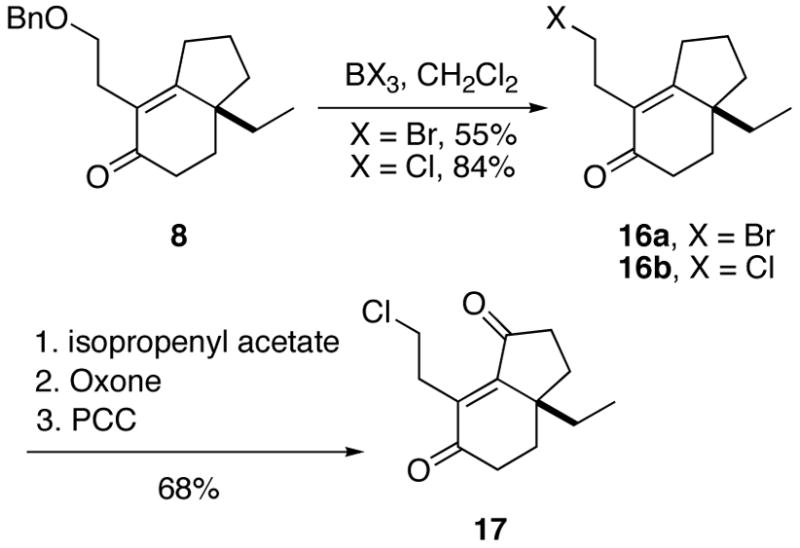
The chemoselective reduction of the double bond in enedione 17 was accomplished by treatment with concentrated HCl and sodium iodide to afford the bicyclic scaffold 18 as a 1:1 diastereomeric mixture (Scheme 6).13 This reduction presumably involves initial 1,4 attack of iodide followed by a second displacementon the α-iodoketone to produce the reduced 1,4-dione. The stage was now set for azide displacement of the chloride; however, the reaction afforded mixtures of the expected product 7 and an undesired cyclopropane 19. The ease of the intramolecular displacement process was confirmed by exposing 18 to base, which cleanly afforded 19 as the only product. This compound proved resistant to all nucleophilic ring opening attempts. In an effort to circumvent this problem, enone 16 was subjected to base, cleanly affording cyclopropane 20 containing a non-conjugated double bond. In contrast to 19, cyclopropane 20 proved to be amenable to ring opening, presumably because of the greater stability of the enolate anion formed upon nucleophilic attack.14 The enone in 21 could be functionalized by γ-oxidation but this route was abandoned due to the difficulty of selective double bond reduction in azide 22.
Scheme 6.
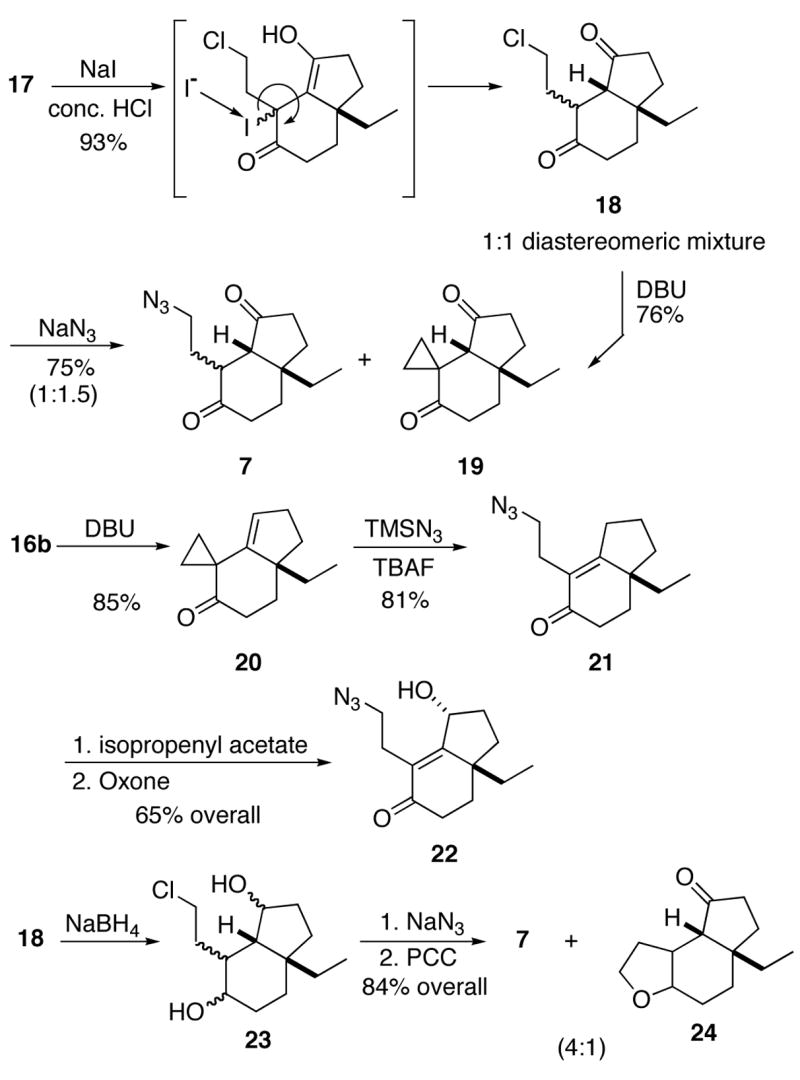
Since enolization/intramolecular trapping was the major hurdle in the functionalization of 18, we decided to reduce both carbonyls to alcohols, followed by an azidation/oxidation sequence. Thus, dione 18 was reduced with NaBH4 to diol 23, which was isolated as an inseparable mixture of products. This mixture was immediately subjected to azidation and oxidation to afford 7. Unfortunately, the interference of another intramolecular process(displacement of the chloride by the proximal alcohol) gave the cyclic ether 24 as an unwanted byproduct (a 4:1 mixture of 7 and 24 was obtained). Compound 7 as obtained in these reactions was valuable as it allowed us to rehearse the critical Schmidt reaction and complete a first-generation synthesis of aspidospermidine (all such experiments are collected in the following section). However, all of the above routes were ultimately rejected for use in the final synthesis due to their inability to cleanly deliver the key intermediate 7.
An effective synthesis of the key bicyclic diketo azide 7
Rethinking our plan, we decided to protect the bicyclic scaffold prior to side chain manipulation. Thus, γ-oxidation of 8 afforded alcohol 25, which was treated with bis(TMS)neopentyl glycol and a catalytic amount of TMSOTf to afford ketal 26 directly (Scheme 7). This “redox ketalization” involves regioselective ketal and concomitant deconjugation, leading to an enol that undergoes tautomerization to ketone 26. This step also rendered the carbon bearing the side chain nonepimerizable. 1H NMR analysis of 26 revealed a 12:1 mixture of diastereomers that contained other inseparable impurities. This material proved to be unstable and so was rapidly subjected to reductive debenzylation followed by a Mitsunobu reaction to afford azide 28. Detailed examination of the NMR spectra, including NOESY and COSY experiments, indicated that the major component of this ca. 12:1 mixture had the stereostructure shown in Scheme 7. For details regarding this and other important structural assignments in this work, see the Supporting Information.
Scheme 7.
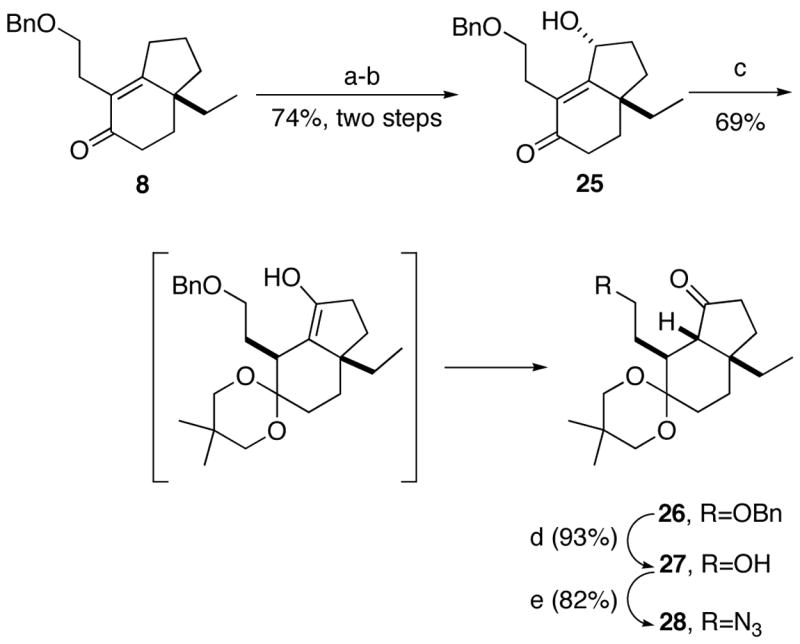
aReagents and conditions: (a) isopropenyl acetate; (b) Oxone, acetone; (c) bis(trimethylsilyl)neopentyl glycol, TMSOTf, CH2Cl2, 0 °C → rt; (d) H2, 10% Pd/C; (e) HN3, PPh3, DEAD, PhH, 0 °C → rt.
Study of the the intramolecular Schmidt reaction
The scaffold was now appropriately functionalized for intramolecular Schmidt reaction with the unprotected carbonyl. Our initial attempts using TFA on 28 provided deketalized compound 6 as the single diastereomer shown, in 21% yield (Scheme 8; see Supporting Information for this structural elucidation). This isomer thus derives from the minor diastereomer of 28, which is present in only a few percent in the starting material. We hypothesize that the minor isomer of
Scheme 8.
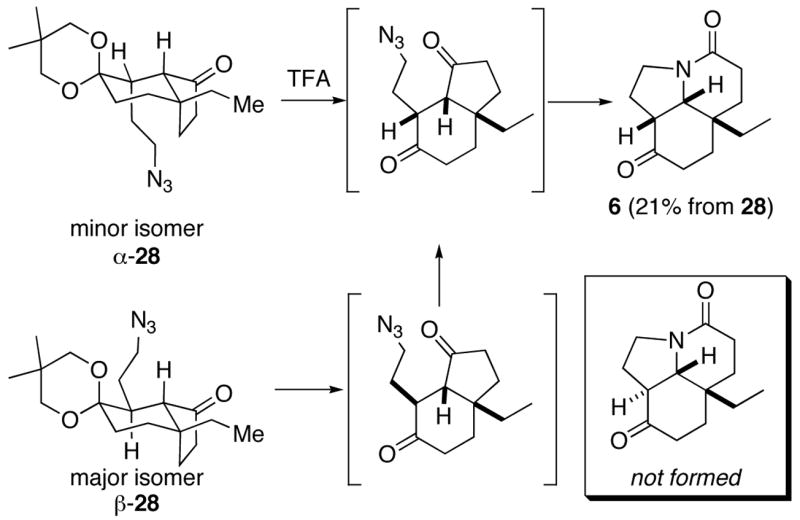
The poor yield in the Schmidt reaction is likely due to competitive pathways occurring in lieu of cyclization. It is possible that the azide in the major isomer β-28 cannot reach the necessary carbonyl group and therefore azide decomposition occurs faster than deketalization or epimerization (note that cyclization of this isomer would afford a product containing a strained trans[4.3.0]bicyclo ring system). Using TFA, only the isomer with the side chain in the axial position (α-28) underwent cyclization to form the all-cis version of 6 (Scheme 8). The fact that the overall yield exceeds the amount of minor azido ketal α-28 present in the reaction mixture probably means that some deketalization can occur prior to Schmidt reaction but that this process is poorly competitive with azide decomposition. (It is also likely that deketalization proceeds prior to Schmidt reaction in α-28 as well – as shown in Scheme 8 – but we have no evidence either in support of or against this.)
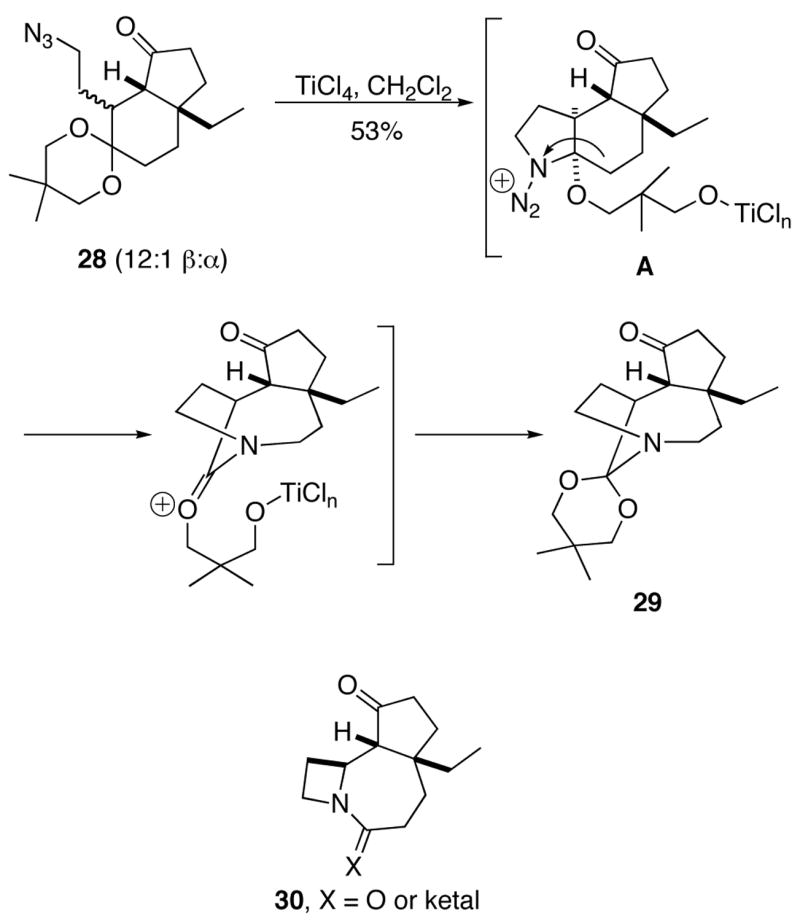
In contrast, treating the 12:1 mixture of 28 with TiCl4 afforded 29 isolated in moderate yieldsThis product apparently arose from Lewis acid activation of the ketal to afford oxenium ion, followed by attack of the azide and C→N migration. Under these conditions, none of the desired compound 6 was isolated.
This result was surprising in several ways. Although reactions of azides with ketals or enol ethers had been previously demonstrated,15 we had expected that the ketal would in this case fulfill its designated role as a protecting group. The preferential reaction of the ketal over the carbonyl could be due to stereochemistry. Thus, carbonyl activation might occur but azide may not attack due to the trans relationship of the side chain to the ketone. Alternatively, TiCl4 activation of the ketal could preferentially effect ketal opening over coordination to the C=O group. Unfortunately, the less stable γ-epimer was not easily prepared to address this question directly. However, we could confirm that β-28 led to 29 by X-ray confirmation of the latter structure (Supporting Information).
Another noteworthy outcome was the isolation of the bridged orthoaminal 29 in this reaction. Prior to this experiment, no bridged product had been isolated in an azido-Schmidt reaction carried out in our laboratory. In the present case, it is likely that ring system 30 containing a 4-membered ring did not form due to strain. It is also possible that the azido-Schmidt reaction of ketals better accommodates bridged product formation than ketones version; in the latter case, the fused amide product enjoys resonance stabilization whereas the bridged (or “twisted”16) amide cannot. Subsequent to this study, we discovered some specialized examples of bridged amides from azido ketones resulting from intramolecular Schmidt reactions of azides and ketones.17 The factors that influence bridged versus fused product formation in intramolecular Schmidt reactions is the subject of current research in this laboratory.
In light of this result, we returned to our original plan of selectively carrying out the ring expansion on diketone 7, postulating that epimerization would preceed Schmidt reaction in this substrate. Gratifyingly, eketalization was easily effected under mild conditions to afford the diketo azide 7, predominantly as the desired isomer (1:10 β/α, Scheme 10; see Supporting Information for structural assignment of 7).
Scheme 10.
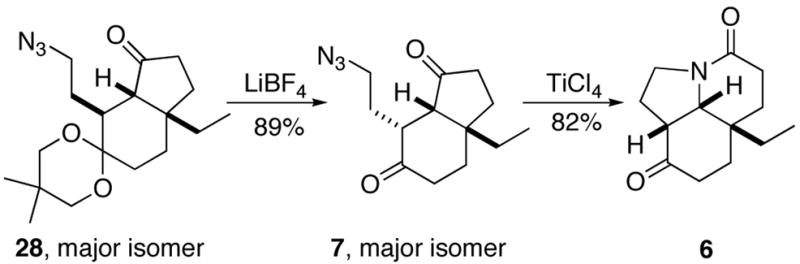
In contrast to the above examples, treatment of 7 with TiCl4gave tricyclic lactam 6 as a single diastereomer in 82% yield and 84% ee. The exclusive formation of this product confirmed our original hypothesis that a regioselective Schmidt reaction was possible in this system. The enantiomerically enriched lactam was then recrystallized from EtOAc/hexanes to enantiomeric purity (50% overall yield after recrystallization, ≥99% ee, Chiralcel OD, 10% EtOH/hexanes).
Conversion of the Stork ketone to aspidospermidine
All that remained was the completion of the synthesis using the Fischer indole protocol. Most literature examples of this particular reaction have been reported to proceed in poor yield (or no yield was reported at all), a fact that lessens the impact of formal syntheses invoking the Stork2 or related intermediates as well.5 We therefore wished to directly address this issue.
As reported for related lactams,5b,c 6 did not prove amenable to a direct Fischer indole synthesis and so was reduced in a three-step process to the corresponding keto amine 3 by selective protection of the ketone carbonyl followed by a reduction/deprotection sequence (Scheme 11). Treatment of 3 with phenylhydrazine, acid, and finally LAH afforded (+)-aspidospermidine (1, 51% overall yield from 3) accompanied by 13% of a side product 31. This side product presumably arose via the Fischer indolization of the less-substituted enamine isomer. A similar product was mentioned in a synthesis of an oxygenated analogue of aspidospermine without ratios or yields.18
Scheme 11.
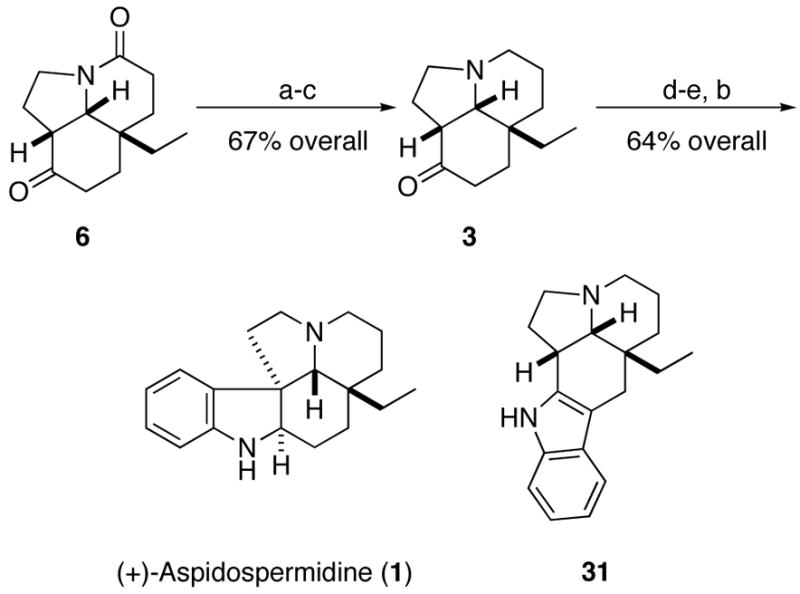
aReagents and conditions: (a) bis(trimethylsilyl)neopentyl glycol, TMSOTf, CH2Cl2, 0 °C→ rt; (b) LAH, THF, reflux; (c) LiBF4, aq CH3CN, reflux; (d) PhNHNH2; (e) AcOH, reflux.
The structural analysis of this latter compound was nontrivial and extensive NMR experiments were needed to deduce the structure (Supporting Information). Interestingly, it was necessary to rigorously rid (+)-aspidospermidine of 31 for analysis as even minor quantities of this “impurity” threw off the optical rotation of 1 substantially on account of large levrorotatory rotation of (−)−31 ([α]D = −100 (c 0.4, EtOH)) relative to (+)−1. Spectral data (1H and 13C NMR, IR, MS), mp (118–119 °C), and specific rotation ([α]D = 20.6 (c 0.64, EtOH)) of purified 1 were fully consistent with reported values (mp 119.5–121 °C, [α]D = 21 (EtOH)).19
Summary
In summary, a total synthesis of (+)-aspidospermidine was completed, with the ultimately crafted version summarized in Scheme 12. The most linear route comprised 22 steps and took place with 1.1% overall yield from butylene glycol. The most noteworthy elements of this synthesis include (1) a highly selective intramolecular Schmidt reaction on a nonsymmetrical diketone, (2) the establishment of the quaternary stereogenic center via a d’Angelo deracemization process (from which all of the rest of the target’s stereocenters derive), (3) the conversion of an γ-hydroxy-α,β-unsaturated ketone to a 1,4-diketone via an internal redox process, and (4) the first observation of a bridged orthoaminal from an intramolecular azide insertion process on a ketal.
Scheme 12.
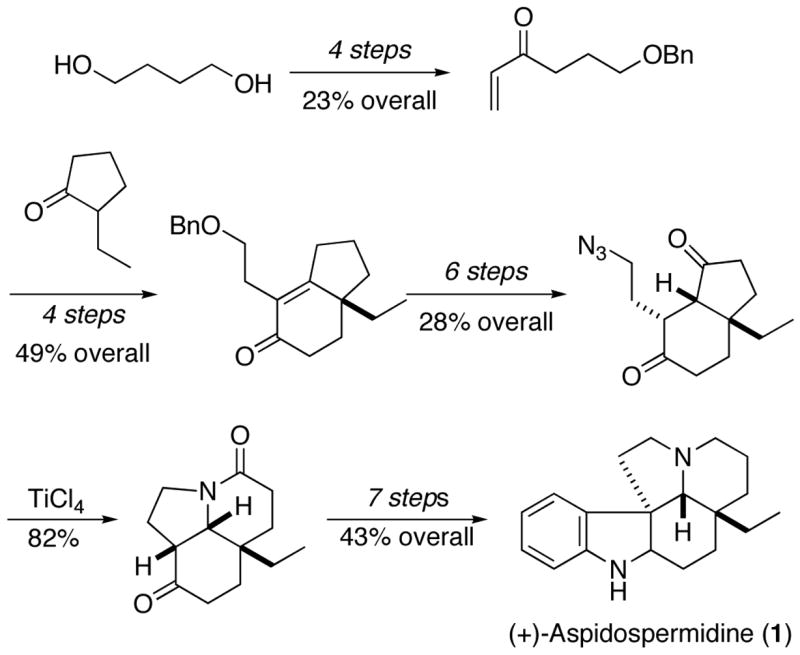
Experimental Section
4-(2′-Benzyloxyethyl)-7a-ethyl-1,2,3,6,7,7a-hexahydroinden-5-one (8)
A solution of 2-ethylcyclopentanone20 (5.36 g, 47.9 mmol), (S)-α-methylbenzylamine (6.38 g, 52.6 mmol) and p-TsOH (0.45 g, 2.4 mmol) in benzene (25 mL) was thoroughly degassed and was allowed to reflux for 17 h using a Dean-Stark trap. After the theoretical amount of H2O had been removed the remaining benzene was distilled off at atmospheric pressure and the crude imine residue was cooled to rt and suspended in Et2O (10 mL).
Fused ZnCl2 (3.26 g, 24 mmol) was suspended in Et2O (15 mL) and was added imine followed by 6-benzyloxy-1-hexen-3-one (11.7 g, 57.4 mmol) and hydroquinone (ca. 100 mg). The resulting solution was heated at reflux for 72 h, then cooled to rt and stirred for an additional 8 h. Aqueous acetic acid (25%, 60 mL) was added and the resulting biphasic solution was stirred vigorously for 3 h. The mixture was poured over 1 M HCl and extracted with Et2O. The combined organic layers were successively washed with saturated aqueous NaHCO3 and brine, dried over MgSO4, and concentrated. The residue was added to a solution of sodium methoxide (119.7 mmol) in methanol (70 mL) and allowed to reflux for 24 h. The reaction mixture was then cooled to rt, quenched with a few drops of acetic acid, and concentrated to yield a brown oil that was dissolved in Et2O, washed with brine, dried over MgSO4, and concentrated in vacuo to afford a brown oil. Baseline impurities were removed by silica gel chromatography, eluting with 10% EtOAc in hexanes, and the crude product distilled (200–220 °C, 1 Torr) to yield 7.00 g (49%) of the title compound 8 (Rf = 0.20, 20% Et2O in hexanes) as a light yellow oil. [α]D = −33.3 (c 3.63, CH2Cl2); IR (film) 1645 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.65 (t, J = 7.5 Hz, 3H), 1.27 (m, 1H), 1.44 (q, J = 7.5 Hz, 2H), 1.63 (td, J = 13.8, 5.4 Hz, 1H), 1.81 (m, 2H), 2.00 (dt, J = 12.6, 4.2 Hz, 1H), 2.18 (ddd, J = 13.3, 5.3, 1.9 Hz, 1H), 2.33 (ddd, J = 18.2, 5.4, 1.9 Hz, 1H), 2.45 (dd, J = 14.2, 5.3 Hz, 1H), 2.55 (m, 2H), 2.64 (t, J = 7.7 Hz, 2H), 3.49 (m, 2H), 4.50 (s, 2H), 7.32 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 9.6, 22.0, 26.0, 27.5, 30.4, 31.3, 33.9, 36.8, 46.7, 69.5, 73.1, 127.78, 127.83, 128.3, 128.7, 139.2, 175.5, 198.9; CIMS m/z (relative intensity) 299 (MH+, 90), 207 (25), 191 (65), 91 (40). Anal. calcd for C20H26O2: C, 80.50; H, 8.78, found: C, 80.22; H, 9.00.
4-(2′-Benzyloxyethyl)-7a-ethyl-3-hydroxy-1,2,3,6,7,7a-hexahydroinden-5-one (25)
A solution of enone 8 (2.0 g, 6.7 mmol) and p-TsOH (0.25 g) in isopropenyl acetate (10 mL) was heated at reflux for 5 h. The reaction mixture was then cooled to rt and diluted with Et2O. The organic layer was washed with saturated NaHCO3 and brine, and dried over MgSO4. The resulting solution was concentrated in vacuo to a light yellow oil. The residue was immediately dissolved in a mixture of acetone (40 mL), saturated aqueous NaHCO3 (20 mL), and H2O (20 mL), and treated with 6 g of Oxone. After 3 h, the reaction mixture was poured into H2O and extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The resulting residue was purified by silica gel chromatography(25% EtOAc/hexanes) to yield 1.85 g (88%) of 25 (Rf = 0.35, 30% EtOAc in hexanes) as a clear oil. [α]D = −7.5 (c 3.25, EtOH); IR (CH2Cl2) 3401, 1657 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.4 Hz, 3H), 1.25–1.40 (m, 2H), 1.67–1.72 (m, 2H), 1.80–1.88 (m, 2H), 1.90–2.00 (m, 1H), 2.15–2.20 (m, 1H), 2.30–2.35 (m, 1H), 2.39–2.47 (m, 2H), 2.78 (dt, J = 2.2, 14.0 Hz, 1H), 3.35 (ddd, J = 2.2, 9.0, 11.5 Hz, 1H), 3.58 (dt, J = 4.0, 9.0 Hz, 1H), 4.47 (d, J = 5.6 Hz, 1H), 4.51 (s, 1H), 4.78 (d, J = 5.6 Hz, 1H), 7.22–7.36 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 9.4, 27.3, 27.7, 30.6, 32.3, 33.2, 33.5, 46.0, 67.0, 70.9, 73.0, 127.8, 127.85, 128.3, 129.7, 137.0, 175.6, 199.3; CIMS m/z (relative intensity) 315 (MH+, 100), 297 (45), 207 (52), 91 (49); HRMS calcd for C20H26O3: 314.1882, found: 314.1902.
7-(2′-Benzyloxyethyl)-3a-ethyl-6-(5′,5′-dimethyl[1,3]dioxan-2′-yl)octahydroinden-1-one (26)
TMSOTf (0.848 g, 3.82 mmol) was added dropwise to a 0 °C solution of allylic alcohol 25 (4.00 g,12.7 mmol) and bis(trimethylsilyl)neopentyl glycol (4.74 g, 19.1 mmol) in CH2Cl2 (30 mL). This mixture was stirred for 10 h while it was allowed to warm to rt. The reaction was then quenched with saturated aqueous NaHCO3 (20 mL), extracted with CH2Cl2, dried over MgSO4, and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (10% EtOAc/hexanes) to yield 3.50 g (69%) of 26 (Rf = 0.7, 25% EtOAc in hexanes) as a yellow oil. [α]D = +30.6 (c 3.6, CH2Cl2); IR (CH2Cl2) 1734 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.77 (s, 3H), 0.86 (t, J = 7.4 Hz, 3H), 1.05 (s, 3H), 1.43–1.95 (m, 11H), 2.12–2.20 (m, 1H), 2.29–2.36 (m, 1H), 2.99 (d, J = 9.4 Hz, 1H), 3.16 (dd, J = 2.0, 11.7 Hz, 1H), 3.26 (dd, J = 2.0, 11.7 Hz, 1H), 3.49–3.61 (m, 4H), 4.50 (d, J = 3.0 Hz, 2H), 7.24–7.36 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 8.17, 22.3, 23.0, 26.7, 28.0, 28.3, 29.5, 30.6, 31.3, 32.0, 34.6, 40.9, 53.2, 69.1, 69.5, 69.6, 70.5, 72.8, 96.5, 127.4, 127.5, 128.1, 128.2, 138.5, 219.0; FAB m/z (relative intensity) 401 (MH+, 35), 307 (32), 207 (49), 154 (100); 136 (100); HRMS calcd for C25H36O4: 400.2599, found: 400.2614.
7-(2′-Hydroxyethyl)-3a-ethyl-6-(5,5-dimethyl-[1,3]dioxan-2-yl)-octahydro-inden-1-one (27)
Benzyl ether 26 (3.10 g, 7.75 mmol) and 10% Pd/C (0.465 g, 15% w/w) were stirred in MeOH (75 mL) under a hydrogen atmosphere (balloon). After 12 h, the reaction mixture was filtered through a pad of Celite, concentrated in vacuo, and the crude product residue purified by chromatography (30% EtOAc/hexanes) to yield 2.05 g (85%) of 27 (Rf = 0.24) as a clear oil. [α]D = +28.1 (c 1.4, CH2Cl2); IR (film) 3429, 1733 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 7.4 Hz, 3H), 0.94 (s, 3H), 0.97 (s, 3H), 1.40–1.49 (m, 3H), 1.55–1.70 (m, 5H), 1.75–1.85 (m, 1H), 1.89 (d, J = 6.2 Hz, 1H), 1.95–2.05 (m, 1H), 2.06–2.15 (m, 1H), 2.17–2.25 (m, 1H), 2.33–2.45 (m, 1H), 2.54 (m, 1H), 3.35–3.41 (m, 2H), 3.48–3.65 (m, 3H), 3.65–3.75 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 8.1, 22.7, 25.7, 27.1, 28.8, 29.7, 30.2, 32.3, 34.6, 35.8, 41.0, 57.3, 62.0, 69.6, 69.7, 98.5, 219.9; CIMS m/z (relative intensity) 310 (M+, 3), 293 (9), 207 (100), 141 (65); 55 (16); HRMS calcd for C18H30O4: 310.2144, found: 310.2131.
7-(2′-Azidoethyl)-3a-ethyl-6-(5′,5′-dimethyl[1,3]dioxan-2′-yl)-octahydroinden-1-one (28)
DEAD (2.58 g, 14.8 mmol) was added to a 0 °C solution of 27 (2.30 g, 7.42 mmol), PPh3 (3.9 g, 14.8 mmol) and HN3 (8.9 mL of a 2.5 M solution in benzene, 22.3 mmol) in benzene (40 mL) and stirred for 11 h while it was allowed to warm up to rt. The reaction was quenched with H2O (100 mL) and extracted with Et2O. The organic extracts were washed with saturated aqueous NaHCO3, water and brine (20 mL each), dried over MgSO4, and concentrated in vacuo, and the resulting residue was purified by chromatography (5% EtOAc/hexanes), to yield 1.81 g (73%) of 28 (Rf = 0.73, 30% EtOAc in hexanes) as a light yellow oil. [α]D = +37.9 (c 5.54, CH2Cl2); IR (CH2Cl2) 2957, 2100, 1733 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 7.4 Hz, 3H), 0.91 (s, 3H), 0.97 (s, 3H), 1.39–1.50 (m, 3H), 1.58–1.70 (m, 4H), 1.77–1.82 (m, 2H), 1.96–2.02 (m, 2H), 2.18–2.25 (m, 1H), 2.32–2.42 (m, 1H), 2.53–2.57 (m, 1H), 3.28–3.55 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 8.1, 22.6, 22.7, 26.5, 26.8, 27.0 (2 peaks), 29.4, 29.6, 32.2, 34.2, 34.6, 50.8, 56.7, 69.6, 69.7, 98.5, 218.9; CIMS m/z (relative intensity) 336 (MH+, 4), 293 (9), 308 (39), 279 (14), 222 (41); 141 (100); HRMS calcd for C18H29N3O3: 335.2209, found: 335.2202.
6-Ethyl-12-(5′,5′-dimethyl[1,3]dioxan-2′-yl)-9-aza-tricyclo[7.2.1.0]dodecan-3-one (29)
TiCl4 (0.07 g, 0.36 mmol) was added dropwise to a solution of 28 (0.040 g, 0.12 mmol) in CH2Cl2 (5 mL) maintained at 0 °C and stirred for 3 h while it warmed to rt. The reaction was quenched with saturated aqueous NaHCO3 and i-PrOH (5 mL), poured into brine, and filtered through a pad of wet Celite. It was then extracted with CH2Cl2, organic extracts washed with saturated aqueous NaHCO3, water and brine (10 mL each), dried over MgSO4, concentrated in vacuo, and the resulting residue purified by chromatography, eluting with EtOAc and then 10% MeOH in EtOAc, to yield 0.020 g (52%) of 29 (Rf = 0.75, 10% MeOH in EtOAc) as a light yellow solid: mp = 74–76 °C; IR (CCl4) 2948, 1737 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.71 (s, 3H), 0.87 (t, J = 7.4 Hz, 3H), 1.10 (s, 3H), 1.25 (m, 1H), 1.36–1.43 (m, 3H), 1.52–1.60 (m, 1H), 1.79 (d, J = 3.6 Hz, 1H), 2.00–2.04 (m, 1H), 2.14–2.20 (m, 2H), 2.23–2.27 (m, 2H), 2.69 (m, 1H), 2.76 (m, 1H), 2.95–3.04 (m, 3H), 3.12 (dd, J = 10.6, 2.4 Hz, 1H), 3.22 (dd, J = 10.3, 2.4 Hz, 1H), 3.61 (d, J = 10.3 Hz, 1H), 3.81 (d, J = 10.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 8.6, 21.7, 22.8, 28.8, 29.3, 29.35, 33.0, 36.6, 38.8, 44.3, 44.8, 47.0, 49.0, 56.0, 68.0, 72.0, 112.0, 213.0; CIMS m/z (relative intensity) 308 (MH+, 100), 307 (M+, 14), 222 (51), 121 (45), 84 (20), 79 (15), 55 (31); HRMS calcd for C18H30NO3: 308.2226, found: 308.2202.
7-(2′-Azidoethyl)-3a-ethylhexahydroindene-1,6-dione (7)
LiBF4 (0.36 g, 3.88 mmol) was added to 28 (0.26 g, 0.77 mmol) in 20% H2O in CH3CN (20 mL total volume) and heated at 70 °C for 18 h. The reaction was then diluted with H2O, extracted with Et2O, dried (MgSO4), concentrated in vacuo and chromatographed to afford 0.17 g (89%) of 7 (Rf = 0.44, 30% EtOAc in hexanes) as a colorless oil. IR (CH2Cl2) 3053, 1737, 1712 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.92 (t, J = 7.4 Hz, 3H), 1.40 (sextet, J = 7.0 Hz, 1H), 1.54–1.60 (m, 2H), 1.81–2.10 (m, 8H), 2.36–2.45 (m, 4H), 2.58–2.62 (m, 1H), 3.26–3.31 (m, 1H), 3.39–3.44 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 8.0, 28.5, 29.3, 29.7, 31.1, 34.8, 35.8, 41.8, 43.6, 49.4, 60.1, 211.1, 217.9; CIMS m/z (relative intensity) 250 (MH+, 2), 222 (44), 192 (12), 165 (17), 43 (100); HRMS calcd for C13H20N3O2: 250.1555, found: 250.1547.
6aS-Ethyloctahydropyrrolo[3,2,1-ij]quinoline-4,9-dione (6)
TiCl4 (0.049 g, 0.256 mmol) was added dropwise to a 0 °C solution of diketo azide 7 (0.058 g, 0.233 mmol) in CH2Cl2 (3 mL) and stirred for 2 h, then allowed to warm to rt and stirred for an additional 15 min. The reaction was quenched with solid NaHCO3/i-PrOH, poured into brine and filtered through a pad of wet Celite. The filtrate was extracted with CH2Cl2, the organic extracts washed with saturated aqueous NaHCO3, dried (MgSO4) and concentrated in vacuo. The brown residue was purified by chromatography, eluting with EtOAc followed by 10% MeOH in EtOAc, to afford 0.042 g (82%) of 6 (Rf = 0.24, EtOAc) as a light yellow crystalline solid. Mp = 139–140 °C; [α]D = −89.1 (c 1.575, CH2Cl2); IR (film) 1705, 1630 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.00 (t, J = 7.5 Hz, 3H), 1.50 (m, 1H), 1.57–1.73 (m, 3H), 1.83 (m, 2H), 2.00 (m, 1H), 2.25–2.50 (m, 4H), 2.63 (m, 1H), 2.86 (t, J = 5.9 Hz, 1H), 3.42 (dd, J = 10.0, 4.7 Hz, 2H), 3.63 (d, J = 4.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 7.4, 22.1, 25.2, 27.9, 29.2, 29.7, 34.1, 36.3, 43.8, 49.4, 68.5, 168.2, 208.6; EIMS m/z (relative intensity) 222 (MH+, 80), 49 (30); HRMS calcd for C13H20NO2: 222.1504, found: 222.1494.
6a-Ethyldecahydropyrrolo[3,2,1-ij]quinolin-9-one (3)
TMSOTf (0.06 g, 0.27 mmol) was added to a mixture of 6 (0.06 g, 0.27 mmol) and bis(trimethylsilyl)neopentyl glycol (0.402 g, 1.62 mmol) in CH2Cl2 (5 mL) maintained at 0 °C. The reaction was stirred for 6 h and warmed to rt. It was quenched with saturated aqueous NaHCO3, extracted with CH2Cl2, dried (MgSO4) and concentrated to a brown oil. Rf = 0.5 (10% MeOH in EtOAc); IR (CCl4) 2951, 1638 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.69 (s, 3H), 0.87 (t, J = 7.4 Hz, 3H), 1.17 (s, 3H), 1.20–1.25 (m, 3H), 1.50–1.56 (m, 2H), 1.72–1.79 (m, 3H), 2.23 (dd, J = 7.0, 5.4 Hz, 1H), 2.29–2.33 (m, 2H), 2.37–2.42 (m, 1H), 2.57 (dt, J = 10.5, 3.6 Hz, 1H), 3.22–3.31 (m, 4H), 3.48 (d, J = 11.4 Hz, 1H), 3.72 (d, J = 11.4 Hz, 1H), 3.80 (q, J = 11.2 Hz, 3H).
The crude ketal was added to LiAlH4 (0.05 g, 1.35 mmol) in THF (5 mL) and heated at reflux for 10 h. The reaction was quenched with water, filtered through wet Celite, and extracted with Et2O. The organic extracts were dried (MgSO4) and concentrated to a brown oil. This oil was charged with LiBF4 (0.126 g, 1.35 mmol) and dissolved in 10 % H2O/CH3CN and heated at 80 °C for 8 h. The reaction was then diluted with water, extracted with Et2O, dried (MgSO4), concentrated and chromatographed to give 0.037 g (66% overall) of 3 as a colorless oil. Rf = 0.51 (10% MeOH in EtOAc); IR (film) 1700 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.95 (t, J = 7.5 Hz, 3H), 1.12 (td, J = 13.4, 4.6 Hz, 1H), 1.34 (m, 1H), 1.51 (m, 2H), 1.59–1.87 (m, 4H), 1.93 (m, 3H), 2.22–2.47 (m, 4H), 2.68 (m, 1H), 3.30 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 7.6, 21.69, 21.72, 26.5, 30.5, 33.3, 35.1, 37.2, 48.6, 53.3, 53.6, 74.0, 211.9; CIMS m/z (relative intensity) 208 (MH+, 100), 178 (30), 82 (50).
(+)-Aspidospermidine 1 and Byproduct 31
Amino ketone 3 (0.036 g, 0.174 mmol) and phenylhydrazine (0.02 g, 0.208 mmol) in 5 mL of PhH were allowed to reflux for 3.5 h. The reaction was then cooled to rt and concentrated in vacuo. The crude phenylhydrazone was allowed to reflux in 5 mL of AcOH for 4 h and then concentrated in vacuo to afford the crude indoline as a dark brown oil. This material was dissolved in THF (5 mL), treated with LiAlH4 (0.066 g, 1.74 mmol) and allowed to reflux for 12 h. After cooling to 0 °C and quenching with H2O/NaOH, the mixture was filtered through a plug of Celite and concentrated in vacuo. The resulting residue was purified by chromatography (EtOAc) to yield 0.025 g (51%) aspidospermidine (1) as a pale yellow oil that crystallized upon standing and 31 (0.0065 g, 13%) as a colorless oil. Data for (+)-aspidospermidine (1): Rf = 0.29 (10% MeOH/EtOAc); mp = 117–119 °C (acetone) (lit.19 119–121 °C) [α]D = +20.6 (c 0.65, EtOH) (lit.2 +21 (c 7.4, EtOH)); IR (film) 3320, 1600 cm−01; 1H NMR (400 MHz, CDCl3) δ 0.66 (t, J = 7.5 Hz, 3H), 0.88 (m, 1H), 1.07 (m, 1H), 1.13 (td, J = 13.5, 4.6 Hz, 1H), 1.36–1.57 (m, 4H), 1.66 (m, 2H), 1.76 (qt, J = 12.8, 3.9 Hz, 1H), 1.97 (m, 2H), 2.24 (s, 1H), 2.22–2.37 (m, 2H), 3.08 (m, 1H), 3.14 (m, 1H), 3.53 (dd, J = 11.0, 6.2 Hz, 1H), 6.66 (d, J = 7.8 Hz, 1H), 6.75 (t, J = 7.3 Hz, 1H), 7.04 (td, J = 7.6, 0.9 Hz, 1H), 7.10 (d, J = 7.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 7.2, 22.2, 23.4, 28.5, 30.4, 34.9, 36.0, 39.2, 53.5, 53.8, 54.3, 66.1, 71.7, 110.7, 119.4, 123.3, 127.5, 136.1, 149.8; CIMS m/z (relative intensity) 283 (MH+, 60), 254 (25), 210 (25), 124 (100).
Data for 5 a-Ethyl-1,3,4,5,5a,6,11,11d-octahydro-2H,11cH-2a,11-diazo-indeno[1,7-ab]fluorene (31) Rf = 0.1 (10% MeOH/EtOAc); [α]D = −100 (c 0.4, EtOH); IR (CH2Cl2) 3459, 2934 cm−1; 1H NMR (400 MHz, MeOH-d4) δ 0.86 (t, J = 7.5 Hz, 3H), 1.23–1.38 (m, 3H), 1.45–1.55 (sextet, J = 7.5 Hz, 1H), 1.68–1.77 (m, 2H), 1.89–2.05 (m, 3H), 2.16 (m, 1H), 2.24 (m, 1H), 2.36 (q, J = 12.0 Hz, 1H), 2.51 (d, J = 15.0 Hz, 1H), 2.79 (d, J = 15.0 Hz, 1H), 3.12 (br t, J = 9.0 Hz, 1H), 3.52 (m, 1H), 6.94 (td, J = 7.0, 1.0 Hz, 1H), 7.04 (td, J = 7.0, 1.0 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H); 13C NMR (100 MHz, MeOH-d4-CDCl3) δ 5.6, 20.5, 22.1, 26.7, 29.2, 31.5, 32.7, 35.2, 52.2, 53.7, 71.5, 103.7, 104.3, 109.3, 116.2, 117.1, 119.3, 126.9, 136.4; EIMS m/z (relative intensity) 281 (MH+, 100), 124 (70), 58 (14). HRMS calcd for C19H25N2: 281.2018, found: 281.2025.
Supplementary Material
Details of stereochemical assignments, additional experimental details, copies of 1H and 13C NMR spectra, and X-ray crystallographic details for compound 29 (cif). This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 9.
Acknowledgments
We thank the National Institutes of Health (GM-49093) for the support of this work, David Vander Velde for NMR spectroscopy consultation, Doug Powell for X-ray crystallography, and Timothy Ribelin for help with compound characterization.
References and Notes
- 1.Saxton JE. In: The Alkaloids: Chemistry and Biology. Cordell GA, editor. Vol. 51. Academic Press; San Diego, CA: 1998. pp. 1–197. [Google Scholar]
- 2.Stork G, Dolfini JE. J Am Chem Soc. 1963;85:2872–2973. [Google Scholar]
- 3.(a) Ban Y, Sato Y, Inoue I, Nagai M, Oishi T, Terashima M, Yonemitsu O, Kanaoka Y. Tetrahedron Lett. 1965:2261–2268. doi: 10.1016/s0040-4039(00)70368-6. [DOI] [PubMed] [Google Scholar]; (b) Barton JED, Harley-Mason J. Chem Commun. 1965:298–299. [Google Scholar]; (c) Kutney JP, Abdurahman N, Le Quesne P, Piers E, Vlattas I. J Am Chem Soc. 1966;88:3656–3657. doi: 10.1021/ja00709a052. [DOI] [PubMed] [Google Scholar]; (d) Harley-Mason J, Kaplan M. Chem Commun. 1967:915–916. [Google Scholar]; (e) Inoue I, Ban Y. J Chem Soc (C) 1970:602–610. doi: 10.1039/j39700000602. [DOI] [PubMed] [Google Scholar]; (f) Kutney JP, Abdurahman N, Gletsos C, Le Quesne P, Piers E, Vlattas I. J Am Chem Soc. 1970;92:1727–1735. doi: 10.1021/ja00709a052. [DOI] [PubMed] [Google Scholar]; (g) Klioze SS, Darmory FP. J Org Chem. 1975;40:1588–1592. [Google Scholar]; (h) Dufour M, Gramain JC, Husson HP, Sinibaldi ME, Troin Y. J Org Chem. 1990;55:5483–5490. [Google Scholar]; (i) Benchekroun-Mounir N, Dugat D, Gramain JC, Husson HP. J Org Chem. 1993;58:6457–6465. [Google Scholar]; (j) Wenkert E, Liu S. J Org Chem. 1994;59:7677–7682. [Google Scholar]; (k) Callaghan O, Lampard C, Kennedy AR, Murphy JA. J Chem Soc, Perkin Trans 1. 1999:995–1002. [Google Scholar]; (l) Callaghan O, Lampard C, Kennedy AR, Murphy JA. Tetrahedron Lett. 1999;40:2225. [Google Scholar]; (m) Patro B, Murphy JA. Org Lett. 2000;2:3599–3601. doi: 10.1021/ol006477x. [DOI] [PubMed] [Google Scholar]; (n) Toczko MA, Heathcock CH. J Org Chem. 2000;65:2642–2645. doi: 10.1021/jo991599s. [DOI] [PubMed] [Google Scholar]; (o) Banwell MG, Smith JA. J Chem Soc, Perkin Trans 1. 2002:2613–2618. [Google Scholar]; (p) Banwell MG, Lupton DW. Org Biomol Chem. 2005;3:213–215. doi: 10.1039/b415977b. [DOI] [PubMed] [Google Scholar]
- 4.(a) Node M, Nagasawa H, Fuji K. J Org Chem. 1990;55:517–521. [Google Scholar]; (b) d’Angelo J, Revial G, Costa PRR, Castro RN, Antunes OAC. Tetrahedron: Asymm. 1991;2:199–202. [Google Scholar]; (c) Desmaële D, d’Angelo J. J Org Chem. 1994;59:2292–2303. [Google Scholar]; (d) Schultz AG, Pettus L. J Org Chem. 1997;62:6855–6861. [Google Scholar]; (e) Callaghan O, Lampard C, Kennedy AR, Murphy JA. Tetrahedron Lett. 1999;40:2225. [Google Scholar]; (f) Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628–4641. doi: 10.1021/ja017863s. [DOI] [PubMed] [Google Scholar]; (g) Marino JP, Rubio MB, Cao G, de Dios A. J Am Chem Soc. 2002;124:13398–13399. doi: 10.1021/ja026357f. [DOI] [PubMed] [Google Scholar]; (h) Tanino H, Fukuishi K, Ushiyama M, Okada K. Tetrahedron Lett. 2002;43:2385–2388. [Google Scholar]; (i) Tanino H, Fukuishi K, Ushiyama M, Okada K. Tetrahedron. 2004;60:3273–3282. [Google Scholar]
- 5.(a) Meyers AI, Berney D. J Org Chem. 1989;54:4673–4676. [Google Scholar]; (b) Fukuda YI, Shindo M, Shishido K. Org Lett. 2003;5:749–751. doi: 10.1021/ol034020s. [DOI] [PubMed] [Google Scholar]; (c) Gnecco D, Vazquez E, Galindo A, Teran JL, Orea L, Bernes S, Enriquez RG. ARKIVOC (Gainesville, FL, United States) xi. 2003. pp. 185–192. [Google Scholar]
- 6.Milligan GL, Mossman CJ, Aubé J. J Am Chem Soc. 1995;117:10449–10459. [Google Scholar]
- 7.d’Angelo J, Desmaële D, Dumas F, Guingant A. Tetrahedron: Asymm. 1992;3:459–505. [Google Scholar]
- 8.Iyengar R, Schildknegt K, Aubé J. Org Lett. 2000;2:1625–1627. doi: 10.1021/ol005913c. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hajos ZG, Parrish DR. J Org Chem. 1974;39:1615–1621. [Google Scholar]; (b) List B. Tetrahedron. 2002;58:5573–5590. [Google Scholar]
- 10.Abel U, Koch C, Speitling M, Hansske Friedrich G. Curr Opinion Chemical Biol. 2002;6:453–458. doi: 10.1016/s1367-5931(02)00338-1. [DOI] [PubMed] [Google Scholar]
- 11.Williams D, Cortez GS. Tetrahedron Lett. 1998;39:2675–2678. [Google Scholar]
- 12.(a) Amrollah-Madjdabadi A, Pham TN, Ashby EC. Synthesis. 1989:614–616. [Google Scholar]; (b) Pelletier JD, Poirier D. Tetrahedron Lett. 1994;35:1051–1054. [Google Scholar]
- 13.(a) Utaka M, Fujii Y, Takeda A. Chem Lett. 1986:1103–1104. [Google Scholar]; (b) Vankar YD, Kumaravel G, Mukherjee N, Rao CT. Synth Commun. 1987;17:181–187. [Google Scholar]; (c) Gondos G, Orr JC. Liebigs Ann Chem. 1990:213–215. [Google Scholar]
- 14.The ring opening of activated cyclopropanes is well established to proceed better when two activating groups are present. Danishefsky S. Acc Chem Res. 1979;12:66–72.
- 15.Mossman CJ, Aubé J. Tetrahedron. 1995;52:3403–3408. [Google Scholar]
- 16.Greenberg A. In: Structure and Reactivity. Liebman JF, Greenberg A, editors. VCH; New York: 1988. pp. 139–178. [Google Scholar]
- 17.(a) Golden JE, Aubé J. Angew Chem, Int Ed. 2002;41:4316–4318. doi: 10.1002/1521-3773(20021115)41:22<4316::AID-ANIE4316>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; (b) Lei Y, Wrobleski AD, Golden JE, Powell DR, Aubé J. J Am Chem Soc. 2005;127:4552–4553. doi: 10.1021/ja050214m. [DOI] [PubMed] [Google Scholar]
- 18.Inoue I, Ban Y. J Chem Soc (C): Org. 1970:602–610. doi: 10.1039/j39700000602. [DOI] [PubMed] [Google Scholar]
- 19.Camerman A, Camerman N, Kutney JP, Piers E, Trotter J. Tetrahedron Lett. 1965:637–642. [Google Scholar]
- 20.Bernady KF, Poletto JF, Nocera J, Mirando P, Schaub RE, Weiss MJ. J Org Chem. 1980;45:4702–4715. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of stereochemical assignments, additional experimental details, copies of 1H and 13C NMR spectra, and X-ray crystallographic details for compound 29 (cif). This material is available free of charge via the Internet at http://pubs.acs.org.