

Abstract
Ex vivo gene therapy approaches can improve the outcome of islet transplantation for treating type I diabetes. We have recently shown improvement in islet survival and function following ex vivo infection of islets with a mixture of adenoviral vectors encoding human vascular endothelial growth factor (Adv-hVEGF) and human interleukin-1 receptor antagonist (Adv-hIL-1Ra). In this study, we constructed a bicistronic vector encoding these two genes (phVEGF-hIL-1Ra) by cloning hIL-1Ra under the cytomegalovirus (CMV) promoter and hVEGF under the elongation factor-1α (EF-1 α) promoter in pBudCE4.1 vector. There was dose and time dependent expression of hVEGF and hIL-1Ra at both mRNA and protein levels after transfection with human islets. Transfected islets were viable, as evidenced by insulin release upon glucose challenge. Co-expression of hVEGF and hIL-1Ra suppressed nitric oxide production, total caspases, apoptosis and necrosis in the presence of inflammatory cytokine cocktail consisting of IL-1β, TNFα and IFNγ. In conclusion, our results indicated that co-expression of growth factor and antiapoptic genes can improve islet survival and function.
Keywords: bicistronic plasmid, hVEGF, hIL-1Ra, human islet, real time RT-PCR, insulin release
INTRODUCTION
Human islet transplantation offers a great potential for treating type I diabetes. However, this approach has limited clinical applications due to the short supply of cadaver donors, host immune rejection, primary non-function and cell death of islets following transplantation [1–3]. To achieve successful islet transplantation, primary non-function of islets has to be eliminated. Islet destruction following transplantation can be prevented by genetically engineering β-cells to (1) promote revascularization, and intercept (2) apoptotic and inflammatory pathways by expressing desired proteins in the vicinity of the islets [4–6].
Pancreatic islets are a complex cluster of heterogeneous cell types with extensive intra-islet vasculature formed of fenestrated capillary endothelial lining, which gets disrupted during islet isolation leading to collapse of vasculature, accumulation of endothelial fragments and compromised perfusion in the core of the islets. Hence, unlike whole pancreas transplantation, islet grafts require the process of revascularization to establish adequate microvascular blood supply [7,8]. We and others have shown that hVEGF expression promotes new blood vessel formation and improves the outcome of islet transplantation [9]. We previously transfected islets with LipofectAMINE®2000/pCAGGS-hVEGF complexes preceding transplantation under the kidney capsules of nonobese diabetic-severe combined immunodeficient (NOD-SCID) mice. We demonstrated that hVEGF gene expression promotes new blood vessel formation after islet transplantation, with the involvement of endothelial cells of both the host (mouse) and the donor (human). Even though the occurrence of revascularization of the transplanted islets could be seen by immunohistochemical staining, transfection efficiency was low [9].
Effective prevention of islet destruction after transplantation requires not only revascularization of islets, but also abrogation of cytokine-mediated islet cell death and dysfunction triggered by immune and inflammatory reactions. Islet injury stimulates resident islet macrophages and passenger leukocytes to produce cytotoxic cytokines IL-1β, TNFα, and IFNγ, which lead to islet dysfunction and death by NFκB/iNOS/NO pathway [10].
In our recently published work [11], we have clearly shown that co-expression of genes that target different insults to transplanted islets can improve the outcome of islet transplantation better than either gene alone. Simultaneous infection of human islets with two adenoviral vectors, one encoding hVEGF and the other encoding hIL-1Ra, synergistically suppress islet dysfunction and NO production in human islets [11]. It also reduced the blood glucose levels and increased the level of blood insulin and c-peptide of mice upon transplantation of human islets tranfected with a mixture of adenoviral vectors encoding hVEGF and hIL-1Ra.
Taking care of this beneficial effect of co-expression of hVEGF and hIL-1Ra, we decided to construct a bicistronic vector encoding these two therapeutic genes. The use of a bicistronic vector not only simplifies the amplification and purification process of plasmid DNA, but also decreases the use of total of plasmid backbone for transfection, which will minimize immunogenic and toxic effect of bacterial plasmid. We evaluated gene expression and cytotoxicity in human islets. In addition, we determined islet function, nitric oxide production, caspases release, apoptosis and necrosis upon incubation with inflammatory cytokine cocktail of IL-1β, TNFα and IFNγ.
MATERIALS AND METHODS
Materials
pBudCE4.1 vector, Zeocin, LipofectAMINE®2000, and TRIZOL reagent were purchased from Invitrogen Corporation (Carlsbad, CA). Terrific broth, Luria agar base, glycerol, agarose, ethidium bromide, ethanol, chloroform, isopropanol, and TBE buffer were purchased from Sigma Chemical, Ltd (St. Louis, MO). Bacterial strain DH5 α, ScaI, XbaI, XhoI, EcoRI, BamHI, AhdI, alkaline phosphatase, and T4 DNA ligase were purchased from New England Biolabs (Ipswich, MA). Fetal bovine serum (FBS), Phosphate Buffered Saline (PBS), penicillin, streptomycin, gentamicin, and L-glutamine were procured from GIBCO-BRL (Gaithersburg, MD). Human insulin ELISA a kit was purchased from Alpco Diagnostics (Windham, NH). hVEGF and hIL-1Ra ELISA kits were purchased from R&D Systems (Minneapolis, MN). SYBR Green real-time PCR master mix, reverse transcriptase reagents, and random hexamers were purchased from Applied Biosystems (Foster City, CA). Custom made primers were procured from Integrated DNA Technologies (Coralville, IA).
METHODS
Construction of phVEGF-hIL-1Ra
To co-express hVEGF and hIL-1Ra, phVEGF-hIL-1Ra was constructed using pBudCE4.1 (Invitrogen Carlsbad, CA), with cloning of hIL-1Ra behind the CMV promoter and that of hVEGF behind the elongation factor-1α (EF-1 α) promoter. The hIL-1Ra gene fragment was excised from pUMVC1-hIL-1Ra vector using ScaI, separated by gel fractionation, and purified using Promega PCR and Clean-up kit (Promega, Madison, WI). pBudCE4.1 was linearized using a single cutting site of ScaI enzyme for ligation of the gene fragment at gene to vector molar ratios of 1:3 and 1:10 using T4 DNA ligase for 18h at 16°C. Following transformation in Top-10 supercompetent cells and amplification in terrific broth media, plasmids were purified using GenElute™ Plasmid Miniprep kit (Sigma, St. Louis, MO) and evaluated by double restriction digestion using AhdI and BamHI enzymes, for single copy gene insertion in the right orientation. hVEGF cDNA was excised from pCI-hVEGF-XhoI linker vector using XhoI restriction digestion and purified as described above. pBudCE4.1-hIL-1Ra was linearized using a XhoI enzyme, and dephosphorylated using alkaline phosphatase. The hVEGF cDNA was ligated with the linearized pBudCE4.1-hIL-1Ra vector at the gene to vector molar ratios of 1:3 and 1:10 using T4 DNA ligase for 16h at 16°C. Following transformation of the ligated mixture in Top-10 supercompetent cells and amplification in terrific broth media, the resulting plasmid, phVEGF-hIL-1Ra, was purified and confirmed by restriction digestion using EcoRI enzyme. Plasmid was characterized in terms of concentration, purity and integrity by UV spectrophotometry at 260/280 nm and agarose gel electrophoresis.
Islet Culture
Human islets were received from one of the several Islet Cell Resource (ICR) Centers through ICR Services for Basic Science Applications. On arrival at our facility, islets were cultured overnight in 162cm2 cell culture flasks in Memphis Serum Free Media (M-SFM) composed of CMRL 1066 medium containing 1% ITS, 1% L-glutamine, 1% penicillin/streptomycin mixture, 1% albumin and 16.8μM zinc sulfate (pH adjusted to 7.40 ± 0.05) at 37°C in a 5% CO2 incubator. After overnight culture, islets were visualized under the microscope for morphological integrity, percent purity and viability by trypan blue exclusion and used for experiments immediately.
Transfection of Human Islets
Human islets were transfected using phVEGF-hIL-1Ra at doses of 1, 2.5 and 5μg DNA equivalent of LipofectAMINE®2000/pDNA (5:1, w/w) complexes per 1000 islets. Non-transfected islets were used as controls. The complexes were prepared at 0.1μg/μl concentration in Opti-MEM medium and incubated at room temperature for 30 min before dilution with serum-free medium for transfection. After 12 h of incubation, 1.7 ml of fresh media was added and the islets were incubated in 24-well plates at 37°C in 5% CO2 incubator. Triplicate samples from each group were withdrawn at days 1, 3, 5, 7 and 10. Supernatants were centrifuged at 1500rpm for 10 min at room temperature and analyzed for the level and duration of hVEGF and hIL-1Ra concentration by ELISA as per vendor protocol (R&D Systems, Minneapolis, MN).
Quantitative real-time RT-PCR
Expression of hVEGF and IL-1Ra in human islets was assessed at mRNA level by real-time RT-PCR. Human islets were collected 48 h post-transfection by centrifugation at 250g for 10 min. Total mRNA was isolated from cell pellets by a guanidine isothiocyanate method using TRIZOL reagent (Invitrogen Corporation, Carlsbad, CA), and RNA concentration was measured by UV Spectrophotometry. Extracted RNA (0.2μg) was converted to cDNA using MultiScribe Reverse Transcriptase reagents and random hexamers (Applied Biosystems, Inc., Branchburg, NJ). Twenty nanograms of the extracted cDNA was amplified by real-time PCR using SYBR Green dye universal master mix on ABI PRISM® 7700 Sequence Detection System (Applied Biosystems, Inc., Foster City, CA) with forward primers from the vector and reverse primers from the genes. For detection of hIL-1Ra, the following forward primer 5′-TGG CTA ACT AGA GAA CCC ACT GCT and gene-specific reverse primer 5′-TTC TGA AGG CTT GCA TCT TGC TGG were used. Similarly for detection of hVEGF, the following reverse primer 5′-AAC AGA TGG CTG GCA ACT AGA AGG, and gene-specific primer 5′-GCG AGG CAG CTT GAG TTA AAC GAA were used. The PCR conditions included denaturation at 95°C for 10 min followed by 40 cycles of amplification by sequential denaturation at 95°C for 15 s and primer annealing as well as strand extension at 60°C for 1 min. Gene expression was normalized to 18s rRNA as internal control using the following primers: forward 5′-GTC TGT GAT GCC CTT AGA TG-3′ and reverse 5′-AGC TTA TGA CCC GCA CTT AC-3′. To confirm amplification specificity, the PCR products were subjected to a melting-curve analysis and agarose gel electrophoresis. Threshold cycle number was compared of three doses (1, 2.5 and 5μg DNA) for phVEGF-hIL-1Ra with the same parameters, using non-transfected samples as controls.
Glucose –stimulated Insulin Secretion
Islets obtained 10 days after transfection with phVEGF-hIL-1Ra and non-transfected islets were washed in fresh CMRL 1066 culture medium and then groups of 500 washed islets were incubated at 37°C with 1 ml of fresh CMRL 1066 culture medium and then containing 5% fetal bovine serum and 60mg/dl or 300mg/dl glucose. After 1 h post incubation, supernatants were collected and analyzed for insulin release by ELISA (AlPCO Diagnostics, Windham, NH).
Effect of Cytokine Treatment on Nitrite Production
Effect of hVEGF and hIL-1Ra gene expression on cytokine-mediated NO production as well as islet function was determined. For all these experiments, six test groups were used consisting of cytokine treated and untreated samples of 1) non-transfected islets, 2) cytokine treated non-transfected islets, 3) islets transfected with LipofectAMINE®2000/phVEGF-hIL-1Ra and treated with cytokines.
For determining NO production, human islets (2000 IE per sample) were transfected in 24-well plates for 12h, and 1.7 ml of fresh media was added. At 24h of transfection, islets were washed, centrifuged at 200g for 10min, and transfered to 24-well plates in 500μl media. Based on the work of Corbett et al.[12], Arnush et al [13], and Eizirik et al [14], cytokine cocktail consisting of IL-1β (375 units/mL), TNFα (5000 units/mL), and IFNγ (5000 units/mL), was prepared in PBS to a total volume of 10μl per well and added to the islets. Incubation was continued for 3 days at 37°C in 5% CO2 incubator. At the end of incubation, NO production in the undiluted media was determined by measuring the concentration of nitrite, a stable, non-volatile metabolite of NO, by colorimetry using Griess assay reagent (Promega, Madison, WI).
Caspase Assay
Groups of 250 islets were transfected with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes (5/1, w/w), using islets transfected with LipofectAMINE®2000/pCMS-EGFP (5/1, w/w) complexes as control. At 12h post transfection, islets were incubated with the cytokine cocktail (TNFα, IFNγ and IL-1β) for 3 days at 37°C. Islets were then transferred to 96-well black plate for determining total caspases activity using Homogeneous Caspases Assay Kit (Roche Applied Sciences, Indianapolis, IN), along with positive, negative and background controls and R-110 standards (free Rhodamine 100) in various concentrations. Substrate was incubated with the lysate for 2h at 37°C followed by determination of relative fluorescence units (RFU) using the excitation filter at 485±20nm (λmax = 499nm) and the emission filter at 528±20nm (λmax = 521nm). Data were corrected for blank absorbance and expressed as the mean ± S.D. of the relative fluorescence units.
Apoptotic and Necrotic Cell Death
Apoptotic and necrotic cell death levels in cytokine treated and untreated samples were determined using non-transfected islets as controls. The relative nucleosome concentration in cytoplasmic lysates was measured to determine apoptosis, and that in the cell culture supernatants to determine necrosis. Human islets were transfected, followed by incubation with or without cytokine cocktail for 3 days. Thereafter, the cell culture supernatants and pellet cytoplasmic fractions were collected and assayed for the content of nucleosomes using Cell Death Detection ELISA Plus kit (Roche Applied Sciences, Indianapolis, IN).
RESULTS
Construction of Bicistronic Vector
As shown in Figure 1, phVEGF-hIL-1Ra was constructed by cloning hIL-1Ra excised from pUMVC1-hIL-1Ra vector using ScaI and inserted into the polylinker site of pBudCE4.1 vector under CMV promoter. hVEGF gene was excised from pCI-hVEGF using XhoI and inserted into another polylinker site of pBudCE4.1 vector under elongation factor-1α (EF-1α) promoter. Linearized vector was dephosphorylated to prevent self-ligation, which may lead to vector recircularization. hIL-1Ra clone was selected by double restriction digestion using AhdI and BamHI enzymes to rule out any interfering patterns of insert polymerization and incorrect gene insertion. The hIL-1Ra gene has one AhdI restriction site, and it generates fragments of 345 and 347 bp, whereas BamHI has no cutting site in the gene. The cutting sites for AhdI and BamHI are at 1050 and 1405, (355 and 4932) bp in the vector. Clone selection of phVEGF-hIL-1Ra was conducted by restriction digestion using EcoRI enzyme, since the hVEGF gene has one restriction site, and the vector has another EcoRI cutting site. The cutting sites for EcoRI are at 1490 and 3790 and showed two bands of 2300 and 3626 bp after digestion with EcoRI. Based on the results of restriction digestion, clones were further sequenced using a pair of gene for hIL-1Ra and hVEGF specific primers, respectively, flanking the gene on the 5′-side. Based on the sequencing data, the right clone was selected and propagated for future applications.
Figure 1.
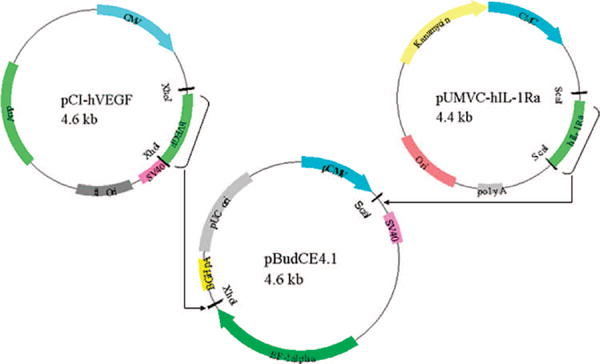
Construction of bicistronic plasmid encoding hVEGF and hIL-1Ra (phVEGF-IL-1Ra)
Co-expression of hVEGF and hIL-1Ra Gene Expression
To determine the levels of hVEGF and hIL-1Ra transcription, real-time RT-PCR was carried out by determining the threshold cycle (Ct), which is defined as the fractional cycle number at which the fluorescene crosses the fixed threshold. The higher the Ct value, the lower the gene expression. Both hVEGF and hIL-1Ra were co-expressed and levels of their expression were dose dependent (Fig. 2). To exclude the possibility of endogenous hIL-1Ra and hVEGF gene expression, we used vector specific forward primer and gene (hVEGF or hIL-1Ra) specific reverse primer for detection of hVEGF and hIL-1Ra. We used 18s RNA as an internal control (Fig. 2C & Fig. 2D). Figure 2A showed significant expression of hIL-1Ra in transfected islets compared to non-detectable expression of controlled samples (Fig. 2C). The Ct values of transfected samples decreased with increase in dose, suggesting a dose dependent expression of hIL-1Ra and hVEGF (Figures 2A and 2B). In case of hVEGF, non-transfected islets also showed amplification in the plot. However, Ct values of transfected samples were much higher, suggesting efficient hVEGF expression. Since we used different primers for hVEGF and hIL-1Ra, the Ct values were different for hVEGF and hIL-1Ra (Fig. 2A and 2B).
Figure 2.

Coexpression of hVEGF and hIL-1Ra after transfection of human islets with LipofectAMINE®2000/phVEGF-hIL-1Ra (5:1, w/w) complexes. hVEGF and hIL-1Ra gene expression levels were measured at mRNA level by real time RT-PCR as described previously [11]. The lower Ct value indicates higher level of mRNA expression. Forward primers were designed from the gene (hVEGF or hIL-1Ra) and the reverse primers from the vector to exclude the detection of endogenuous expression. Islets were harvested at day 2, followed by RNA extraction, reverse transcription and real time PCR using SYBR Green I Dye.
Time Course of Gene Expression in Human Islets
We also determined the levels of hVEGF and hIL-1Ra secreted from human islets upon transfection with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes (Fig. 3). The levels of secreted hVEGF (Fig. 3A) and hIL-1Ra (Fig. 3B) proteins increased with increase in dose and time. There was no decrease in hVEGF and hIL-1Ra protein levels even at 10 days post-transfection, as determined by ELISA (Fig. 3). This phenomenon may be explained by the fact that hVEGF and hIL-1Ra are naturally secreted proteins by the islets. However, increase in the levels of hVEGF and hIL-1Ra proteins was only modest compared to non-transfected controls, suggesting low transfection efficiency of LipofectAMINE®2000/phVEGF-hIL-1Ra into human islets.
Figure 3.
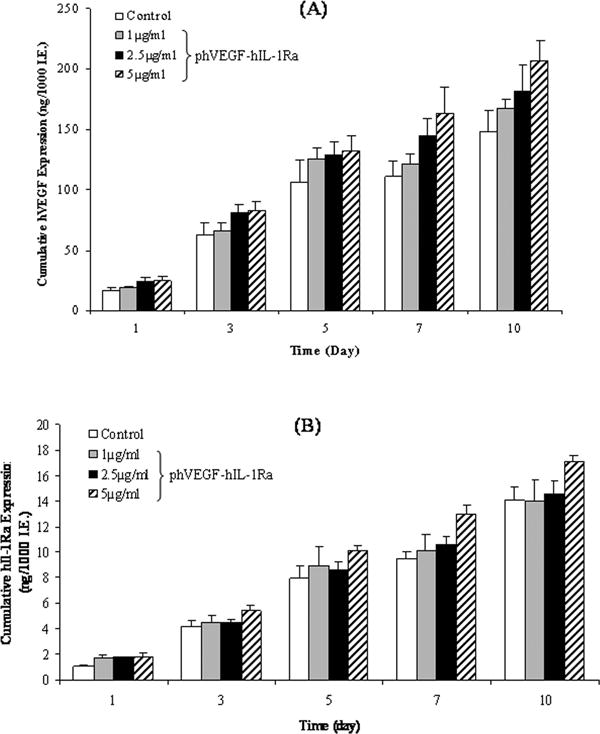
Time profile of hVEGF and hIL-1Ra gene expression at protein levels in human islets transfected with the bicistronic plasmid, phVEGF-hIL-1Ra, as compared to non-transfected control. ELISA of cell culture supernatants of islets transfected with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes (5:1, w/w) at doses of 1, 2 and 5 μg DNA/1000 islets was peformed at days 1, 3, 5, 7 and 10 to give the cumulative expression levels presented in this figure. The data show consistent hVEGF and hIL-1Ra gene expressions over the time period studied. A) hVEGF, B) hIL-1Ra.
Effect of Gene Expression on Islet Function
To determine whether insulin secretion was adversely affected by transfection with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes, in vitro islet function tests by static incubation were carried out at 10 days after transfection. Insulin secretion in response to glucose challenge was determined by quantifying glucose-stimulated insulin release at basal (60mg/dl) and stimulated (300mg/dl) glucose concentrations. As shown in Fig. 4A, transfection of human islets with LipofectAMINE®2000/phVEGF-IL-1Ra complexes did not affect the insulin stimulation index compared to un-transfected human islets.
Figure 4.
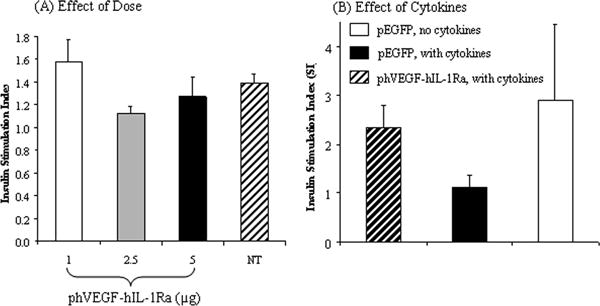
Stimulation Index (SI) of human islets with (B) or without (A) cytokine incubation after transfection with phVEGF-hIL-1Ra. Non-transfected islets and islets transfected with pEGFP were used as controls. SI was determined as the ratio of the amount of insulin secretion at 300mg/dL concentration of glucose in the media to that at 60mg/dL concentration upon incubation for 1h period. Insulin concentrations were determined by insulin enzyme immunoassay (EIA).
There was significant decrease in the stimulation index upon incubation of human islets transfected with LipofectAMINE®2000/pEGFP complexes with inflammatory cytokine cocktail (TNF-α, IL-1β and IFN-γ) (Fig. 4). However, transfection of islets with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes prevented such decrease in the stimulation index, suggesting protective effect of hVEGF and hIL-1Ra co-expression (Fig. 4B).
Effect of Cytokines on Nitrite Levels
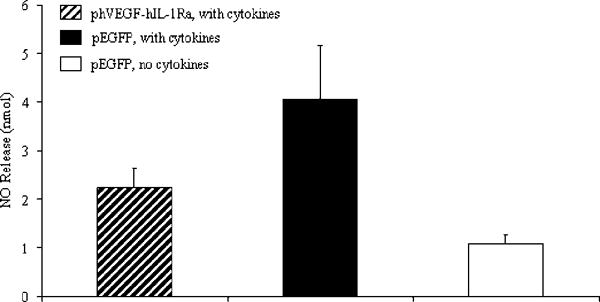
To determine the cytoprotective effect of hIL-1Ra and hVEGF expression on the islets, NO production was determined in terms of nitrite by Griess assay using islets incubated with a cocktail of inflammatory cytokine like Il-1β, TNFα, and IFNγ after transfection with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes (Fig. 5). Compared to untreated human islets, islets transfected with LipofectAMINE®2000/pEGFP complexes produced higher amount of NO in presence of inflammatory cytokine cocktail. In contrast, pre-transfection of islets with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes prevented this increase to some extent.
Figure 5.
Effect of hVEGF and hIL-1Ra co-expression on nitric oxide (NO) production by human pancreatic islets upon incubation with cytokine cocktail, consisting of IL-1β, TNFα and IFNγ for 2 days at 37ºC in 5% CO2 incubator. Non-transfected and Lipofetamine/p-EGFP complexes (5:1, w/w) were used a controls. NO production was determined in the form of nitrite in the cell culture supernatant by Griess assay and presented as mean ± SD.
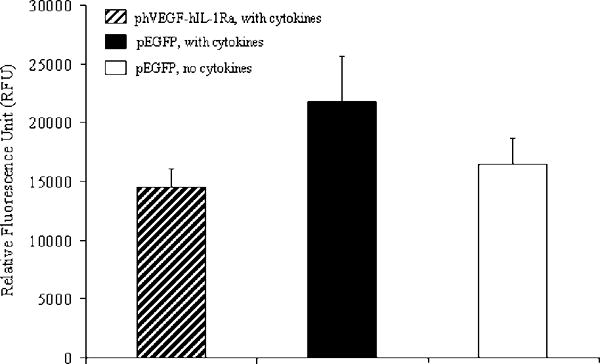
Total Caspase Activity
Caspases not only play an important role in apoptotic cell death, but also associated with immune response. Therefore, we determined total caspase activity in transfected as well as non-transfected human islets at 3 days post-incubation of islets with cytokines. As shown in Fig. 6, the total caspase activity of cell extracts increased with addition of the cytokine cocktail of TNF-α, IL-1β and IFN-γ in the islet culture media. However, transfection of islets with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes reduced caspase activity, suggesting hVEGF and hIL-1Ra gene expression suppressing cytokine-mediated caspase upregulation.
Figure 6.
Effect of hVEGF and hIL-1Ra co-expression on the activation of caspases in human islets upon treatment of cytokines. Islets were transfected with phVEGF-hIL-1Ra using pEGFP as control. At 12h post transfection, islets were incubated with the cytokine cocktail (TNFα, IFNγ and IL-1β) for 3 days at 37°C. Islets were then transferred to 96-well black plate for determining total caspases activity using Homogeneous Caspases Assay Kit. Relative total caspase activity was determined by fluorimetric assay, and expressed as Relative Fluorescence Unit (RFU).
Apoptosis and Necrosis of Human Islets
We determined the relative level and type of cell death, and whether coexpression of hIL-1Ra and hVEGF had any beneficial effect on islet survival. Apoptotic cells have nucleosome accumulation in the cytoplasm of dying cells, while necrotic cells undergo membrane disruption leading to nucleosome release in the cell culture medium. Hence, the levels of DNA fragmentation were determined in terms of relative nucleosome concentration in the cytoplasmic fraction of islet cell lysates compared to non-transfected islets. Similarly, necrotic cell death was determined in terms of nucleosome concentration in the cell culture supernatants. Nucleosome concentration in the control group was considered the background level of cell death in the islets. Absorbance in the test samples was divided by this value to determine the enrichment factor (EF) which could vary from 0 to 5 in the presented experiments. Thus, higher EF indicated greater cell death, relative to background levels.
We observed an increase in islet apoptosis and necrosis after cytokine treatment of islets transfected with LipofectAMINE®2000/pEGFP complexes (Fig. 7). In contrast, transfection of islets with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes significantly reduced both apoptosis and necrosis, confirming the beneficial effect of hVEGF and hIL-1Ra co-expression.
Figure 7.
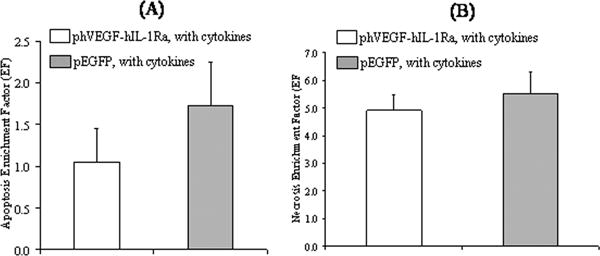
Apoptotic (A) and necrosis (B) cell death in the islets with cytokine exposure for 4 days after transfection with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes. LipofectAMINE®2000/pEGFP complex transfected human islets were used as controls. Relative cell death was determined by measuring nucleosome concentration in the cell lysates and culture supernatants for apoptosis and necrosis, respectively by ELISA (Cell Death Detection ELISAPlus, Roche Applied Sciences). Data were normalized to untreated controls and expressed as a ratio of nucleosome concentration, called enrichment factor. Reported values are the mean ± S.E. of n = 4.
DISCUSSION
Isolation and purification of islets are known to disrupt their microvasculatures, making them dependent upon diffusion of oxygen and nutrients from the islet periphery. The lack of establishment of capillary networks within the islets, exacerbates immune destruction of islets upon transplantation. We and others have shown that hVEGF expression promotes new blood vessel formation and improve the outcome of islet transplantation [9,15]. However, effective prevention of islet destruction after transplantation requires not only revascularization of islets, but also abrogation of cytokine-mediated islet cell death and dysfunction triggered by immune and inflammatory reactions. Islet injury stimulates resident islet macrophages and passenger leukocytes to produce cytotoxic cytokines IL-1β, TNF-α and IFN-γ [16]. The release of these inflammatory cytokines induces the production of nitric oxide (NO), which leads to islet cell death [17].
Inhibition of IL-1β binding using IL-1Ra has been shown to prevent suppressing of islet function and NO production in mouse, rat and human islets. Therefore, we recently simultaneously infected human islets with Adv-hVEGF and Adv-hIL-1Ra, which suppressed cytokine induced iNOS gene expression and NO production [11]. Similarly, Bertera et al. (2004)[16] reported a synergistic effect after simultaneous adenoviral infection of mouse islets with two individual genes, such as IL-1Ra and indoleamine 2,3-dioxygenase or hIL-1Ra and superoxide dismutase (MuSOD).
The objective of this study was to evaluate a bicistronic plasmid vector for multiple gene delivery to human islets for suppressing inflammatory cytokine-mediated destruction and improving islet function. The use of a bicistronic vector not only simplifies the amplification and purification process of plasmid DNA, but also the use of a bicristronic vector decreases the use of total plasmid backbone. This is expected to minimize the immunogenic and toxic effects of bacterial plasmids.
To determine hVEGF and hIL-1Ra co-expression after transfection of human islets with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes, while excluding hVEGF and hIL-1Ra expression resulting from endogenous production, we designed vector-specific forward primers and gene (hVEGF or hIL-1Ra)-specific reverse primers. In the real time PCR experiment, lower cycle number for amplification to the threshold copy number of the target sequence indicates higher level of gene expression Since two different forward and reverse primers and separate standard calibration curves for real time PCR experiment were used, we cannot directly correlate the results for hVEGF and hIL-1Ra. Figure 2A and 2B show rapid and dose-dependent amplification of hIL-1Ra and hVEGF at 2 days after transfection of human islets with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes. Amplification specificity was confirmed by melting curve analysis of all the PCR products, with single predominant distinct melting temperatures (Tm) for hIL-1Ra and hVEGF, respectively.
Since the presence of two identical promoters in a bicistronic vector may cause some interruption of the transcription of two genes, we wanted to use two different promoters in the same vector. Therefore, we used pBudCE4.1 vector, which contains CMV and EF-1α promoters. Islets undergo inadequate revascularization upon transplantation due to the disruption of their microvasculature during their isolation and culture. Therefore, we selected EF-1α promoter to express hVEGF, since EF-1α promoter is known to be more potent than CMV promoter. As can be seen in Figure 2A and 2B, hVEGF expression level was much higher compared to hIL-1Ra, which can be due to the following two reasons: i) hVEGF is driven by EF-1α promoter while hIL-1Ra is driven by CMV promoter, which has been reported to be less potent than EF-1α promoter [18–20]. ii) hVEGF is known to be upregulated under hypoxic conditions [21] as discussed above. However, at this stage we do not know whether we need hVEGF expression higher than IL-1Ra, which inhibits apoptosis.
Ideally, the total RNA should be treated with DNase I following isolation to avoida any possible DNA contamination. However, in this study we did not treat the total RNA with DNase, since the effect of the trace amount of DNA in the total RNA will be minimal. Moreover, our main objective of this RT-PCR study was to compare the gene expression at different doses instead of quantifying how many copies of gene was expressed.
Concentrations of hIL-1Ra and hVEGF protein were determined by ELISA of the culture supernatant of human islets transfected with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes. The results indicated persistent and dose-dependent expression of hVEGF and hIL-1Ra over a period of 10 days, which were higher than the control. In Figure 3A, increase in the levels of hVEGF proteins was only modest compared to non-transfected controls. This is likely due to the endogenous production and/or immunostimulation of CpG motifs of bacterial plasmids and cationic liposomes. VEGF is a naturally secreted protein by the islets. Both VEGF and its receptors, flt-1 and flk-1, are upregulated under hypoxic conditions. Moreover, isolated islets in vitro are dependent upon diffusion of oxygen. Because 1000 islets were incubated in 2 ml, these islets most likely experienced hypoxia. These are the possible reasons for only a modest increase in the levels of hVEGF protein after islet transfection with LipofectAMINE/phVEGF-hIL-1Ra complexes. We used vector specific forward primers, and hVEGF gene specific reverse primers for the real time RT-PCR to exclude endogenous hVEGF gene expression. Therefore, the mRNA level of hVEGF significantly increased after transfection of islets with LipofectAMINE/phVEGF-hIL-1Ra complexes. In contrast, there was little increase in hVEGF mRNA levels for the non-transfected controls (Figure 2A). These results are in good agreement with our previously published results [9].
Similar to hVEGF protein levels, there is only a modest difference in the levels of hIL-1Ra protein of transfected islet samples compared to their non-transfected controls (Figure 3B). We could not use more than 5μg DNA dose for islet transfectiion due to the high toxicity of LipofectAMINE at higher doses, which impaired insulin secretion from the islets. Unlike real time RT-PCR which allows us to exclude the endogenous production of hIL-1Ra mRNA, the ELISA assay does not offer this possibility. The relative difference in the level of hIL-1Ra protein at day 1 was more significant than that at day 3, 5, 7 and 10. This difference at early stage is more important for islet survival and function.
Since we had used pEGFP as control in our previous studies, we tried to use the same control in this study. Another advantage of pEGFP is that we can monitor transfection efficiency using a fluorescence microscope. Since transfection of islets with LipofectAMINE/pDNA complexes influences inflammatory cytokine production, insulin secretion and other properties of islets, we did not include non-transfected controls in this study.
Transfection process should not affect the normal insulin secretion capability of human islets, which varies from batch to batch. Therefore, we measured insulin release in response to glucose challenge to assess the islet functional viability. The stimulation index of insulin secretion by the islets was measured as a ratio of insulin at higher (300 mg/dl) glucose as compared to lower, basal (60 mg/dl) glucose concentration. The stimulation index of LipofectAMINE®2000/phVEGF-hIL-1Ra transfected islets was compared to non-transfected islets. As shown in Fig. 4, there was only slight decrease in the stimulation index when we increased the DNA dose from 1μg to 5μg per 1000 islets. These data indicated that transfection with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes had only minimal adverse effect on the islet β cell response to glucose challenge.
To mimic the in vivo situation of inflammatory cytokine-mediated insults, we incubated islets with a cocktail of IL-1β, TNFα and IFN-γ. Incubation with these cytokines significantly decreased the insulin stimulation index when islets were transfected with LipofectAMINE®2000/pEGFP complexes, but had little effect when the islets were transfected with LipofectAMINE®2000/phVEGF-hIL-1Ra complexes (Fig. 4B), suggesting the protective effect of hVEGF and hIL-1Ra co-expression.
Islets produced low level of nitric oxide (NO), but its amount significantly increased when islets were transfected with LipofectAMINE®2000/pEGFP complexes and then incubated with the cytokine cocktail (Fig. 5). However, the increase in NO production significantly suppressed for LipofectAMINE®2000/phVEGF-hIL-1Ra transfected samples (Fig, 5). These results also correlated with islet function (Fig. 4B). Our results are in good agreement with the literature when adenoviral vector encoding hIL-1Ra has been shown to suppress NO production and IL-1β induced impairement of islet function [5]. Unlike adenoviral vector, phVEGF-hIL-1Ra-based formulations showed only partial suppression of NO production, due to their poor transfection into human islets.
We used caspase assay and the envichment factor (EF) values to determine the islet cell death in order to keep our analytical techniques consistent with our previous studies. In addition, the mechanism of Cell Death Detection ELISA Plus kit we used in the present study is similar to the TUNEL assay, since it detects DNA strand breaks during apoptosis. Because of unknown differences in relative importance of different caspases, we determined total caspase activity in islets. We observed increase in total caspase activity in islets transfected with LipofectAMINE®2000/pEGFP complexes, followed by incubation with the inflammatory cytokine cocktail. In contrast, co-expression of hVEGF and hIL-1Ra reduced the total caspase activity (Fig. 6), suggesting the beneficial effect of these two therapeutic genes.
We determined the relative level of apoptosis and necrosis in human islets incubated with cytokines cocktail at 3 days post-transfection by estimation of nucleosome concentration. The enrichment factor (EF) values were calculated for all transfected samples by using 1 as the EF of the non-transfected islets without cytokine. As can be seen in Fig. 7, there was significant increase in the enrichment factor for the islets tansfected with LipofectAMINE®2000/pEGFP complexes and then incubated with the cytokine cocktail. Co-expression of hVEGF and hIL-1Ra significantly suppressed this increase.
In conclusion, we constructed a bicistronic plasmid vector phVEGF-hIL-1Ra and determined whether co-expression of hVEGF and hIL-1Ra shows any beneficial effect on human islet viability and function. We demonstrated that co-expression of hVEGF and hIL-1Ra can minimize the effect of inflammatory cytokines on human islet viability and function. The quality and purity of human islets vary from batch to batch, which often complicates the data analysis and correlation of our results from one experiment to another. However, we have seen consistently the beneficial effect of hVEGF and hIL-1Ra coexpression on islet viability and function.
Acknowledgments
We thank the National Institute of Health (R01 DK069968) financial support, and the Islet Cell Resource (ICR) Centers for providing human islets.
References
- 1.Garcia-Ocana A, Takane KK, Reddy VT, Lopez-Talavera JC, Vasavada RC, Stewart AF. J Biol Chem. 2003;278:343–51. doi: 10.1074/jbc.M207848200. [DOI] [PubMed] [Google Scholar]
- 2.Ryan EA, et al. Diabetes. 2002;51:2148–57. doi: 10.2337/diabetes.51.7.2148. [DOI] [PubMed] [Google Scholar]
- 3.Embury J, et al. Diabetes. 2001;50:1706–13. doi: 10.2337/diabetes.50.8.1706. [DOI] [PubMed] [Google Scholar]
- 4.Contreras JL, et al. Transplantation. 2001;71:1015–23. doi: 10.1097/00007890-200104270-00001. [DOI] [PubMed] [Google Scholar]
- 5.Giannoukakis N, Rudert WA, Ghivizzani SC, Gambotto A, Ricordi C, Trucco M, Robbins PD. Diabetes. 1999;48:1730–6. doi: 10.2337/diabetes.48.9.1730. [DOI] [PubMed] [Google Scholar]
- 6.Gorden DL, Mandriota SJ, Montesano R, Orci L, Pepper MS. Transplantation. 1997;63:436–43. doi: 10.1097/00007890-199702150-00018. [DOI] [PubMed] [Google Scholar]
- 7.Menger MD, Vajkoczy P, Beger C, Messmer K. J Clin Invest. 1994;93:2280–5. doi: 10.1172/JCI117228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukinius A, Jansson L, Korsgren O. Am J Pathol. 1995;146:429–35. [PMC free article] [PubMed] [Google Scholar]
- 9.Narang AS, Cheng K, Henry J, Zhang C, Sabek O, Fraga D, Kotb M, Gaber AO, Mahato RI. Pharm Res. 2004;21:15–25. doi: 10.1023/b:pham.0000012147.52900.b8. [DOI] [PubMed] [Google Scholar]
- 10.Narang AS, Mahato RI. Pharmacol Rev. 2006;58:194–243. doi: 10.1124/pr.58.2.6. [DOI] [PubMed] [Google Scholar]
- 11.Narang AS, Sabek O, Gaber AO RIM. Pharm Res. 2006 doi: 10.1007/s11095-006-9065-7. [DOI] [PubMed] [Google Scholar]
- 12.Corbett JA, Sweetland MA, Wang JL, Lancaster JR, Jr, McDaniel ML. Proc Natl Acad Sci U S A. 1993;90:1731–5. doi: 10.1073/pnas.90.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. J Clin Invest. 1998;102:516–26. doi: 10.1172/JCI844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eizirik DL, et al. J Clin Invest. 1994;93:1968–74. doi: 10.1172/JCI117188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang N, et al. Diabetes. 2004;53:963–70. doi: 10.2337/diabetes.53.4.963. [DOI] [PubMed] [Google Scholar]
- 16.Bertera S, Alexander AM, Crawford ML, Papworth G, Watkins SC, Robbins PD, Trucco M. Exp Diabesity Res. 2004;5:201–10. doi: 10.1080/15438600490486822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welsh N, Bendtzen K, Welsh M. J Clin Invest. 1995;95:1717–22. doi: 10.1172/JCI117848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teschendorf C, Warrington KH, Jr, Siemann DW, Muzyczka N. Anticancer Res. 2002;22:3325–30. [PubMed] [Google Scholar]
- 19.Elliott MD, Kapoor A, Parker MA, Kaufman DB, Bonow RO, Gheorghiade M. Circulation. 2001;104:563–9. doi: 10.1161/hc3001.093434. [DOI] [PubMed] [Google Scholar]
- 20.Chung S, Andersson T, Sonntag KC, Bjorklund L, Isacson O, Kim KS. Stem Cells. 2002;20:139–45. doi: 10.1634/stemcells.20-2-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ristimaki A, Narko K, Enholm B, Joukov V, Alitalo K. J Biol Chem. 1998;273:8413–8. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]