

Abstract
Sepsis is associated with increased expression of TNF-α with subsequent activation of nuclear factor-kappa B (NF-κB). The glucocorticoid receptor (GR) and NF-κB function as mutual antagonists and induction of the latter is believed to play a major role in the acquired glucocorticoid resistance that occurs in some septic patients. GR expression and function has been reported to be elevated in septic muscle suggesting a limited effect of the activated NF-κB on GR function in this context. In this study, the L6 myocyte cell line was used as an in vitro model for a sepsis-like condition in skeletal muscle. While short or long term treatment with TNF-α had no effect on GR expression, glucocorticoid-dependent downregulation of GR occurred with a kinetic profile that is accelerated relative to that observed in most cells. This downregulation was not affected by co-treatment or prior priming of L6 cells with TNF-α. The synthetic glucocorticoid, dexamethasone (DEX) blunted TNF-α-stimulated NF-κB activation in L6 cells. However, although effective at activating an NF-κB transcriptional response, TNF-α treatment exerted a minimal effect in myoblasts and no effect in myotubes on GR transcriptional activity. This limited impact of TNF-α on GR activity was not universal as TNF-α and DEX exerted an additive effect on the reduction in myosin heavy chain (MHC) protein expression caused by either agent alone. Thus, the selective perseverance of GR function in the presence of increased levels of glucocorticoids and TNF-α during sepsis or other inflammatory states may exacerbate muscle protein breakdown.
Keywords: TNF-α, Glucocorticoid receptor, Myosin heavy chain
1. Introduction
Glucocorticoid responsiveness is regulated at the cellular level by alterations in expression and accumulation of the glucocorticoid receptor (GR) protein. In many situations, a direct correlation exists between the number of receptors and the magnitude of glucocorticoid response [1,2]. Specifically, the magnitude of the transcriptional response elicited by GR in vitro has been shown to be directly proportional to the number of receptor molecules per cell [3]. Regulation of GR expression by its ligand has been documented in a variety of cell lines, intact animals, and humans. Chronic GC treatment typically leads to downregulation of GR levels both in cell culture and in intact tissue [4]. This homologous downregulation of the receptor reflects GC effects on both GR gene transcription [5,6], and protein turnover [7,8].
Sepsis is associated with tissue-specific changes in GR responsiveness. It's been shown that GR levels declined by about 40, 56, and 40% in septic liver, brain, and muscle cytosol, respectively [9]. However, other studies [10] reported increased expression and binding activity of GR in septic muscle tissue. Tian and co-workers recently showed that GR gene transcription in L6 myocytes in vitro is upregulated by treatment with sera from septic rats in a manner similar to that measured in septic rats in vivo [11]. Furthermore, Hasselgren and Fischer [12] showed that dexamethasone (DEX) stimulates proteasome- and calcium-dependent proteolysis in the same cells [13]. Other studies [12,14] confirmed that sepsis induces proteolysis by activation of the ubiquitin–proteasome system, and that this activation was regulated by GCs [15]. These findings represent a unique situation where the increased GC levels described with sepsis are associated with upregulated GR levels in muscle cells, in contrast to most cells, where increased GCs levels are associated with GR downregulation.
Nuclear factor (NF-κB) is the major transcription factor that regulates the expression of the genes encoding proinflamma-tory mediators and molecules that are produced excessively in sepsis. GR and NF-κB function as mutual transcriptional antagonists [16] modulating the effects of each other especially on the immune system. Tumor necrosis factor-α (TNF-α), a cytokine product of monocytes and macrophages is a rapid and potent activator of NF-κB. In previous in vitro experiments [17], treatment of cultured L6 muscle cells with TNF-α resulted in activation of the transcription factor NF-κB.
Sepsis, cancer, and many inflammatory conditions are associated with multiple metabolic changes in skeletal muscles with subsequent muscular protein loss and muscle cachexia. Numerous studies showed that glucocorticoids and cytokines play a major role in this protein loss particularly the myosin heavy chain (MHC) [18,19]. Thus MHC expression changes follow the same pattern as muscle mass, providing an attractive molecular model to study the effects of glucocorticoids and cytokines on muscle protein metabolism [20]. Effects of interaction between increased GCs and cytokines (such as seen in sepsis) on muscle protein regulation have not been examined in detail.
In order to model the impact of sepsis in skeletal muscle in vitro, we used the L6 rat skeletal muscle cell line. The L6 line expresses many features of myoblasts and can be differentiated to form myotubes in vitro. Furthermore, TNF-α treatment of L6 cells results in activation of the NF-κB transcription factor, which is one of the responses that accompanies a septic state in skeletal muscle. L6 cells also express GR and therefore, provide a suitable model to examine the impact of cytokines on glucocorticoid action in muscle [10,11].
2. Methods and materials
2.1. Cultured L6 cells
L6 rat skeletal muscle cells were purchased from the American Type Culture Collection (Rockville, MD). Cells were thawed and maintained at low density in 10-cm diameter culture dishes and were used between passages 2 and 10. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% charcoal stripped fetal bovine serum (C/S FBS) [proliferation medium, PM]. For myotube studies, differentiation was induced by changing medium to DMEM with 2% C/S FBS [differentiation medium, DM]. After 3 days, when approximately 90% of the cells formed myotubes, the cells were treated with 10 μM cytosine arabinoside (Sigma–Aldrich, Inc., Saint Louis, MO) for 48 h to eliminate residual dividing myoblasts.
2.2. Luciferase and renilla assay in transiently transfected cells
Myoblasts were plated at 50 × 104/well in 6-well plates. The following day, the medium was changed to Opti-MEM I. Precipitated plasmid was prepared 30 min before addition to cells and consisted of a glucocorticoid-responsive luciferase reporter plasmid [pMMTV-luc] 0.4 μg or a NF-κB-responsive luciferase reporter plasmid [NF-κB-luc] 0.5 μg, and a renilla reporter plasmid 0.05 μg. Plasmid was diluted in Opti-MEM I and lipofectamine (Invitrogen Corp., Carlsbad, CA) was added. Cells were incubated for 3 h after with the DNA/lipofectamine mixture and then additional Opti-MEM I with 20% C/S FBS was added.
For myoblast studies, medium was changed the next day to PM. For myotubes studies, medium was changed to DM and cells were allowed to differentiate as discussed above.
Whole-cell extracts, prepared using the Luciferase Assay Buffer (Promega Corp., Madison, WI), were assayed for luciferase and renilla activity at various times after treatment with DEX or mouse recombinant TNF-α (Sigma–Aldrich, Inc., Saint Louis, MO). To correct for variation in transfection efficiency, values of the luciferase assay were normalized with measured renilla activities. The promoter driving renilla expression is not regulated by glucocorticoids or TNF-α.
2.3. Western blot analysis
For myoblast studies, cells were plated at a density of 50 × 104 in 35 mm dish for 2–3 days to reach confluence before starting the experiment. For myotube studies, cells were plated similarly but were allowed to differentiate into myotubes as described above. Cells were treated with DEX or TNF-α at different concentrations for various periods as indicated below. Cells were washed three times with PBS and whole-cell extracts were obtained by scrapping the cells after adding protein sample buffer. Equal amounts of total lysate protein were loaded onto sodium dodecyl sulfate 7.5% polyacrylamide gels and subjected to electrophoresis. Separated proteins were transferred to Immobilon-P membranes (Milipore Corp., Bedford, MA) and subjected to Western blot analysis to detect GR, actin, or myosin heavy chain (MHC). Actin levels were determined to control for equal loading of the lanes. The BuGR2 anti-GR monoclonal antibody was used to detect GR, whereas C-2 actin monoclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used to detect actin. MHC was detected using MHC (fast) antibody (Vector Lab, Inc., Burlingame, CA). Peroxidase-conjugated goat anti-mouse IgG was used as a secondary antibody (BioRad). Immunoreactive bands were visualized on Western blots using enhanced chemiluminescence (ECL) kit (NEN Life Science Products, Boston, MA). In some cases, ECL signals were quantified by densitometry (Personal Densitometer SI, Molecular Dynamics) and analyzed using Image Quant 5.2 software.
3. Results
3.1. Dexamethasone-induced GR downregulation
Chronic exposure of cells in vitro and in vivo to glucocorticoid hormone is typically associated with downregulation of GR protein levels that can reflect negative effects on both GR gene transcription [5,6] and protein turnover [7,8]. Previous studies in L6 cells suggest that increased levels of GCs seen in sepsis are associated with increased level of GR protein and mRNA in septic muscles [11]. We therefore, used Western blot analysis to examine the glucocorticoid effects on steady state levels of GR protein. As shown in Fig. 1, DEX treatment of L6 myotubes or myoblasts with 10−6M DEX for up to 24h (Fig. 1A) or up to 72h (Fig. 1B) resulted in downregulation of GR reaching its nadir after 24 h. This downregulation of GR protein expression required the continuous presence of hormone as GR levels were restored following the removal of DEX (data not shown). A dose response analysis established that a significant reduction of GR protein occurred with physiological doses of glucocorticoid (Fig. 2A). Thus, muscle cells in vitro do not appear to be immune to glucocorticoid-induced GR downregulation. In fact, the downregulation of GR protein by DEX treatment of L6 muscle cells was more rapid than other cultured cells typically examined for GR downregulation [21,22]. Significant reductions in GR protein levels were noted within 1h (Fig. 1C). This apparent heightened sensitivity of GR to hormone-induced downregulation does not limit the transactivation activity of the receptor, at least from a transiently transfected glucocorticoid responsive MMTV promoter (Fig. 2B). A similar pattern of GC-induced transcription of GR in L6 myocytes has been reported by Sun et al. [11].
Fig. 1.

Effects of DEX and TNF-α on GR expression in L6 cells. Total protein in whole cells lysates was separated by SDS-PAGE and subjected to Western blot analysis to detect GR or actin. Representative blots are shown. Treatment with DEX 10−6M for various intervals, 1–24h in (A) or 24–72h in (B) led to downregulation of GR. In (C) Western blots were quantified by densitometry and GR levels were expressed relative to actin. Results shown are the mean of three independent experiments ± S.E.M. *P < 0.02 when compared to no hormone sample.
Fig. 2.
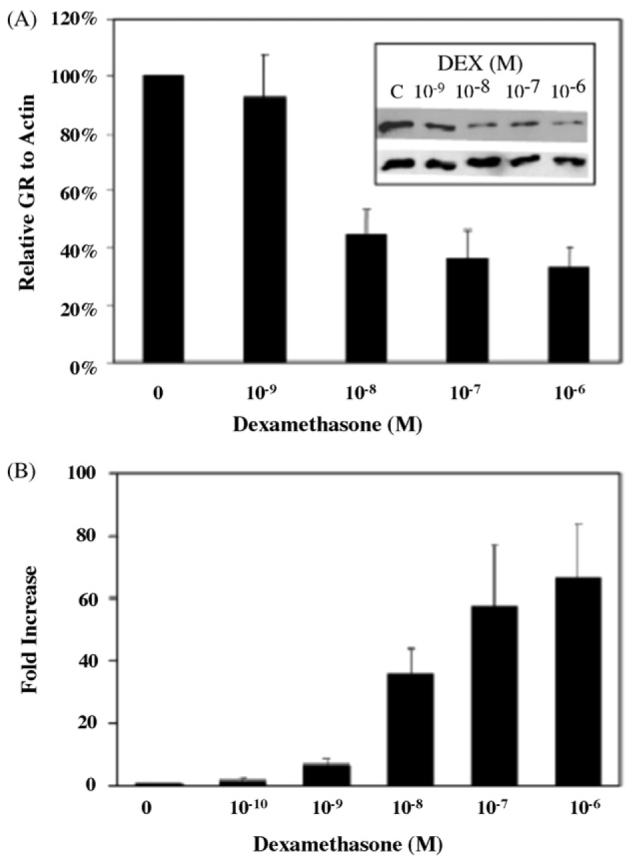
(A) Effects of DEX on GR expression in L6 myoblasts. Treatment of L6 myoblasts with increasing doses of DEX for 6h led to a gradual increase of GR downregulation. Total protein in whole cells lysates was separated by SDS-PAGE and subjected to Western blot analysis to detect GR or actin. Blots were quantified by densitometry. Results shown are the mean of three independent experiments ± S.E.M. (B) Effects of DEX on GR transcription in L6 myoblasts. Cells were transfected with a GR-dependent reporter plasmid (pMMTV-Luc) and Renilla control plasmid. After transfection, cells were exposed to increasing concentrations of DEX (10−10 to 10−6M) for 8h. To correct for variation in transfection efficiency, values of the luciferase assay were normalized with measured renilla activities. Luciferase assays in individual transfections were performed in duplicate. Results shown are the mean of three independent experiments ± S.E.M.
3.2. TNF-α effect on GR function
In many cell culture models, activation of NF-κB, particularly by cytokines like TNF-α inhibits GR-induced transcription [23]. Given the recent reports of sepsis-induced increased expression and transcription of GR in skeletal muscle cells [10,11], we examined whether GR function was affected upon TNF-α activation of NF-κB in both L6 myoblasts and myotubes. Treatment of L6 myotubes or myoblasts with increasing doses of TNF-α for 12h (Fig. 1A) or 72h (Fig. 1B) did not result in a significant decrease of GR protein levels. Simultaneous treatment of L6 myoblasts with DEX and TNF-α did not affect the extent or kinetics of GR downregulation that was observed upon treatment with DEX alone (Fig. 1B).
Furthermore, priming of L6 myoblasts with increasing doses of TNF-α for 12h prior to treatment with DEX did not alter the profile of DEX-dependent downregulation of GR (Fig. 3). This priming was done to ensure adequate activation of NF-κB system by TNF-α prior to DEX exposure. We chose to focus our studies on cells primed with TNF-α since DEX effects on GR downregulation in L6 cells were quite rapid (Fig. 1).
Fig. 3.
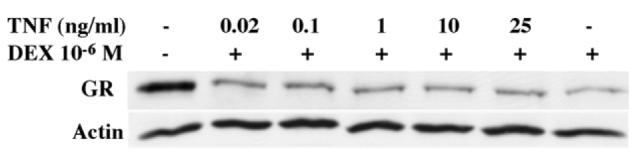
L6 myoblasts were primed with increasing doses of TNF-α for 12h before adding DEX 10−6M for 6h. Total protein in whole cells lysates was separated by SDS-PAGE and subjected to Western blot analysis to detect GR or actin. Representative blot of two independent experiments is shown.
Since hormone-induced downregulation of GR protein was not affected by TNF-α in L6 cells, we assessed the impact of this cytokine on DEX-induced GR transactivation. L6 myocytes were transfected with a GR-dependent reporter plasmid (pMMTV-Luc) and a Renilla control plasmid. After transfection, cells were exposed to DEX 10−6M for 6h following a 12h priming period with increasing doses of TNF-α. As anticipated, DEX induced transcription in L6 myotubes and myoblasts (Fig. 4). This transcription was not affected when myotubes were primed with increasing doses of TNF-α for 12h (Fig. 4A). On the other hand, such priming led to some decrease (∼32%, P 0.01) of the DEX-induced transcription in myoblasts (Fig. 4B).
Fig. 4.
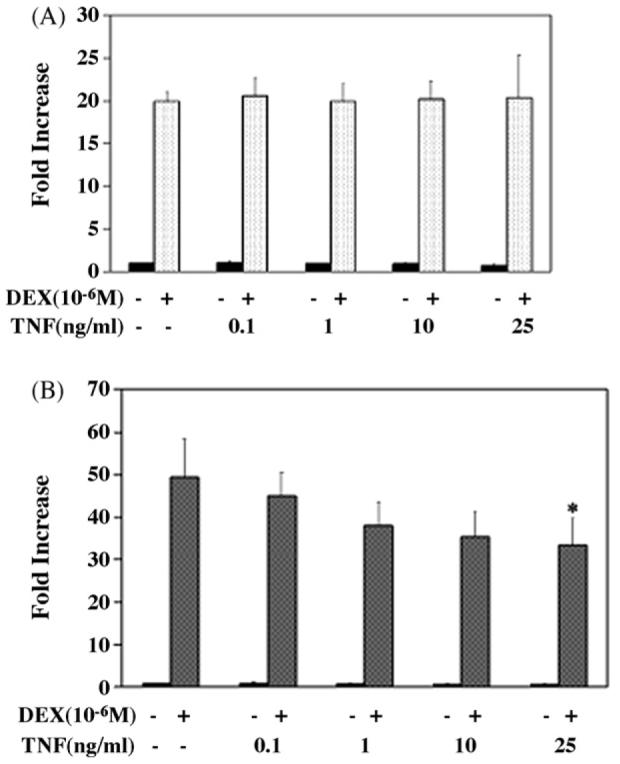
Effects of TNF-α on GR transcription in L6 myotubes (A) and myoblasts (B). L6 myoblasts were transfected with a GR-dependent reporter plasmid (pMMTV-Luc) and Renilla control plasmid. After transfection, cells were exposed to DEX 10−6M for 6h after 12h priming period with increasing doses of TNF-α. Differentiation of myoblasts into myotubes was performed following transfection. To correct for variation in transfection efficiency, values of the luciferase assay were normalized with measured renilla activities. Luciferase assays in individual transfections were performed in duplicate. Results shown are the mean of three independent experiments ± S.E.M. *P < 0.01 when compared with DEX treatment in absence of TNF-α.
3.3. Glucocorticoids interfere with NF-κB function in L6 myoblasts and myotubes
To ensure that NF-κB was appropriately induced by TNF-α, L6 myotubes and myoblasts were transiently transfected with an NF-κB-responsive reporter plasmid and then treated with increasing doses of TNF-α for 12 h. Fig. 5A and B illustrates the increased transcription from the NF-κB reporter consistent with induction of NF-κB by TNF-α in L6 myotubes and myoblasts, respectively. In contrast to the lack of effect (in myotubes) or limited effect (in myoblasts) of a TNF-α-mediated NF-κB induction on GR function in L6 cells (see above), treatment of L6 myotubes (Fig. 6A) or myoblasts (Fig. 6B) with 10−6M of DEX for 6h leads to a significant reduction of NF-κB transactivation (48 and 44%, respectively). Thus, glucocorticoids are effective inhibitors of NF-κB activation in L6 cells.
Fig. 5.
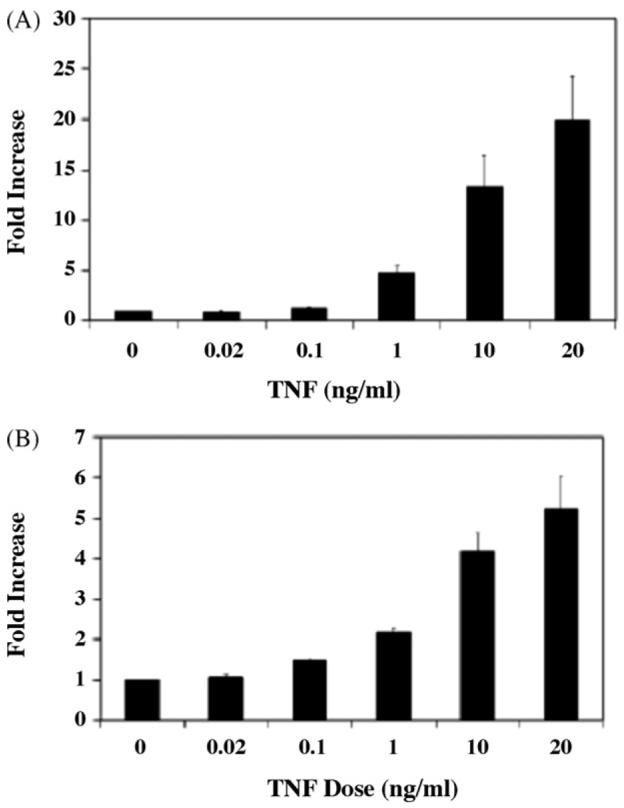
Effects of TNF-α on NF-κB transcription in L6 myotubes (A) and myoblasts (B). L6 myoblasts were transfected with NF-κB-dependent reporter plasmid and Renilla control plasmid. After transfection, cells were exposed to increasing concentrations of TNF-α for 12 h. Differentiation of myoblasts into myotubes was performed following transfection. To correct for variation in transfection efficiency, values of the luciferase assay were normalized with measured renilla activities. Luciferase assays in individual transfections were performed in duplicate. Results shown are the mean of three independent experiments ± S.E.M.
Fig. 6.

Effects of TNF-α and DEX on NF-κB transcription in L6 myotubes (A) and myoblasts (B). L6 myoblasts were transfected with a NF-κB-dependent reporter plasmid and Renilla control plasmid. After transfection, cells were exposed to 20 ng/ml of TNF-α for 12h followed by DEX 10−6M for 6h. Differentiation of myoblasts into myotubes was performed following transfection. To correct for variation in transfection efficiency, values of the luciferase assay were normalized with measured renilla activities. Luciferase assays in individual transfections were performed in duplicate. Results shown are the mean of three independent experiments ± S.E.M.
3.4. The effects of TNF-α and DEX on MHC expression
Degradation of MHC is a major contributor to skeletal muscle protein catabolism seen in sepsis [24]. GCs and cytokines play a major role in muscle protein loss seen in different pathological conditions with selective loss of MHC [18,19]. Since DEX treatment of L6 cells limits the action of TNF-α as assessed by NF-κB activation, we assessed the impact of DEX and TNF-α, either alone or in combination on steady state levels of MHC. As shown in Fig. 7A and B, both DEX and TNF treatment leads to reduced levels of MHC in both L6 myotubes and myoblasts, respectively. Interestingly, a combined treatment with DEX and TNF-α generates a more significant loss of MHC expression than any treatment alone. Thus, although glucocorticoids can limit one aspect of TNF-α action in L6 cells (i.e. TNF-α induced activation of NF-κB), a combined exposure of L6 cells to these inflammatory mediators heightens the muscle wasting phenotype of either agent alone, as reflected in a dramatic loss of MHC protein.
Fig. 7.
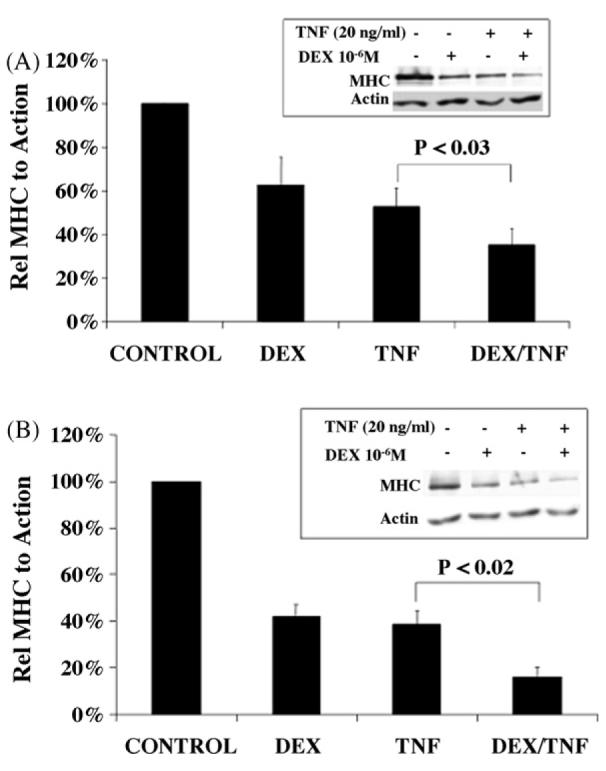
Effects of TNF-α and DEX on MHC expression in L6 myotubes (A) and myoblasts (B). Cells were treated 10–6M of DEX, 20 ng/ml of TNF-α or both for 48 h (TNF-α was added at 0 and 24 h to ensure its continuous effect). Total protein in whole cell lysates was separated by SDS-PAGE and subjected to Western blot analysis to detect MHC or actin. Results were quantified by densitometry. Data shown are the mean of eight (myotubes) and three (myoblasts) independent experiments ± S.E.M.
4. Discussion
In contrast to previous studies [10] where increased expression of GR in septic muscle accompanied an increase in serum GCs, our present study shows that DEX at different concentrations and for different periods induced downregulation of GR in L6 muscle cells. This hormone-dependent downregulation of GR is consistent with reports in numerous cell types and confirms that in the L6 cell model of skeletal muscle, glucocorticoid responsiveness has the capacity to be limited under conditions of chronic hormone treatment via reduced expression of GR. The presence of TNF-α did not affect hormone-dependent downregulation of GR in L6 cells suggesting that in vitro cytokine-mediated NF-κB activation partially mimicking a septic state may not alter GR processing.
The L6 muscle cells are unique in the rapid induction of GR downregulation following hormone treatment. Glucocorticoids enhance GR ubiquitylation and delivery of GR to the proteasome [25], but the mechanisms responsible for linking hormone activation to receptor ubiquitylation and protea-some degradation are unknown. The association of hormone activated steroid receptors with the transcriptional machinery participates in the ultimate delivery of the receptor to the proteasome for degradation [26-28]. GR is an effective trans-activator in L6 cells but may be more efficiently linked to the ubiquitin–proteasome system at various stages. The L6 cells may provide a useful model for more detailed analysis of the coupling between steroid receptor transactivation and degradation.
TNF-α exerted a limited effect in myoblasts and no effect in myotubes on the ability of GR to activate transcription from a transiently transfected reporter plasmid. This was unexpected since TNF-α is a known stimulant of NF-κB, a transcription factor that is known to be a strong antagonist of GR transactivation activity. Thus, in the L6 cells, GR escapes the antagonism by NF-κB. This limited effect of TNF-α on GR trans-activation was similarly seen in a recent study when HeLa cells were transiently transfected with GC-responsive MMTV11-Luc reporter plasmid, and then treated for 16 h with DEX in the presence or absence of increasing concentrations of TNF-α [29].
Transfection studies with an NF-κB-responsive promoter confirmed appropriate induction of NF-κB by TNF-α in L6 muscle cells. However, treatment of L6 cells with DEX significantly suppressed transcription of TNF-α-induced NF-κB activation. This effect of DEX is mediated through the action of GR against NF-κB by different mechanisms [30]. This includes induction of gene transcription and synthesis of the NF-κB inhibitor IκB, which binds to NF-κB in the cytoplasm and prevents its activation and nuclear translocation. Activated GR also antagonizes NF-κB by directly complexing with, and inhibiting NF-κB binding to DNA (simple model), or through associations with NF-κB bound to an NF-κB DNA site (composite model). In addition, GR may also compete with NF-κB for nuclear coactivators (competition model). The mechanism responsible for GR antagonism of NF-κB action in L6 cells awaits further studies.
Proinflammatory cytokines, especially TNF-α play a key role in the pathophysiology of sepsis [31], including adrenal insufficiency and acquired glucocorticoid resistance. Previous studies [10,11] and our study suggest that this may not hold true in all tissues especially skeletal muscle cells where GR shows normal processing and transcription activities. In fact, the increased GCs levels seen in sepsis could lead to increased GR transcription in muscle cells. Furthermore, enhanced GR activation in muscle cells may be insensitive to feedback control by NF-κB the pathway. This may contribute to the metabolic derangements seen with sepsis, such as hyper-glycemia, insulin resistance, increased free fatty acids, and proteolysis.
MHC is an important muscle contractile protein. Loss of MHC leads to decreased functional contractile units in the muscle with subsequent muscle wasting. Degradation of MHC is a major contributor to skeletal muscle proteolysis seen in sepsis [25]. Similarly, its loss has been described recently to play a significant role in cancer-related muscle cachexia [18,32]. This muscle wasting, which is resistant to improved nutrition, is also seen in other inflammatory conditions and plays a major role in their morbidity and mortality. In our study, we showed for the first time that DEX and TNF-α cooperatively suppressed MHC expression in L6 myotubes and myoblasts. DEX inhibited TNF-α-induced activation of NF-κB. Yet, at the same time DEX acted in synchrony with TNF-α to aggravate further decrease of MHC expression. The synchronized inhibitory effect of DEX and TNF-α on MHC expression suggests that the antagonism between glucocorticoids and TNF-α is not universal; glucocorticoids and TNF-α were also shown to regulate cooperatively toll-like receptor 2 gene expression [33]. Glucocorticoids as NF-κB antagonist appear to modulate the action of different inflammatory mediators. Yet, at the same time glucocorticoids may act in synchrony with TNF-α to aggravate muscle protein loss.
Recent studies suggest that GCs and TNF-α may utilize distinct pathways to induce muscle protein loss. Specifically, in C2C12 mouse myoblast cell line TNF-α treatment leads to a NF-κB mediated induction of the E2 ubiquitin-conjugating enzyme UbcH2 [34]. Furthermore, DEX-induced muscle atrophy in the C2C12 cell line is mediated by induction of the ubiquitin ligase atrogin-1, brought about by the relief of AKT induced inhibition of the Foxo transcription factors [35]. If these mechanisms operate to bring about the additional effects of DEX and TNF-α on MHC protein loss in L6 myoblasts and myotubes, both GR and NF-κB mutual antagonism may be directed towards these targets.
Cytokines especially TNF-α may not be the only culprit in the muscle cachexia seen in inflammatory conditions, and may have indirect effect through stimulation of increased secretions of glucocorticoids. The latter may lead to further muscle loss through actions of GR, especially when pharmacological doses are added to control the inflammation. Thus, cell-specific or gene-specific positive interactions between GR and NF-κB may operate in some pathological conditions to enhance muscle loss.
Acknowledgement
This work has been supported by a RAC grant from Children's Hospital of Pittsburgh.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration.
REFERENCES
- 1.Bourgeois S, Newby RF. Correlation between glucocorticoid receptor and cytolytic response of murine lymphoid cell lines. Cancer Res. 1979;39:4749–51. [PubMed] [Google Scholar]
- 2.Gehring U, Mugele K, Ulrich J. Cellular receptor levels and glucocorticoid responsiveness of lymphoma cells. Mol Cell Endocrinol. 1984;36:107–13. doi: 10.1016/0303-7207(84)90089-3. [DOI] [PubMed] [Google Scholar]
- 3.Vanderbilt JN, Miesfeld R, Maler BA, Yamamoto KR. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol. 1987;1:68–74. doi: 10.1210/mend-1-1-68. [DOI] [PubMed] [Google Scholar]
- 4.Oakley RH, Cidlowski JA. Homologous downregulation of the glucocorticoid receptor: the molecular machinery. Crit Rev Eukaryot Gene Expr. 1993;3:63–88. [PubMed] [Google Scholar]
- 5.Burnstein KL, Jewell CM, Sar M, Cidlowski JA. Intragenic sequences of the human glucocorticoid receptor complementary DNA mediate hormone-inducible receptor messenger RNA down-regulation through multiple mechanisms. Mol Endocrinol. 1994;8:1764–73. doi: 10.1210/mend.8.12.7708063. [DOI] [PubMed] [Google Scholar]
- 6.Rosewicz S, McDonald AR, Maddux BA, Goldfine ID, Miesfeld RL, Logsdon CD. Mechanism of glucocorticoid receptor down-regulation by glucocorticoids. J Biol Chem. 1988;263:2581–4. [PubMed] [Google Scholar]
- 7.Dong Y, Poellinger L, Gustafsson JA, Okret S. Regulation of glucocorticoid receptor expression: evidence for transcriptional and posttranslational mechanisms. Mol Endocrinol. 1988;2:1256–64. doi: 10.1210/mend-2-12-1256. [DOI] [PubMed] [Google Scholar]
- 8.McIntyre WR, Samuels HH. Triamcinolone acetonide regulates glucocorticoid-receptor levels by decreasing the half-life of the activated nuclear-receptor form. J Biol Chem. 1985;260:418–27. [PubMed] [Google Scholar]
- 9.Ali M, Allen HR, Vedeckis WV, Lang CH. Depletion of rat liver glucocorticoid receptor hormone-binding and its mRNA in sepsis. Life Sci. 1991;48:603–11. doi: 10.1016/0024-3205(91)90534-i. [DOI] [PubMed] [Google Scholar]
- 10.Sun X, Fischer DR, Pritts TA, Wray CJ, Hasselgren PO. Expression and binding activity of the glucocorticoid receptor are upregulated in septic muscle. Am J Physiol Regul Integr Comp Physiol. 2002;282:R509–18. doi: 10.1152/ajpregu.00509.2001. [DOI] [PubMed] [Google Scholar]
- 11.Sun X, Mammen JM, Tian X. Sepsis induces the transcription of the glucocorticoid receptor in skeletal muscle cells. Clin Sci (Lond) 2003;105:383–91. doi: 10.1042/CS20030087. [DOI] [PubMed] [Google Scholar]
- 12.Hasselgren PO, Fischer JE. The ubiquitin–proteasome pathway: review of a novel intracellular mechanism of muscle protein breakdown during sepsis and other catabolic conditions. Ann Surg. 1997;225:307–16. doi: 10.1097/00000658-199703000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Luo GJ, Wang JJ, Hasselgren PO. Dexamethasone stimulates proteasome- and calcium-dependent proteolysis in cultured L6 myotubes. Shock. 1998;10:298–306. doi: 10.1097/00024382-199810000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Tiao G, Hobler S, Wang JJ, Meyer TA, Luchette FA, Fischer JE, et al. Sepsis is associated with increased mRNAs of the ubiquitin–proteasome proteolytic pathway in human skeletal muscle. J Clin Invest. 1997;99:163–8. doi: 10.1172/JCI119143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiao G, Fagan J, Roegner V, Lieberman M, Wang JJ, Fischer JE, et al. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest. 1996;97:339–48. doi: 10.1172/JCI118421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–59. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 17.Sen CK, Khanna S, Reznick AZ, Roy S, Packer L. Glutathione regulation of tumor necrosis factor-alpha-induced NF-κB activation in skeletal muscle-derived L6 cells. Biochem Biophys Res Commun. 1997;237:645–9. doi: 10.1006/bbrc.1997.7206. [DOI] [PubMed] [Google Scholar]
- 18.Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest. 2004;114:370–8. doi: 10.1172/JCI20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seene T, Kaasik P, Pehme A, Alev K, Riso EM. The effect of glucocorticoids on the myosin heavy chain isoforms' turnover in skeletal muscle. J Steroid Biochem Mol Biol. 2003;86:201–6. doi: 10.1016/j.jsbmb.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Hickson RC, Czerwinski SM, Wegrzyn LE. Glutamine prevents downregulation of myosin heavy chain synthesis and muscle atrophy from glucocorticoids. Am J Physiol. 1995;268:E730–4. doi: 10.1152/ajpendo.1995.268.4.E730. [DOI] [PubMed] [Google Scholar]
- 21.Hoeck W, Rusconi S, Groner B. Down-regulation and phosphorylation of glucocorticoid receptors in cultured cells. Investigations with a monospecific antiserum against a bacterially expressed receptor fragment. J Biol Chem. 1989;264:14396–402. [PubMed] [Google Scholar]
- 22.Wang X, Pongrac JL, DeFranco DB. Glucocorticoid receptors in hippocampal neurons that do not engage proteasomes escape from hormone-dependent down-regulation but maintain transactivation activity. Mol Endocrinol. 2002;16:1987–98. doi: 10.1210/me.2001-0287. [DOI] [PubMed] [Google Scholar]
- 23.Franchimont D, Martens H, Hagelstein MT, Louis E, Dewe W, Chrousos GP, et al. Tumor necrosis factor alpha decreases, and interleukin-10 increases, the sensitivity of human monocytes to dexamethasone: potential regulation of the glucocorticoid receptor. J Clin Endocrinol Metab. 1999;84:2834–9. doi: 10.1210/jcem.84.8.5931. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad S, Karlstad MD, Choudhry MA, Sayeed MM. Sepsis-induced myofibrillar protein catabolism in rat skeletal muscle. Life Sci. 1994;55:1383–91. doi: 10.1016/0024-3205(94)00752-7. [DOI] [PubMed] [Google Scholar]
- 25.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276:42714–21. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 26.Kang Z, Pirskanen A, Janne OA, Palvimo JJ. Involvement of proteasome in the dynamic assembly of the androgen receptor transcription complex. J Biol Chem. 2002;277:48366–71. doi: 10.1074/jbc.M209074200. [DOI] [PubMed] [Google Scholar]
- 27.Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–48. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 28.Shao W, Keeton EK, McDonnell DP, Brown M. Coactivator AIB1 links estrogen receptor transcriptional activity and stability. Proc Natl Acad Sci USA. 2004;101:11599–604. doi: 10.1073/pnas.0402997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szatmary Z, Garabedian MJ, Vilcek J. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J Biol Chem. 2004;279:43708–15. doi: 10.1074/jbc.M406568200. [DOI] [PubMed] [Google Scholar]
- 30.Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol. 2002;71:9–15. [PubMed] [Google Scholar]
- 31.Marik PE. Nuclear factor-kappa B inhibition in sepsis: steroids versus specific nuclear factor-kappa B inhibitors? Crit Care Med. 2002;30:2393–4. doi: 10.1097/00003246-200210000-00042. [DOI] [PubMed] [Google Scholar]
- 32.Chamberlain JS. Cachexia in cancer—zeroing in on myosin. N Engl J Med. 2004;351:2124–5. doi: 10.1056/NEJMcibr042889. [DOI] [PubMed] [Google Scholar]
- 33.Hermoso MA, Matsuguchi T, Smoak K, Cidlowski JA. Glucocorticoids and tumor necrosis factor alpha cooperatively regulate toll-like receptor 2 gene expression. Mol Cell Biol. 2004;24:4743–56. doi: 10.1128/MCB.24.11.4743-4756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li YP, Lecker SH, Chen Y, Waddell ID, Goldberg AL, Reid MB. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 2003;17:1048–57. doi: 10.1096/fj.02-0759com. [DOI] [PubMed] [Google Scholar]
- 35.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]