

Abstract
The possible role of Ca2+ as a second messenger mediating regulatory volume decrease (RVD) in osmotically swollen cells was investigated in murine neural cell lines (N1E-115 and NG108-15) by means of novel microspectrofluorimetric techniques that allow simultaneous measurement of changes in cell water volume and [Ca2+]i in single cells loaded with fura-2. [Ca2+]i was measured ratiometrically, whereas the volume change was determined at the intracellular isosbestic wavelength (358 nm). Independent volume measurements were done using calcein, a fluorescent probe insensitive to intracellular ions. When challenged with ∼40% hyposmotic solutions, the cells expanded osmometrically and then underwent RVD. Concomitant with the volume response, there was a transient increase in [Ca2+]i, whose onset preceded RVD. For hyposmotic solutions (up to ∼−40%), [Ca2+]i increased steeply with the reciprocal of the external osmotic pressure and with the cell volume. Chelation of external and internal Ca2+, with EGTA and 1,2-bis-(o -aminophenoxy) ethane-N,N,N ′,N ′-tetraacetic acid (BAPTA), respectively, attenuated but did not prevent RVD. This Ca2+-independent RVD proceeded even when there was a concomitant decrease in [Ca2+]i below resting levels. Similar results were obtained in cells loaded with calcein. For cells not treated with BAPTA, restoration of external Ca2+ during the relaxation of RVD elicited by Ca2+-free hyposmotic solutions produced an increase in [Ca2+]i without affecting the rate or extent of the responses. RVD and the increase in [Ca2+]i were blocked or attenuated upon the second of two ∼40% hyposmotic challenges applied at an interval of 30–60 min. The inactivation persisted in Ca2+-free solutions. Hence, our simultaneous measurements of intracellular Ca2+ and volume in single neuroblastoma cells directly demonstrate that an increase in intracellular Ca2+ is not necessary for triggering RVD or its inactivation. The attenuation of RVD after Ca2+ chelation could occur through secondary effects or could indicate that Ca2+ is required for optimal RVD responses.
Keywords: calcium, volume regulation, neuroblastoma, regulatory volume decrease
introduction
Neurons, like all animal cells, possess mechanisms for maintaining constant volume in isosmotic media in the face of the Donnan effect or changes in intracellular solute content, such as those produced by neurotransmitters, hormones, repetitive nerve impulses, or upon sodium pump inhibition (Serve et al., 1988; Alvarez-Leefmans et al., 1992). In addition, neurons, like most cells, regulate their volume when exposed to anisosmotic media (Hoffmann and Simonsen, 1989). Upon exposure to hyposmotic media, cells initially swell, and then return to their initial volume, a phenomenon termed regulatory volume decrease (RVD).1 This regulatory response is accomplished through the loss of intracellular solutes along with osmotically obligated water. Three components are involved in this negative feedback loop: (a) the volume sensor, which detects a change in cell volume and transduces this change into an intracellular signal; (b) the effector mechanism that causes the loss of osmolytes and water, thereby correcting the aberrant volume; and (c) the signal or second messenger that couples the sensor to the effector. Chief among the candidates for second messenger is intracellular Ca2+ (for review see McCarty and O'Neil, 1992). However, the evidence supporting a signaling role for Ca2+ is fragmentary and controversial (Grinstein and Smith, 1990; Foskett, 1994). For instance, RVD measured in cell populations of neurons or astrocytes has been claimed to be unaffected by external Ca2+ removal, although internal Ca2+ was neither chelated nor measured (Pasantes-Morales et al., 1993, 1994). In neuroblastoma cells, RVD occurred in the absence of external Ca2+ in cells loaded with the Ca2+ chelator 1,2-bis-(o -aminophenoxy) ethane-N,N,N ′,N ′-tetraacetic acid (BAPTA; Lippmann et. al., 1995). However, intracellular Ca2+ was not measured to verify the effectiveness of Ca2+ buffering. A role for Ca2+ as mediator of RVD has been claimed for various cell types (McCarty and O'Neil, 1992) including astrocytes (O'Connor and Kimelberg, 1993) and cortical neurons (Churchwell et al., 1996). More recently, it was concluded that Ca2+ signaling by Ca2+ release or Ca2+ entry appears to play no role in the activation mechanism for the RVD responses in Ehrlich cells (Jørgensen et al., 1997). However, changes in intracellular free Ca2+ concentration, [Ca2+]i, were measured in cell populations or on single cells, in which changes in volume were not measured simultaneously. Clearly, part of the above controversy is due to the fact that changes in [Ca2+]i and volume have not been measured simultaneously in single cells and a causal relationship between these two variables has not been definitively established in the above studies.
In the present study, we unify the above conflicting claims in the literature by directly demonstrating that RVD can proceed independently of a rise in intracellular Ca2+, but that there is a component of the response that is sensitive to external and internal Ca2+ chelation. We used novel optical techniques to simultaneously measure changes in [Ca2+]i and cell volume in response to hyposmotic challenges in two neuronal cell lines (N1E-115 and NG108-15). Changes in cell water volume (CWV) were estimated from the change in intracellular concentration of trapped fluorescent dyes (Alvarez-Leefmans et al., 1995). By using the Ca2+-sensitive dye fura-2 and recording at the Ca2+-sensitive (380 nm) and -insensitive (isosbestic, 358 nm) wavelengths, we monitored simultaneous changes in CWV and [Ca2+]i in single cells (Muallem et al., 1992). CWV changes were also measured using calcein as the fluorescent probe (Crowe et al., 1995), thus providing independent measurements of CWV with a dye having different chemical properties than fura-2.
We find that upon hyposmotic swelling there is a rise in [Ca2+]i whose onset precedes RVD. However, chelation of external and internal Ca2+ only attenuates, but does not prevent, RVD. Moreover, this Ca2+-independent RVD proceeds even when [Ca2+]i decreases below resting level. We also find that upon multiple hyposmotic challenges the RVD response is lost. We call this effect inactivation. This phenomenon is also independent of Ca2+. Two models are consistent with our observations. In the first model, a single sequential process underlies RVD. In this model, a rise in [Ca2+]i cannot be the triggering signal for RVD. The attenuation of RVD after Ca2+ chelation could occur through secondary effects or could indicate that Ca2+ is required for optimal responses. In the second model, RVD could be the consequence of the operation of two parallel processes, one of which is Ca2+ independent. Some of these results have been reported in preliminary form (Altamirano et al., 1996).
materials and methods
Cell Culture
Two murine neural cell lines maintained in culture, N1E-115 neuroblastoma and neuroblastoma × glioma NG108-15, were used in the present experiments. N1E-115 is a clone of cells derived from mouse neuroblastoma C-1300 (Amano et al., 1972), and expresses characteristics of neuronal cells. The neuroblastoma × glioma hybrid cells NG108-15 are hybrid cells obtained from a rat glioma cell line and a mouse neuroblastoma line, and express characteristics of both glial and neuronal cells (Hamprecht, 1977). Cells were grown at 37°C on culture dishes containing 90% Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS, 1% hypoxanthine-aminopterin-thymidine (HAT) and 1% L-glutamine, in a 5% CO2/95% air atmosphere. Cells from passages 9–25 were plated on 25-mm-diameter glass coverslips (Bellco Glass, Inc., Vineland, NJ), previously treated with poly-d-lysine. Differentiation was induced 24 h after plating by supplying the cells with a low serum growth medium composed of 98% DMEM, 1% FCS, 1% HAT, 1% L-glutamine, 1 mM theophylline, and 10 μM prostaglandin E1 (Kasai and Neher, 1992). Cells were used from 1 to 14 d after the differentiation treatment.
Saline Solutions
The standard external solution (SES) contained (mM): 130 NaCl, 5.5 KCl, 2.5 CaCl2, 1.25 MgCl2, 20 HEPES, 10 dextrose, 13 sucrose. The pH was adjusted to 7.3 with NaOH and the osmolality was 312 ± 3 mosmol/kg water. The control isosmotic solution was prepared by substituting 65 mM NaCl for sucrose to match the osmolality of the culture medium (312 mosmol/kg water). Anisosmotic solutions were prepared by sucrose addition or removal to get the desired osmolality, thus maintaining constant the ionic strength and at the same value as the isosmotic control. Anisosmotic solutions were expressed as the decrement (or increment) percentage with respect to the control isosmotic solutions. Thus, a solution referred to as a “44% hyposmotic” means that its measured osmolality was 56% of the control isosmotic solution. The zero Ca2+ solutions were made by substituting CaCl2 for MgCl2 and adding 0.5 mM EGTA. The osmolality of all solutions was measured with a vapor pressure osmometer (5100 B; Wescor Inc., Logan, UT).
Dye Loading and Bath Chamber
Each coverslip with the cells attached was mounted in a Leiden chamber (Medical Systems Corp., Greenvale, NY) filled with SES and placed on the stage of an epifluorescence inverted microscope. Cells were loaded with the fluorophores fura-2 or calcein as described previously (Alvarez-Leefmans et al., 1995). In brief, they were incubated at room temperature (22°C) in SES containing either 5 μM fura-2 acetoxymethyl ester (AM), or 2 μM calcein/AM. To prepare the fura-2-loading solution, we used a stock containing 50 μg fura 2/AM dissolved in 4 μl DMSO plus 4 μl pluronic (10% wt/wt in DMSO). The final concentration of fura-2/AM for this stock solution was 6.23 mM. The calcein loading solution was prepared from a stock containing 50 μg calcein/AM dissolved in 4 μl DMSO plus 4 μl pluronic (10% wt/wt in DMSO). The final calcein/AM concentration of this stock solution was 6.28 mM. Dye loading was monitored fluorometrically by sampling a single cell every 30 or 60 s until reaching the desired level of fluorescence. For calcein, the level was ≈14.5× the initial fluorescence (i.e., the cell fluorescence without dye), and for fura-2 the level was ≈8, at 358 nm. The loading time was 37 ± 3 min for calcein (n = 22) and 43 ± 3 min for fura-2 (n = 26). The loading solution was then washed out with SES for at least 1 h before starting the experimental measurements. All solutions, except those used for loading (see below), were perfused into the chamber at a rate of 3 ml/min. The chamber fluid was exchanged with a time constant of 3.6 ± 0.3 s.
BAPTA Loading
To buffer intracellular Ca2+ near resting levels, in some experiments we used the Ca2+ chelator BAPTA. In this series of experiments, after the cells were loaded with either calcein or fura-2 as described above, they were briefly washed with SES and loaded with BAPTA. This was accomplished by incubating the cells at room temperature for 2 h in SES containing 100 μM BAPTA-AM. The BAPTA loading solution was prepared from a stock containing 3.82 mg of BAPTA/AM dissolved in 1 ml DMSO. After BAPTA loading, cells were superfused with SES for 1 h before starting the experimental measurements. The chelating action of intracellular BAPTA was verified by ratiometric measurements with fura-2.
Measurement of Cell Volume with Fluorescent Dyes
Basic principle.
Changes in CWV can be assessed by measuring changes in concentration of impermeant substances (volume markers) introduced into cells (Alvarez-Leefmans et al., 1995). Intracellularly trapped fluorescent dyes such as calcein (Crowe et al., 1995) and fura-2 (Muallem et al., 1992) have been successfully used as volume indicators. These optical techniques provide superior time resolution and sensitivity relative to other techniques and, when using ratiometric dyes, they allow parallel measurements of changes in CWV and intracellular ion concentrations in single cells. Changes in CWV are estimated from changes in fluorescence intensity of the dye resulting in turn from changes in its intracellular concentration. By using the Ca2+-sensitive dye fura-2 and recording at the Ca2+-sensitive (380 nm) and -insensitive (isosbestic) wavelengths, we recorded simultaneously changes in CWV and [Ca2+]i in single cells. CWV changes were inferred from changes in the concentration of fura-2, recorded at the isosbestic wavelength (see below). The simultaneous changes in [Ca2+]i were monitored by taking the ratio 358/380. Independent and parallel measurements of CWV were provided by using calcein, a dye that is more intensely fluorescent than fura-2 and insensitive to changes in the concentration of native cellular ions within physiological ranges. The validation, pitfalls, and limitations of these techniques have been discussed in detail in previous publications (Alvarez-Leefmans et al., 1995; Crowe et al., 1995, 1996).
Fluorescence measurements.
Total fluorescence from a small region of fluorophore-loaded single cells was measured with a customized microspectrophotometry system that has been described in detail elsewhere (Alvarez-Leefmans et al., 1995). The system included an inverted, epi-fluorescence microscope (Diaphot-TMD; Nikon, Tokyo, Japan) equipped with a fluor oil-immersion objective lens (40×, 1.3 NA; Nikon). The excitation light coming from a 150 W xenon arc lamp passed through a water filter, and was then divided by a beam splitter. Each beam passed through a computer controlled high speed shutter (Uniblitz; Vincent Assoc., Rochester, NY). Attached to each shutter was a filter wheel holding a selection of customized excitation filters (Omega Optical, Brattleboro, VT). For fura-2 measurements, beam 1 passed through a filter centered at 380 ± 6 nm. For exciting fura-2 at its isosbestic wavelength, beam 2 passed through a filter centered at 362 or 358 ± 5 nm. The 358-nm wavelength was preferred over the 362-nm because it was closer to the intracellular isosbestic wavelength measured in vivo with a monochromator and the same objective lens used in the present work (Crowe et al., 1996). In some experiments using the monochromator, measurements were done at wavelengths of 350 and 358 nm (slit = 8 nm) to optimize the sampling rate.
Optical systems may distort spectral responses of fluorescent dyes through various components in the light path; e.g., the objective lens having different spectral light transmission properties or the bandwidth of the excitation light. Fig. 1 shows a bath calibration of fura-2 performed in the recording chamber with the optical system used in the present experiments. This calibration shows that at 358 nm, the fura-2 signal was insensitive to changes in [Ca2+] in the range between 0 and 39.80 μM. As expected, the dye showed adequate sensitivity for Ca2+ at the 380-nm wavelength. The latter wavelength was preferred over 340 nm because the light transmission properties of the objective used were better at 380 nm. Light transmission of this objective is 74% at 380 nm, 68% at 360 nm, and 55% at 340 nm.
Figure 1.

Common mode of rejection test. Fura-2 fluorescence recorded in the experimental bath chamber in response to a series of 0–10 mM calcium-EGTA buffer solutions, using a fluor 40×/1.3 oil-immersion objective. F, fluorescence in arbitrary units (a.u.). The free [Ca2+] (micromolar) is indicated by vertical arrows. Fura-2 was excited at 358 and 380 nm. Emission was measured at 510 nm. Note that at 358 nm, the isosbestic wavelength, the dye does not respond to Ca2+.
For CWV measurements using calcein, beam 1 passed through a filter centered at 495 ± 10 nm to excite this fluorophore at its peak excitation wavelength. When dual excitation wavelength measurements were done (fura-2), the filtered light beams were recombined by means of a bifurcated, fused silica-fiber bundle. The combined light beam was then collimated and coupled to the microscope epi-illuminator. To minimize photobleaching and photodynamic damage, the intensity of the excitation light beam was attenuated with a 10% neutral density filter and the shutter was opened only during data sampling (see below). The field diaphragm was closed to its smallest opening to limit the excitation area to a single cell. For fura-2 measurements, a cube holding a dichromatic mirror (400 nm) and a 510 ± 20-nm emission filter was positioned underneath the objective lens in the filter cassette holder of the microscope. For calcein, a dichromatic mirror (515 nm) and a 535 ± 13 emission filter was used instead. The emitted light passed through a one-times relay lens coupled to a turret diaphragm accessory (Microflex PFX; Nikon) containing a series of seven circular pinhole openings of varying diameter (0.1–10 mm).
The emitted light exiting through a selected pinhole (see below) was measured with a photometer system as described previously (Alvarez-Leefmans et al., 1995). Once amplified, the photomultiplier signal was digitized with an analog to digital converter, displayed, and stored on an IBM-compatible PC computer using customized software. The fluorescence data were collected at a frequency of either 0.1 or 0.2 Hz during shutter openings of 130-ms duration. Subsequent data analysis and presentation were performed using commercial software.
As CWV changes, the concentration of osmotically active fluorophore molecules changes in inverse proportion. To observe changes in fluorescence intensity due to changes in intracellular fluorophore concentration, it is necessary to record from a small region of the loaded cell. With epi-illumination, the image of the region of the cell from which light is measured is a volume element determined by the pinhole aperture placed at the image plane and the numerical aperture of the objective. The pinhole aperture used for collecting calcein fluorescence was 0.2 mm in diameter and, with the objective lens used, the combination gave a total recording area of 19.6 μm2. The mean area of the cells, at the image plane, was between 1,250 and 1,950 μm2. The pinhole used for measuring fura-2-emitted fluorescence was 0.5 mm in diameter, giving a total recording area of 122.7 μm2. The larger pinhole is necessary to improve the signal/noise ratio in fura-2 recordings. This is because the fura-2 fluorescence is dimmer than that emitted by calcein, as expected from their corresponding quantum yields and extinction coefficients (Haugland, 1996), and also because of the spectral transmission properties of most objective lenses (Keller, 1995), including the one we used (see above). We have studied the effects of pinhole size and location on the signals recorded at 358 nm in fura-2-loaded cells. It was found that there is a compromise between pinhole size, resolution, and signal-to-noise (S/N) ratio. We have determined that optimal S/N ratio without loss of resolution is obtained with a pinhole located well within the cell boundaries and comprising no more than 10% of the total area of the cell body (Crowe et al., 1996).
Calculation of cell water volume changes.
Normalized cell water volume changes (V t/V o) were computed from monitored changes in relative fluorescence (F t/F o) resulting from exciting fura-2 at 358 nm or calcein at 495 nm, according to the following equation (Alvarez-Leefmans et al., 1995):
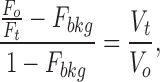 |
1 |
where F o is the fluorescence from a pinhole region of the cell equilibrated with an isotonic SES or in a solution isosmotic and isotonic with the SES, having an osmotic pressure π0; F t is the fluorescence of the same region of the cell in a solution of osmotic pressure πt; F bkg is the background fluorescence (see below); V o is the water volume of the cell in a solution having an osmotic pressure π0 and V t is the corresponding volume of the cell in a solution of osmotic pressure πt. In the present study, F bkg includes the intrinsic cell fluorescence plus the osmotically insensitive component of the fluorescence in dye-loaded cells. Both components of F bkg are insensitive to changes in external osmolality. F bkg was estimated from the y-intercept of plots of F o/F t vs. π0/πt, as described previously (Alvarez-Leefmans et al., 1995). It is worth noting that F bkg determined graphically is similar to the fluorescence remaining after controlled plasma membrane permeabilization with α-toxin (Crowe et al., 1995). Moreover, the two approaches have been validated by direct morphometric measurements of cell volume using differential interference contrast (Alvarez-Leefmans et al., 1995) or confocal image reconstruction (our unpublished observations).
We characterized the relation between F o/F t vs. π0/πt for each individual cell. Steady state changes in F o/F t were measured in response to brief (5–10 min) exposures to anisosmotic solutions having nominal osmolalities in the range between −10 and +10% relative to the isosmotic solution. Within this range of external osmolalities and exposure times, the cells exhibited osmometric behavior and did not regulate their volume. Moreover, the peak response to hyposmotic solutions having osmolalities ∼40% was also osmometric before the cells started undergoing RVD. All experiments were done at room temperature (22°C).
Drift Correction and Statistical Analysis
When necessary, the drift of the fluorescence signal resulting from dye leakage and photobleaching was corrected by fitting a linear regression to the base line and multiplying the slope of this regression line by the time at which each data point was sampled. This process yields point by point drift values that, in turn, are subtracted from the record yielding the corrected trace.
RVD response variables were measured as defined in Fig. 2. The extent of RVD (Fig. 2 f ) was measured 20 min after the onset of the regulatory response and is presented as
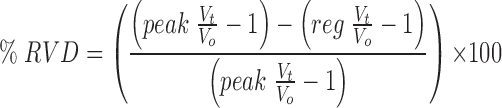 |
2 |
Figure 2.

Schematic drawing illustrating measured variables of osmotic swelling and RVD responses elicited by a 40% hyposmotic challenge. Ordinate: relative cell volume (Vt/Vo). Abscissa: time. (a) Maximum (peak) swelling, (b) initial rate of swelling, (c) time to peak swelling, (d) RVD delay, (e) initial rate of RVD, (f ) extent of RVD at t = 20 min, and (g) regulated relative cell volume at t = 20 min.
where peak V t/V o and reg V o/V t are the peak and regulated relative volumes, respectively. Therefore,% RVD denotes the magnitude of the return from the peak swollen volume back toward the isosmotic initial volume, V o, such that 0% RVD indicates no volume regulation and 100% RVD indicates complete volume recovery. Referring to Fig. 2, % RVD is simply [(a − g)/a] × 100. All measurements are expressed as mean ± SEM. The one-tailed unpaired t test was used to establish the significance of differences between means at the 95% confidence limit.
Chemicals
All chemicals used to prepare bathing media came from Sigma Chemical Co. (St. Louis, MO). Tissue culture media (Dulbecco's modified Eagle's medium), L-glutamine, and hypoxanthine-aminopterin-thymidine came from GIBCO BRL (Gaithersburg, MD), FCS from Hyclone (Logan, UT), theophylline and prostaglandin E1 from Sigma Chemical Co., and poly-d-lysine from ICN (Costa Mesa, CA). Fura-2/AM, fura-2-free acid, calcein/AM, BAPTA/AM pluronic, and calcium buffers came from Molecular Probes, Inc. (Eugene, OR).
results
Regulatory Volume Decrease in Single Cells Loaded with Calcein or Fura-2
Regulatory volume decrease was studied in cultured N1E-115 and NG108-15 cells loaded with either calcein or fura-2. This strategy provided an independent measurement of CWV with dyes having different chemical properties, thus giving more reliability to our observations. In particular, calcein fluorescence is independent of changes in the concentration of native intracellular ions within the physiological range. The parallel use of calcein eliminates possible intracellular Ca2+ buffering effects of fura-2, which may attenuate putative Ca2+-dependent components of the RVD response pattern. On the other hand, the use of fura-2 allowed for the simultaneous measurement of changes in [Ca2+]i and CWV in single cells.
Fig. 3 shows a typical RVD pattern in response to a hyposmotic challenge recorded in a single NG108-15 cell loaded with calcein. The cell was initially superfused with an isosmotic solution to obtain a stable baseline. Upon exposure to a 44% hyposmotic solution, the calcein relative fluorescence (F t/F o) decreased due to intracellular dye dilution resulting from net osmotic influx of water. After reaching a minimum, the F t/F o signal recovered close to initial values, reflecting a gradual increase in intracellular dye concentration resulting from regulatory net water efflux. When the hyposmotic solution was replaced with the isosmotic solution, the F t/F o signal increased beyond control levels, reached a peak, and recovered to initial values. This last sequence of fluorescence signals was also expected because, upon returning to the isosmotic solution from a hyposmotic one, the isosmotic solution is hypertonic with respect to the cell that shrinks, and then recovers its initial water volume. Fluorescence signals like these were transformed into relative cell water volume changes (V t/V o) using Eq. 1. This transformation yielded traces like the one shown in the bottom of Fig. 3. Before testing the 44% hyposmotic solution, the cell was exposed to calibration test solutions (Fig. 3, inset). These calibration solutions had osmolalities of +10 and −12% with respect to the isosmotic solution. The inset includes a plot that was routinely made for each cell, showing the relationship between steady state values of changes in V t/V o and the reciprocal of the relative osmotic pressure of the medium (πo/πt). The line denotes the predicted behavior of a perfect osmometer according to the equation:
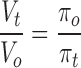 |
3 |
Figure 3.

RVD monitored in a calcein-loaded NG108-15 cell in response to a 44% hyposmotic challenge. (top) Percent change in relative fluorescence (Ft/Fo). (bottom) Relative cell water volume changes (Vt/Vo) computed using Eq. 1. The box indicates the time of application of isosmotic (iso) and hyposmotic (hypo) solutions. The cell swelled to a maximum (83% of its initial volume) and then underwent RVD. Maximal volume recovery (87%) was achieved 22.7 min after RVD onset. As expected, upon returning to the isosmotic solution, the cell shrank and eventually recovered its initial volume. (inset) V t/V o in response to calibration test anisosmotic solutions having nominal osmolalities of ±10% with respect to the isosmotic solution (actual osmolalities were −12 and +10%). Steady state V t/V o changes in response to the test solutions are plotted against the reciprocal of the relative osmotic pressure of the medium (πo/πt) for the calibration pulses (•). The peak V t/V o produced by the 44% hyposmotic solution is also plotted (○). The solid line denotes the theoretical behavior of a perfect osmometer according to Eq. 3. This kind of plot was made for all cells and is shown in subsequent figures.
where V t, V o, πo, and πt, have been defined. (Fig. 3, •) Osmotic calibration pulses. Note that the measurements fall close to the theoretical line for a perfect osmometer. (Fig. 3, ○) The peak amplitude of the osmotic response produced by the 44% hyposmotic solution, which also falls close to the theoretical line denoting ideal behavior, before RVD ensues.
Experiments like the one described were performed in seven NG108-15 cells. Upon exposure to a solution ∼40% hyposmotic (41.3 ± 1%), the cells swelled to a maximum of 69 ± 5% above their initial volume, which is not significantly different from the value of 71 ± 3% predicted for ideal behavior. The time to reach the maximum swelling was 4.4 ± 0.5 min. After a delay of 47 ± 6 s measured from the peak of the osmotic swelling, RVD ensued at an average initial rate of −3.4 ± 0.6% min−1 with a partial volume recovery at 20 min (percent RVD recovery) of 57 ± 11%. Upon returning to the control isosmotic solution (which would now be hypertonic with respect to the cells), the cells shrank and their volume started to recover. In two cells, complete recovery occurred at 37 and 103 min, respectively, upon returning to the isosmotic solution. The above results are summarized in Table I. Similar results were obtained in three N1E-115 neuroblastoma cells (see Crowe et al., 1995).
Table I.
Osmotic Swelling and RVD in Cells Loaded with Calcein or Fura-2
| Variable | Calcein | Fura-2 | P | |||
|---|---|---|---|---|---|---|
| n = 7 | n = 10 | |||||
| Osmolality (%) | −41.3 ± 1 | −41.4 ± 0.6 | NS | |||
| Peak swelling (%) | ||||||
| Observed | 69 ± 5 | 59 ± 5 | NS | |||
| Osmometric | 71 ± 3 | 71 ± 2 | NS | |||
| Time to peak (min) | 4.4 ± 0.5 | 5.0 ± 0.4 | NS | |||
| RVD latency (s) | 47 ± 6 | 90 ± 23 | NS | |||
| RVD rate (% min−1) | −3.4 ± 0.6 | −3.0 ± 0.5 | NS | |||
| % RVD | 57 ± 11 (n = 6) | 45 ± 8 (n = 7) | NS |
Values are means ± SEM.
The prevailing controversy about the possible role of intracellular Ca2+ as the signal that couples the activation of the volume sensor and the transport mechanisms for RVD prompted us to measure simultaneously [Ca2+]i and volume changes in fura-2-loaded cells. Fig. 4 shows that a 42% hyposmotic solution produced an increase in [Ca2+]i accompanied by RVD. When exposed to an ∼40% hyposmotic solution (41.4 ± 0.6%), fura-2-loaded NG108-15 cells (n = 10) swelled to a maximum of 59 ± 5% above their initial volume. The time to reach the maximum swelling was 5.0 ± 0.4 min. After a delay of 90 ± 23 s, RVD ensued at an average initial rate of −3.0 ± 0.5% min−1 with partial recovery (percent RVD) of 45 ± 8%. The ratio 358/380, which signals changes in [Ca2+]i, started to increase with a latency of 29.5 ± 12 s measured from the onset of the volume change produced by the hyposmotic challenge. The ratio peaked at 7.6 ± 1.4 min after the onset of the volume change produced by the hyposmotic challenge; i.e., ∼1 min after the onset of RVD. Similar results were obtained in N1E-115 cells (n = 4) following the same protocol. However, in cells loaded with fura-2, the mean values of some variables of RVD responses with respect to those recorded in calcein-loaded cells were slightly attenuated (e.g., maximum swelling and percentage of RVD) or slowed down (e.g., time to peak swelling), but the differences were not statistically significant (Table I).
Figure 4.

Relative volume changes (Vt/Vo) and [Ca2+]i recorded simultaneously in response to a 42% hyposmotic challenge in a fura-2-loaded NG108-15 cell. (top) V t/V o obtained from changes in fluorescence measured at 358-nm excitation wavelength, the isosbestic point for fura-2. (bottom) Changes in [Ca2+]i measured as the ratio 358/380. Dashed vertical lines are traced at the onset (1) and peak (2) of the volume response, and at the peak of the intracellular Ca2+ signal (3). (inset) Calibration plot obtained as explained in Fig. 3.
These results show that fura-2-loaded cells behave like those loaded with calcein in terms of their response to anisosmotic solutions and that the change in cell volume is parallel to a rise in [Ca2+]i. Moreover, the temporal relationship between changes in [Ca2+]i and RVD is appropriate for a putative signaling role of internal Ca2+. However, the results do not prove a causal relation between the rise in [Ca2+]i and RVD.
Relationship between External Osmolality, [Ca2+]i, and Cell Volume
To test further the Ca2+ hypothesis for RVD, we recorded simultaneously changes in volume and [Ca2+]i in cells loaded with fura-2 that were exposed to solutions of various osmolalities. Fig. 5 A shows an example in an NG108-15 cell. No measurable changes in the [Ca2+]i signal were produced by a 14% hyperosmotic solution that caused an osmometric shrinkage. For hyposmotic solutions, [Ca2+]i increased steeply with πo/πt (Fig. 5 B, ○) and cell volume (Fig. 5 C ). The 10% hyposmotic solution resulted in an osmometric increase in cell volume accompanied by a modest rise in [Ca2+]i (Fig. 5 A). There was no sign of RVD response. The 26 and 40% hyposmotic solutions resulted in respective transient increases in [Ca2+]i as a function of πo/πt and V t/V o, and clear RVD responses in Fig. 5 A, I and II . The rate of RVD increased with [Ca2+]i and V t/V o. The peak of the responses I and II fall close to the value predicted for the behavior of a perfect osmometer (Fig. 5 B, I and II ), thus confirming that RVD started after the cell had increased its water volume in an osmometric manner. The above findings show again a close parallel between changes in [Ca2+]i, cell volume, and rate of RVD. However, they do not prove that the increase in [Ca2+]i upon osmotic swelling is a necessary causal signal for RVD and not just an independent parallel process.
Figure 5.

Relationship between external osmolality, [Ca2+]i, and relative cell volume (Vt/Vo) in a fura-2-loaded NG108-15 cell. (A) Changes in V t/V o and [Ca2+]i (measured as the ratio 362/380) in response to anisosmotic solutions. The percent osmolalities of each solution relative to the isosmotic control are indicated at the bottom of each box. Note that the rate of RVD (I and II ) increased together with [Ca2+]i and cell volume. (B) Estimated changes in [Ca2+]i, nanomoles (○), and V t/V o (•) plotted as a function of the reciprocal of the relative osmotic pressure of the medium (πo/πt). (C ) Estimated changes in [Ca2+]i (nanomoles) as a function of V t/V o.
Changes in [Ca2+]i and RVD upon Repeated Hyposmotic Challenges
The close parallel between changes in [Ca2+]i and RVD was further shown by exposing the cells to two 40% hyposmotic challenges, each lasting ∼40 min, applied with an interval of 30–60 min. RVD and the concomitant increase in [Ca2+]i did not occur or were significantly attenuated upon exposure to the second hyposmotic challenge. Fig. 6 illustrates one of these experiments performed in an N1E-115 cell. The first hyposmotic challenge produced the expected transient increase in [Ca2+]i accompanied by RVD. However, the second hyposmotic challenge, applied 55 min after the end of the first one, produced a measurable increase in neither [Ca2+]i nor RVD. In some experiments, the second hyposmotic challenge produced an increase in [Ca2+]i, albeit smaller than the one evoked by the first hyposmotic challenge, without a concomitant RVD response. The disappearance of the RVD response upon the second hyposmotic shock was also observed in calcein-loaded cells in both cell lines (Fig. 7).
Figure 6.

Inactivation of RVD response upon repeated hyposmotic challenges. Changes in relative cell volume (Vt/Vo) and [Ca2+]i in response to two identical hyposmotic challenges in a fura-2-loaded N1E-115 cell. The cell was exposed to two 40% hyposmotic challenges, each lasting ∼40 min applied with an interval of 55 min. (top) V t/V o. (bottom) Changes in [Ca2+]i. (inset) Calibration plot obtained as explained in Fig. 3. (○) Peak amplitude of the first pulse, and (▵) peak amplitude of the second pulse. Boxes indicate the time of application of isosmotic (iso) and hyposmotic (hypo) solutions.
Figure 7.

Inactivation of RVD response upon repeated hyposmotic challenges in the virtual absence of external Ca2+. Changes in relative cell volume (V t/V o) in response to two hyposmotic challenges of identical osmolality, in a single NG108-15 cell loaded with calcein. Except for the calibration test solutions, the isosmotic and hyposmotic solutions were Ca2+-free and contained 0.5 mM EGTA. After testing the anisosmotic calibration pulses, the cell was superfused with a Ca2+-free isosmotic solution for 10 min before the first Ca2+-free 40% hyposmotic challenge. The second Ca2+-free hyposmotic challenge was applied 44 min after the end of the first one. During the interval between hyposmotic challenges, the cell was superfused with Ca2+-free isosmotic solution. The inset shows a calibration plot obtained as explained in Fig. 3. (○) Peak amplitude of the first pulse, and (▵) peak amplitude of the second pulse. Boxes indicate the time of application of isosmotic (iso) and hyposmotic (hypo) solutions.
To test for the external Ca2+ dependence of RVD inactivation, calcein-loaded cells were exposed to Ca2+-free isosmotic and hyposmotic solutions containing 0.5 mM EGTA. The cells were first superfused with the Ca2+-free isosmotic solution for at least 10 min, and then the Ca2+-free 40% hyposmotic solution was applied twice with an interval of ∼50 min. The results show that both RVD and its inactivation upon the second hyposmotic challenge proceeded in the virtual absence of external Ca2+ (Fig. 7).
We conclude that RVD inactivation is independent of (a) the type of dye with which the cells are loaded, (b) the cell type (NG108-15 or N1E-115), and (c) the presence of external Ca2+.
The fact that RVD disappearance coincides with suppression or attenuation of the concomitant change in [Ca2+]i confirms the parallel between a rise in [Ca2+]i and RVD.
Effect of Extracellular and Intracellular Ca2+ Chelation on RVD and [Ca2+]i
The experiments so far described demonstrate a tight correlation between changes in [Ca2+]i and RVD. Demonstration of this correlation is necessary but not sufficient to suggest a causal role for Ca2+ as a second messenger mediating RVD. A crucial test for the Ca2+ hypothesis is to show directly that RVD is abolished when the rise in intracellular Ca2+ upon osmotic swelling is inhibited. Elevations of [Ca2+]i produced by hyposmotic solutions could result either from Ca2+ influx across the plasma membrane or Ca2+ release from internal stores (McCarty and O'Neil, 1992; Wu et al., 1997). Thus, we studied the effect of extracellular and intracellular Ca2+ chelation on RVD. To remove the external source of Ca2+, these experiments were done with Ca2+-free EGTA isosmotic and hyposmotic solutions. Intracellular Ca2+ was chelated with BAPTA. For this purpose, cells were incubated with 100 μM BAPTA/AM for 2 h, loaded with calcein, and then superfused with the Ca2+-free EGTA isosmotic solution for at least 10 min before the Ca2+-free EGTA hyposmotic solution. The use of calcein was important in these experiments because this dye does not affect intracellular Ca2+ and does not have the side effects of fura-2. We compared a control population of cells with those treated with BAPTA and Ca2+-free EGTA solutions. Such Ca2+ chelation did not abolish RVD; however, there was a significant (P < 0.01) decrease in the rate (53%) and extent (56%) of RVD responses (Table II).
Table II.
Effect of Ca2+ Chelation with EGTA and BAPTA on Osmotic Responses Elicited in Calcein-loaded Cells
| Variable | Control | Ca2+-free | P | |||
|---|---|---|---|---|---|---|
| n = 7 | n = 7 | |||||
| Osmolality (%) | −41.3 ± 1.0 | −41.3 ± 0.8 | NS | |||
| Peak swelling (%) | ||||||
| Observed | 69 ± 5 | 68 ± 4 | NS | |||
| Osmometric | 71 ± 3 | 71 ± 2 | NS | |||
| Time to peak (min) | 4.4 ± 0.5 | 5.6 ± 0.9 | NS | |||
| RVD latency (s) | 47 ± 6 | 73 ± 31 | NS | |||
| RVD rate (% min−1) | −3.4 ± 0.6 | −1.6 ± 0.3 | <0.01 | |||
| % RVD | 57 ± 11 (n = 6) | 25 ± 4 (n = 6) | <0.01 |
Values are means ± SEM.
The persistent RVD could be due to incomplete intracellular Ca2+ chelation. The ability of BAPTA to chelate intracellular Ca2+, like that of other Ca2+ chelators, depends on the [Ca2+]i, the ionic strength and composition of the cytosol, and the [BAPTA]i. Unless one knows these factors, it is not possible to be sure that BAPTA is adequately chelating internal Ca2+. In the absence of such knowledge, it is essential to verify that BAPTA is adequately chelating intracellular Ca2+. To verify that BAPTA was actually damping any hyposmotically induced increases in [Ca2+]i, another series of experiments was done with fura-2-loaded cells. Under these conditions, the hyposmotic challenge resulted in a decrease in [Ca2+]i below resting levels, but the RVD response persisted (Fig. 8).
Figure 8.

Effect of extracellular and intracellular Ca2+ chelation on RVD and [Ca2+]i. RVD elicited by a 43% hyposmotic solution, under external and internal Ca2+ chelation, in a fura-2-loaded NG108-15 cell. The cell was incubated with BAPTA/AM (100 μM) for 2 h and kept in a Ca2+-free isosmotic solution containing EGTA (0.5 mM) for 11 min before the Ca2+-free 43% hyposmotic challenge. Note that the apparent [Ca2+]i, measured as the ratio 358/ 380, decreases upon exposure to the hyposmotic solution but the RVD response persists.
In seven fura-2-loaded cells treated following the above protocol, upon exposure to the Ca2+-free ∼40% hyposmotic (42.6 ± 0.4%) solution, the cells swelled to a maximum of 62 ± 4% above their initial volume. The time to reach the maximum swelling was 4.9 ± 0.5 min. After a delay of 54 ± 8 s, RVD ensued at an initial average rate of −1.6 ± 0.5% min−1 with 30 ± 6% partial recovery (percent RVD). The mean values of all these variables were smaller than those measured in fura-2-loaded control cells without BAPTA, but the differences were statistically significant only for the initial rate of RVD (Table III and Fig. 9 A). However, the differences in the average rate and extent of RVD in the Ca2+-free EGTA-BAPTA-treated cells loaded with fura-2 compared with the calcein-loaded control cells were statistically significant (P < 0.05). The Ca2+ chelation effects on the extent of RVD responses were not statistically significant when compared with the control cells loaded with fura-2 (Table III), probably because the latter indicator was already exerting some chelating or other side effects in the control group. In other words, the control responses in the fura-2-loaded cells were slightly blunted from the beginning. This can be appreciated by comparing the “control” parameters between calcein-loaded and fura-2-loaded cells (Table I and Fig. 9). Again, although the differences between controls are not statistically significant, the tendencies are obvious.
Table III.
Effect of Ca2+ Chelation with EGTA and BAPTA on Osmotic Responses Elicited in Fura-2-loaded Cells
| Variable | Control | Ca2+-free | P | |||
|---|---|---|---|---|---|---|
| n = 10 | n = 7 | |||||
| Osmolality (%) | −41.4 ± 0.6 | −42.6 ± 0.4 | NS | |||
| Peak swelling (%) | ||||||
| Observed | 59 ± 5 | 62 ± 4 | NS | |||
| Osmometric | 71 ± 2 | 74 ± 1 | NS | |||
| Time to peak (min) | 5.0 ± 0.4 | 4.9 ± 0.5 | NS | |||
| RVD latency (s) | 90 ± 23 | 54 ± 8 | NS | |||
| RVD rate (% min−1) | −3.0 ± 0.5 | −1.6 ± 0.5 | <0.05 | |||
| % RVD | 45 ± 8 (n = 7) | 30 ± 6 (n = 6) | NS |
Values are means ± SEM.
Figure 9.

Effect of extracellular and intracellular Ca2+ chelation on the rate (A) and extent (B) of RVD in cells loaded with calcein or fura-2. The initial rate of RVD (percent at min−1) and the extent of RVD (percent RVD at 20 min) were measured as described in Fig. 2. There is a significant difference in the initial average rate of RVD (A), between control and Ca2+-chelated (0 Ca2+-EGTA/BAPTA) conditions in both calcein-loaded (P < 0.01) and fura-2-loaded cells (P < 0.05). There is a significant difference (P < 0.01) in the extent of RVD (B), between control and Ca2+-chelated (0 Ca2+-EGTA/ BAPTA) conditions in calcein-loaded cells, but not in fura-2-loaded cells.
These results prove that RVD can be produced in the absence of a rise in internal Ca2+, suggesting that if a single sequential process underlies RVD, a rise in [Ca2+]i cannot be the triggering signal. The effects of Ca2+ removal suggest that Ca2+ may be acting as a modulator affecting RVD kinetics, or chelation may affect RVD through secondary mechanisms.
The Rate of RVD Responses Elicited with Ca2+-free Hyposmotic Challenges Is Not Affected by Restoring External Ca2+
An increase in [Ca2+]i in isosmotic media produces shrinkage of N1E-115 cells (Crowe et al., 1995). This suggests that these cells are endowed with Ca2+-sensitive mechanisms for net solute efflux, which can be activated by Ca2+ in isosmotic media. The question arises as to whether such Ca2+-triggered mechanisms can be activated during osmotic swelling. To examine this question, experiments were done in which [Ca2+]i was increased during ensuing RVD responses elicited by exposure to a Ca2+-free EGTA hyposmotic solution. The rationale was that if Ca2+-dependent processes are important, then the rate of cell volume recovery would be enhanced by an increase in [Ca2+]i. In these experiments, we took advantage of the fact that after superfusion with a Ca2+-free solution, restoration of [Ca2+]o results in an increase in [Ca2+]i. Fig. 10 A shows one such experiment in a fura-2-loaded cell. Note that upon removal of external Ca2+ in the isosmotic solution, the basal level of [Ca2+]i decreased. When a Ca2+-free 40% hyposmotic solution was applied, the relative cell volume increased and the cell started to downregulate its volume. A Ca2+-containing hyposmotic solution was then applied while the cell was still undergoing RVD. This resulted in the expected transient increase in [Ca2+]i. However, the rate of RVD (Fig. 10 A, top) did not increase. Similar experiments were conducted in calcein-loaded cells (n = 7). An example is shown in Fig. 10 B in which readmission of external Ca2+ during an RVD response elicited by exposure to a 40% Ca2+-free hyposmotic solution did not alter its time course.
Figure 10.

Restoration of extracellular Ca2+ during RVD elicited by a 0 Ca2+-EGTA hyposmotic solution. (A) NG108-15 cell loaded with fura-2. (top) V t/V o, (bottom) apparent [Ca2+]i, measured as the ratio 358/380. (B) NG108-15 cell loaded with calcein. The cells were superfused with a Ca2+-free isosmotic solution containing 0.5 mM EGTA (0 Ca2+ EGTA) for at least 10 min before applying the 0 Ca2+-EGTA–40% hyposmotic challenge. External Ca2+ (2.5 mM) was restored at the time indicated by the boxes. Restoration of external Ca2+ did not increase the slope of RVD, although this maneuver did produce a transient increase in [Ca2+]i, shown in the fura-2-loaded cell (A, bottom).
The results obtained in six calcein-loaded cells are summarized in Fig. 11. External Ca2+ was readmitted in a time window between 8 and 14 min after RVD ensued. The time at which external Ca2+ was readmitted in each experiment is indicated by the large gray circles. The filled circles correspond to mean values ± SEM of the six RVD responses. No sign of enhancement of RVD rate upon external Ca2+ readmission was observed.
Figure 11.

Lack of effect of extracellular Ca2+ restoration during RVD responses elicited by 0 Ca2+-EGTA hyposmotic solutions. The same experimental protocol was shown in Fig. 10. Ordinate, percent RVD. Abscissa, time. (○) Individual NG108-15 cells. (•) Mean ± SEM RVD response recorded in six cells. External Ca2+ was restored at the interval delimited by the parallel dashed lines. The time at which external Ca2+ was restored in each individual cell is signaled by larger shaded circles.
Other experiments were done (n = 3) in which [Ca2+]i was increased during osmotic swelling by adding 1 μM ionomycin in fura-2-loaded cells. The results were similar; i.e., no significant enhancement of RVD rate could be demonstrated. On the basis of these results, we suggest that Ca2+ is not a triggering signal for RVD in osmotically swollen cells, at least within the explored time window.
discussion
Calcium-dependent and Calcium-independent RVD
The nature of the signal transducing the change in cell volume to the activation of osmolyte efflux pathways for RVD in osmotically swollen cells is not clear. Two major models have been proposed, one involving Ca2+ as an intracellular second messenger and the other in which RVD is mediated through Ca2+-independent mechanisms. According to the first model, cell swelling causes an increase in cytosolic Ca2+ that activates volume regulatory mechanisms. The increase in [Ca2+]i may result from Ca2+ influx, via plasma membrane channels (Christensen, 1987), or Ca2+ release from intracellular stores (McCarty and O'Neil, 1992; Wu et al., 1997). The proposed effectors are mostly Ca2+-activated cation and anion channels and carriers. This model has been widely accepted as a paradigm for the involvement of Ca2+ as a transducing signal for RVD. Nevertheless, the existing evidence backing up this model is often weak and fragmentary, and the comprehensive analysis necessary to establish an active role of cytosolic Ca2+ for RVD is lacking in many of the cells in which it has been implicated (Foskett, 1994). One of the experimental facts that is missing in the work claiming a signaling role for Ca2+ in RVD is the simultaneous measurement of changes in [Ca2+]i and cell volume, and the demonstration that the Ca2+ signal is the cause of RVD and not just a parallel phenomenon. In the second model, RVD is effected by Ca2+-independent mechanisms (Cahalan and Lewis, 1988; Grinstein and Smith, 1990; Foskett et al., 1994; Lippmann et al., 1995). According to this model, hyposmotic swelling opens anion channels, causing Cl− efflux with consequent cell membrane depolarization towards the equilibrium potential for Cl−. This depolarization activates voltage-gated K+ channels and increases the driving force for K+ loss. The end result is the electrically coupled exit of K+ and Cl−, accompanied by water. The volume-activated anion channels may be sufficiently nonselective to permit efflux of organic anions, as well as inorganic anions such as Cl− and HCO3 − (Strange et al., 1996). The mechanism whereby these anion channels are activated upon cell swelling remains to be elucidated. However, with the exception of a recent study in human neuroblastoma cells (Basavappa et al., 1995), there is general agreement that Ca2+ is not the activating signal of these anion channels (Doroshenko and Neher, 1992; Strange et al., 1996). In addition to channel-mediated solute efflux, there are carrier-mediated efflux pathways such as electroneutral KCl cotransport that can be activated in swollen cells in the virtual absence of Ca2+ (Thornhill and Laris, 1984; Kramhøft et al., 1986).
Unlike previous reports, in the present study we measured changes in CWV and [Ca2+]i simultaneously in the same cells. We found that in single N1E-115 neuroblastoma cells and in NG108-15 hybrid neuroblastoma × glioma cells, there is a close parallel between osmotic swelling, increase in [Ca2+]i, and RVD. The extent and rate of rise of [Ca2+]i and RVD were proportional to the degree of medium hypotonicity and the change in cell volume. Moreover, the correlation between the increase in cell volume, the initiation of the rise of [Ca2+]i, and subsequent [Ca2+]i changes, and the onset of RVD show the appropriate time sequence to suggest that a rise in [Ca2+]i might, in principle, mediate RVD. However, cells bathed in Ca2+-free EGTA solutions, loaded with both the Ca2+ chelator BAPTA and the Ca2+ indicator fura-2, showed a robust RVD without a concomitant rise in [Ca2+]i. Moreover, the persistent RVD occurred even when [Ca2+]i decreased below resting levels. Thus, no cause–effect relation exists between the rise in intracellular Ca2+ and the RVD response. These results directly prove for the first time that if a single process underlies RVD, a rise in [Ca2+]i is not the triggering signal. The notion that Ca2+ plays no role in the activation of the normal RVD response has been suggested by other investigators based on less direct evidence from experiments on cell populations in which no simultaneous measurements of intracellular Ca2+ and volume were made (e.g., Jørgensen et al., 1997). Cell populations may or may not be homogeneous and may or may not respond synchronously to osmotic challenges. Therefore, experiments in cell populations do not allow reliable inferences and definitive conclusions about what is happening in single cells, particularly when the relevant variables (changes in cell volume and [Ca2+]i) are not recorded simultaneously.
The rate and extent of the RVD responses that persisted after Ca2+ chelation were reduced with respect to control responses. External and internal Ca2+ chelation may attenuate the RVD response through secondary effects; e.g., membrane depolarization leading to a decreased driving force for Cl− efflux or side effects of BAPTA such as intracellular acidification or loss of K+ and Cl− (Jørgensen et al., 1997). Alternatively, the results could be the consequence of the operation of two parallel processes activated by osmotic swelling, one of which is Ca2+ independent. In this model, the rate and extent of RVD would be affected by Ca2+ chelation simply by the elimination of the Ca2+-sensitive component. The role of Ca2+ in the latter component is not clear at this time. It could be that a certain resting level of intracellular (and extracellular) Ca2+ may be necessary for optimal RVD responses, as has been suggested for a medullary thick ascending limb cell line derived from rabbit kidney (Montrose-Rafizadeh and Guggino, 1991). According to this view, Ca2+ might play a permissive role, for example acting as a cofactor or maintaining cell integrity, rather than acting as a second messenger.
Ca2+ Regulatory Mechanisms in Anisosmotic and Isosmotic Media
Our conclusion for a Ca2+-independent RVD is strengthened by the fact that the rate of cell volume recovery was not enhanced by restoring external Ca2+ within an interval of 8–14 min after RVD onset. Readmission of external Ca2+ always resulted in an increase in [Ca2+]i, indicating that the lack of effect of external Ca2+ readdition on the rate of RVD was not due to block of Ca2+ entry during the regulatory response. Similar results were obtained by adding the Ca2+ ionophore ionomycin (1 μM) during RVD (not shown). These experiments do not disprove that an increase in [Ca2+]i could have a role at earlier times during the RVD response, as has been suggested for cell populations of renal proximal tubules (McCarty and O'Neil, 1992). Unfortunately, we cannot reintroduce Ca2+ at earlier times and have a reliable measurement of the change in RVD rates because there is not sufficient sampling time to assess the change in the slope. The experiment is also not feasible using a two-hyposmotic-challenges protocol because the second response exhibits inactivation. Interestingly, we (Crowe et al., 1995) and others (Hoffmann et al., 1984; Foskett et al., 1994) have shown that elevating [Ca2+]i in cells maintained in isosmotic medium produces cell shrinkage. On the basis of these results, we suggest that in addition to the Ca2+-independent volume-regulatory machinery that is activated by cell swelling in hyposmotic media, cells also possess Ca2+-activated transport pathways that result in cell shrinkage in isosmotic media.
Ca2+ Signals in Osmotically Swollen Cells Loaded with Fura-2
The apparent increase in [Ca2+]i upon cell swelling produced by hyposmotic solutions could be due to a change in the K d of fura-2 for Ca2+ that accompanies a decrease in internal ionic strength (Uto et al., 1991), rather than a genuine increase in [Ca2+]i. Such a change in internal ionic strength might be expected to accompany a dilution of the internal constituents with the hyposmotic challenge, but we have no independent measure of such changes. We have two arguments to suggest that the monitored Ca2+ signals are not spurious. First, we note that they disappear when the cells are pretreated with BAPTA (Fig. 8). This argument is somewhat weakened by the fact that the BAPTA might essentially remove the free internal Ca2+ so that the contention that the fura-2 affinity for Ca2+ becomes higher is irrelevant since there would not be sufficient [Ca2+]i to react with the dye. However, the argument becomes stronger for the case in which external Ca2+ was chelated with EGTA. In these cells, as in those loaded with BAPTA, the apparent [Ca2+]i does not increase upon osmotic swelling, but actually decreases as a consequence of intracellular dilution (Fig. 10). The second argument is the demonstration that the Ca2+ signal is often absent in the second of two identical hyposmotic challenges even though the dilution of the cell contents is the same as in the first hyposmotic challenge, which always results in a transient increase in intracellular Ca2+ (Fig. 6). The role, if any, of the rise of [Ca2+]i observed during osmotic swelling in the presence of extracellular Ca2+ is not clear at this time. This change in [Ca2+]i may be a modulating influence or just an epiphenomenon in the RVD process.
A Signaling Role for Ca2+ in RVD?
Four pieces of evidence are usually given to propose an active role for Ca2+ as the signal coupling cell volume expansion to RVD responses (McCarty and O'Neil, 1992; Foskett, 1994). Since these arguments are crucial for the interpretation of the results of the present study, it is worth examining them. First, abolition of RVD when a cell is swollen with a Ca2+-free hyposmotic solution containing chelators is often given as proof of a signaling role for Ca2+ (e.g., O'Connor and Kimelberg, 1993). However, chelation in conjunction with the osmotic shock is known to depolarize cells (Berman et al., 1994). In addition, the osmotic challenge results in dilution of cell contents. All these factors, individually or in concert, could alter the driving forces for ion efflux or shift the activation curves for voltage-sensitive channels involved in RVD as a consequence of changes in surface potential. On the other hand, persistence of RVD upon external Ca2+ removal does not exclude the possibility of an osmotically induced release of Ca2+ from intracellular stores (McCarty and O'Neil, 1992), which could be acting as the second messenger.
The second argument generally offered to propose a signaling role for Ca2+ on RVD is the disappearance of the response upon external and internal Ca2+ chelation with EGTA and BAPTA, respectively (e.g., Adorante and Cala, 1995). The conclusions from this kind of experiment must also be treated with caution for the same reasons discussed for external Ca2+ chelation. Conversely, persistence of RVD in cells loaded with a Ca2+ chelator has been interpreted as proof of a Ca2+-independent RVD (Lippmann et al., 1995). However, unless the adequacy of internal Ca2+ chelation is verified, as was done in the present study, this conclusion is not warranted.
The third piece of evidence often given as proof for Ca2+-dependent RVD is the block of the response with Ca2+ entry blockers. In particular, these include blockers of voltage-gated Ca2+ channels (e.g., dihydropyridines) or blockers of mechanogated channels (e.g., gadolinium). However, such blockers may have effects on mechanisms other than those involved in Ca2+ entry (Foskett, 1994; Sánchez-Olea et al., 1995; Hamill and McBride, 1996).
The fourth argument usually given in favor of the Ca2+ hypothesis for RVD signaling is that hyposmotic swelling induces an increase in [Ca2+]i. Although a necessary logical condition for postulating a signaling role for Ca2+, it must be proved that the temporal relation between the change in [Ca2+]i and the onset of RVD are appropriate and, more importantly, that the Ca2+ increase is necessary for RVD and not just a parallel phenomenon.
Some investigators (e.g., Hoffmann et al., 1984; Cala, 1986) have suggested that swelling causes a change in the Ca2+ affinity of a putative intracellular element of the RVD machinery, and that therefore a rise in Ca2+ is not necessary for Ca2+ to act as a trigger of the RVD response. First, if there was such a change in the Ca2+ sensitivity of some element of the RVD machinery, then the second messenger or triggering signal would not be Ca2+, but the change in affinity for Ca2+ of this putative element of the RVD machinery. Second, we think that this possibility is unlikely to explain our results because [Ca2+]i decreases below resting levels, yet hyposmotic challenges elicit vigorous RVD responses.
Inactivation of RVD upon Sequential Hyposmotic Challenges
We show that the RVD response and the concomitant increase in [Ca2+]i were abolished or significantly attenuated upon the second of two identical hyposmotic challenges applied with an interval of 30–60 min. We demonstrated that the RVD inactivation persists in the nominal absence of external Ca2+ and is independent of changes in internal Ca2+. Moreover, we show that inactivation is independent of the dye used to measure cell volume changes. At least three explanations can be offered for these observations. First, if the RVD is mediated by mechanogated channels, then it is conceivable that these channels adapt and do not recover in time for the second hyposmotic challenge (Hamill and McBride, 1994). If there are mechanogated channels present in neuroblastoma cells that mediate Ca2+ influx in addition to those mediating volume regulatory osmolyte effluxes (Falke and Misler, 1989), then both processes (i.e., the rise in intracellular Ca2+ and RVD) would decrease in parallel without being interdependent. Second if RVD is mediated by the efflux of internal solutes from a finite store, then the apparent inactivation could represent the depletion of that store (Larson and Spring, 1984). Third, inactivation could be due to damage of RVD machinery as a consequence of excessive mechanical stress (Hamill and McBride, 1997). We have recently shown that RVD inactivation can be reversed by loading the cells with taurine before the second hyposmotic challenge. On the basis of this observation, we suggest that inactivation is the consequence of depletion of some internal osmolyte store such as taurine (Brodwick et al., 1997).
In summary, our simultaneous measurements of intracellular Ca2+ and volume in single neuroblastoma cells directly demonstrate that an increase in intracellular Ca2+ is not necessary for triggering RVD. In addition, we demonstrate that the RVD response “inactivates” upon repeated challenges, and that this process is also independent of Ca2+. Our results unify conflicting claims in the literature, based on indirect measurements in cell populations, by showing that: (a) RVD can be independent of a rise in intracellular Ca2+, and (b) there is a component of the response that is sensitive to Ca2+ chelation. The role of Ca2+ in the Ca2+- dependent component, if any, is not clear at this time. Ca2+ chelation may attenuate RVD through secondary effects. Nevertheless, it is clear that RVD itself can be triggered without a rise of [Ca2+]i in the neuronal cell lines investigated in this paper.
Acknowledgments
The authors thank Drs. Luis Reuss and Simon Lewis for their thoughtful comments on the manuscript, Mrs. Lynette Durant and Alicia Maldonado for secretarial work, Mr. José R. Fernández, Mr. Sergio Márquez Baltazar, and Miss Carrie Preite for skilled technical assistance, and Dr. William E. Crowe for participating in some experiments. Both cell lines used in this work were kindly provided by Dr. Marshall Nirenberg, Department of Biochemical Genetics, National Heart, Lung and Blood Institute (Bethesda, MD).
This work was supported by the National Institute of Neurological Disorders and Stroke grant NS29227, and National Council of Science and Technology (Mexico) grant F-285-N9209 to F.J. Alvarez-Leefmans.
Abbreviations used in this paper
- AM
acetoxymethyl ester
- BAPTA
1,2-bis-(o -aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid
- CWV
cell water volume
- RVD
regulatory volume decrease
- SES
standard external solution
references
- Adorante JS, Cala PM. Mechanisms of regulatory volume decrease in nonpigmented human ciliary epithelial cell. Am J Physiol. 1995;268:C721–C731. doi: 10.1152/ajpcell.1995.268.3.C721. [DOI] [PubMed] [Google Scholar]
- Altamirano J, Brodwick MS, Alvarez-Leefmans FJ. Regulatory volume decrease is independent of internal calcium changes in N1E115 and NG108 cells. Biophys J. 1996;70:A413. . (Abstr.) [Google Scholar]
- Alvarez-Leefmans FJ, Gamiño SM, Reuss L. Cell volume changes upon sodium pump inhibition in Helix aspersaneurones. J Physiol (Camb) 1992;458:603–619. doi: 10.1113/jphysiol.1992.sp019436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Altamirano J, Crowe WE. Use of ion-selective microelectrodes and fluorescent probes to measure cell volume. Methods Neurosci. 1995;27:361–391. [Google Scholar]
- Amano T, Richelson E, Nirenberg M. Neurotransmitter synthesis by neuroblastoma clones. Proc Natl Acad Sci USA. 1972;69:258–263. doi: 10.1073/pnas.69.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavappa S, Chartouni V, Kirk K, Prpic V, Ellory C, Mangel AW. Swelling-induced chloride currents in neuroblastoma cells are calcium dependent. J Neurosci. 1995;15:3662–3666. doi: 10.1523/JNEUROSCI.15-05-03662.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DM, Peña-Rasgado C, Rasgado-Flores H. Changes in membrane potential associated with cell swelling and regulatory volume decrease in barnacle muscle cells. J Exp Zool. 1994;268:97–103. doi: 10.1002/jez.1402680205. [DOI] [PubMed] [Google Scholar]
- Brodwick MS, Altamirano J, Alvarez-Leefmans FJ. Inactivation of the regulatory volume decrease in neuronal cell lines examined with fluorescent probes. Soc Neurosci Abstr. 1997;23:647. . (Abstr.) [Google Scholar]
- Cahalan, M.D., and R.S. Lewis. 1988. Role of potassium and chloride channels in volume regulation by T lymphocytes. In Cell Physiology of Blood. R.B. Gunn and J.C. Parker, editors. The Rockefeller University Press, New York. 281–301. [PubMed]
- Cala PM, Mandel LJ, Murphy E. Volume regulation by Amphiumared blood cells: cytosolic free Ca and alkali metal-H exchange. Am J Physiol. 1986;250:C423–C429. doi: 10.1152/ajpcell.1986.250.3.C423. [DOI] [PubMed] [Google Scholar]
- Churchwell KB, Wright SH, Emma F, Rosenberg PA, Strange K. NMDA receptor activation inhibits neuronal volume regulation after swelling induced by veratridine-stimulated Na+influx in rat cortical cultures. J Neurosci. 1996;16:7447–7457. doi: 10.1523/JNEUROSCI.16-23-07447.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen O. Mediation of cell volume regulation by Ca2+influx through stretch-activated channels. Nature. 1987;330:66–68. doi: 10.1038/330066a0. [DOI] [PubMed] [Google Scholar]
- Crowe WE, Altamirano J, Huerto L, Alvarez-Leefmans FJ. Cell volume changes in single N1E-115 neuroblastoma cells measured with a fluorescent probe. Neuroscience. 1995;69:283–296. doi: 10.1016/0306-4522(95)00219-9. [DOI] [PubMed] [Google Scholar]
- Crowe WE, Altamirano J, Alvarez-Leefman FJ. Monitoring changes in [Ca2+]iand cell volume using fura-2. Soc Neurosci Abstr. 1996;22:323. . (Abstr.) [Google Scholar]
- Doroshenko P, Neher E. Volume-sensitive chloride conductance in bovine chromaffin cell membrane. J Physiol (Camb) 1992;449:197–218. doi: 10.1113/jphysiol.1992.sp019082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke LC, Misler S. Activity of ion channels during volume regulation by clonal N1E115 neuroblastoma cells. Proc Natl Acad Sci USA. 1989;86:3919–3923. doi: 10.1073/pnas.86.10.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett, J.K. 1994. The role of calcium in the control of volume-regulatory transport pathways. In Cellular and Molecular Physiology of Cell Volume Regulation. K. Strange, editor. CRC Press, Boca Raton, FL. 259–277.
- Foskett JK, Wong MMM, Sue-A-Quan G, Robertson MA. Isosmotic modulation of cell volume and intracellular ion activities during stimulation of single exocrine cells. J Exp Zool. 1994;268:104–110. doi: 10.1002/jez.1402680206. [DOI] [PubMed] [Google Scholar]
- Grinstein S, Smith J. Calcium-independent cell volume regulation in human lymphocytes. J Gen Physiol. 1990;95:97–120. doi: 10.1085/jgp.95.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, McBride DW. Molecular mechanisms of mechanoreceptor adaptation. NIPS (News Physiol Sci) 1994;9:53–59. [Google Scholar]
- Hamill OP, McBride DW. The pharmacology of mechanogated membrane ion channels. Pharmacol Rev. 1996;48:231–252. [PubMed] [Google Scholar]
- Hamill OP, McBride DW. Induced membrane hypo/ hyper-mechanosensitivity: a limitation of patch-clamp recording. Annu Rev Physiol. 1997;59:621–631. doi: 10.1146/annurev.physiol.59.1.621. [DOI] [PubMed] [Google Scholar]
- Hamprecht B. Structural, electrophysiological, biochemical and pharmacological properties of neuroblastoma-glioma hybrid cells in culture. Int Rev Cytol. 1977;49:99–170. doi: 10.1016/s0074-7696(08)61948-8. [DOI] [PubMed] [Google Scholar]
- Haugland, R.P. 1996. Handbook of Fluorescent Probes and Research Chemicals. 6th edition. Molecular Probes, Inc., Eugene, OR. 679 pp.
- Hoffmann EK, Simonsen LO. Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev. 1989;69:315–382. doi: 10.1152/physrev.1989.69.2.315. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Simonsen LO, Lambert IH. Volume-induced increase of K+ and Cl− permeabilities in Ehrlich ascites tumor cells. Role of internal Ca2+ . J Membr Biol. 1984;78:211–222. doi: 10.1007/BF01925969. [DOI] [PubMed] [Google Scholar]
- Jørgensen NK, Christensen S, Harbak H, Brown AM, Lambert IH, Hoffmann EK, Simonsen LO. On the role of calcium in the regulatory volume decrease (RVD) response in Ehrlich mouse ascites tumor cells. J Membr Biol. 1997;157:281–299. doi: 10.1007/s002329900236. [DOI] [PubMed] [Google Scholar]
- Kasai H, Neher E. Dihydropyridine-sensitive and ω-conotoxin-sensitive calcium channels in a mammalian neuroblastoma-glioma cell line. J Physiol (Camb) 1992;448:161–188. doi: 10.1113/jphysiol.1992.sp019035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, H.E. 1995. Objective lenses for confocal microscopy. In Handbook of Biological Confocal Microscopy. J.B. Pawley, editor. Plenum Publishing Corp., New York. 111–126.
- Kramhøft B, Lambert IH, Hoffmann EK, Jørgensen F. Activation of Cl-dependent K transport in Ehrlich ascites tumor cells. Am J Physiol. 1986;251:C369–C379. doi: 10.1152/ajpcell.1986.251.3.C369. [DOI] [PubMed] [Google Scholar]
- Larson M, Spring KR. Volume regulation by Necturusgallbladder: basolateral KCl exit. J Membr Biol. 1984;81:219–232. doi: 10.1007/BF01868715. [DOI] [PubMed] [Google Scholar]
- Lippmann BJ, Yang R, Barnett DW, Misler S. Pharmacology of volume regulation following hypotonicity-induced cell swelling in clonal N1E115 neuroblastoma cells. Brain Res. 1995;686:29–36. doi: 10.1016/0006-8993(95)00447-x. [DOI] [PubMed] [Google Scholar]
- McCarty NA, O'Neil RG. Calcium signaling in cell volume regulation. Physiol Rev. 1992;72:1037–1061. doi: 10.1152/physrev.1992.72.4.1037. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Guggino WB. Role of intracellular calcium in volume regulation by rabbit medullary thick ascending limb cells. Am J Physiol. 1991;260:F402–F409. doi: 10.1152/ajprenal.1991.260.3.F402. [DOI] [PubMed] [Google Scholar]
- Muallem S, Zhang B-X, Loessberg PA, Star RA. Simultaneous recording of cell volume changes and intracellular pH or Ca2+concentration in single osteosarcoma cells UMR-106-01. J Biol Chem. 1992;267:17658–17664. [PubMed] [Google Scholar]
- O'Connor ER, Kimelberg HK. Role of calcium in astrocyte volume regulation and in the release of ions and amino acids. J Neurosci. 1993;13:2638–2650. doi: 10.1523/JNEUROSCI.13-06-02638.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasantes-Morales H, Maar TE, Morán J. Cell volume regulation in cultured cerebellar granule neurons. J Neurosci Res. 1993;34:219–224. doi: 10.1002/jnr.490340209. [DOI] [PubMed] [Google Scholar]
- Pasantes-Morales H, Murray RA, Lilja L, Morán J. Regulatory volume decrease in cultured astrocytes. I. Potassium- and chloride-activated permeability. Am J Physiol. 1994;266:C165–C171. doi: 10.1152/ajpcell.1994.266.1.C165. [DOI] [PubMed] [Google Scholar]
- Sánchez-Olea R, Morales-Mulia M, Morán J, Pasantes-Morales H. Inhibition by dihydropyridines of regulatory volume decrease and osmolyte fluxes in cultured astrocytes is unrelated to extracellular calcium. Neurosci Lett. 1995;193:165–168. doi: 10.1016/0304-3940(95)11691-o. [DOI] [PubMed] [Google Scholar]
- Serve G, Endres W, Grafe P. Continuous electrophysiological measurements of changes in cell volume of motoneurones in the isolated frog spinal cord. Pflügers Arch. 1988;411:410–415. doi: 10.1007/BF00587720. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol. 1996;270:C711–C730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Thornhill WB, Laris PC. KCl loss and cell shrinkage in the Ehrlich ascites tumor cell induced by hypotonic media, 2-deoxyglucose and propranolol. Biochim Biophys Acta. 1984;773:207–218. doi: 10.1016/0005-2736(84)90084-1. [DOI] [PubMed] [Google Scholar]
- Uto A, Arai H, Ogawa Y. Reassessment of fura-2 and the ratio method for determination of intracellular Ca2+concentrations. Cell Calc. 1991;12:29–37. doi: 10.1016/0143-4160(91)90082-p. [DOI] [PubMed] [Google Scholar]
- Wu X, Yang H, Iserovich P, Fischbarg J, Reinach PS. Regulatory volume decrease by SV 40-transformed rabbit corneal epithelial cells requires ryanodine-sensitive Ca2+-induced Ca2+release. J Membr Biol. 1997;158:127–136. doi: 10.1007/s002329900250. [DOI] [PubMed] [Google Scholar]