

Abstract
The rate constants of acetylcholine receptor channels (AChR) desensitization and recovery were estimated from the durations and frequencies of clusters of single-channel currents. Diliganded-open AChR desensitize much faster than either unliganded- or diliganded-closed AChR, which indicates that the desensitization rate constant depends on the status of the activation gate rather than the occupancy of the transmitter binding sites. The desensitization rate constant does not change with the nature of the agonist, the membrane potential, the species of permeant cation, channel block by ACh, the subunit composition (ε or γ), or several mutations that are near the transmitter binding sites. The results are discussed in terms of cyclic models of AChR activation, desensitization, and recovery. In particular, a mechanism by which activation and desensitization are mediated by two distinct, but interrelated, gates in the ion permeation pathway is proposed.
Keywords: single-channel, kinetics, electrophysiology
introduction
Acetylcholine receptors (AChR)1 are ion channels that open transiently after binding two agonist molecules. In the continuous presence of agonist, AChR become refractory to the stimulus and the cellular response declines. This process, called desensitization, occurs because AChR adopt liganded, stable conformations through which ions cannot permeate (reviewed by Ochoa et al., 1989; Scuka and Mozrzymas, 1992). At the vertebrate neuromuscular junction, desensitization is slow and may not play a significant role in shaping the endplate current or in synaptic depression. However, currents generated by other synaptic receptors often decline rapidly and desensitization is likely to be an important determinant of the amplitude, time-course, and stability of these responses. It is therefore of some physiological importance to understand the molecular events that constitute the desensitization of AChR and other synaptic receptor channels.
Katz and Thesleff (1957) observed that at the frog neuromuscular junction, the steady application of ACh virtually abolished the endplate response within seconds, but upon the removal of the agonist, sensitivity recovered rapidly. Because there was no detectable depolarization during recovery, they proposed a cyclic model for AChR activation and desensitization: agonists bind, AChR open and then “desensitize,” and upon washout agonists dissociate and AChR return to their resting condition without reopening. A cyclic reaction scheme implies that unliganded AChR can desensitize.
The affinity of desensitized AChR for agonists (Weber et al., 1975; Boyd and Cohen, 1980; Sine and Taylor, 1982) is ∼10,000-fold higher than that of resting AChR (Akk and Auerbach, 1996; Wang et al., 1997), but may be similar to that of open AChR (Colquhoun and Sakmann, 1985). It is important to note that “desensitization” describes a host of inactivation phenomena that may arise from a spectrum of molecular and cellular processes. There are multiple components to the desensitization time course (Heidmann and Changeux, 1979). The main component that was first studied in detail by Katz and Thesleff (1957) occurs on a time scale of seconds, but faster (milliseconds; Sakmann et al., 1980; Magleby and Palotta, 1981; Dilger and Brett, 1990) and slower (minutes; Feltz and Trautman, 1982; Chestnut, 1993) components have been identified. Here, we focus on the component that occurs on the 0.1–1-s time scale.
Several electrophysiological studies have been done regarding the kinetics of this component of AChR desensitization and recovery. Cachelin and Colquhoun (1989; frog muscle) confirmed the cyclic reaction mechanism and speculated that desensitization occurs exclusively from the diliganded, open conformation. They proposed that the rate limiting step to recovery upon washout is the agonist-independent isomerization of the receptor. Dilger and Liu (1992; mouse BC3H1 cells) found that desensitization closely paralleled the open probability of the channel and used the cyclic scheme to estimate molecular rate constants for the desensitization of open AChR (20 s−1) and the recovery of unliganded AChR (3 s−1). Franke et al. (1993; single-channel currents from embryonic mouse muscle) concluded that the recovery from desensitization is rate limited by agonist dissociation rather than an agonist-independent conformational change. They observed that the probability of opening during washout was extremely low, ∼10−4.
Despite a wealth of information on the phenomenology of AChR desensitization, the molecular basis of the reaction remains mysterious. It is not known whether desensitization reflects a global change in the protein structure or more local changes in the conformation of the binding site and/or pore domains. While the above-mentioned kinetic studies have shown that diliganded receptors desensitize faster than vacant receptors, the rate constants for the desensitization of diliganded open vs. closed receptors are not known. This distinction is significant because it illuminates whether desensitization depends on the occupancy of the binding sites or the status of the activation gate. Mutagenesis experiments have not clarified this issue because desensitization is altered by mutations to both binding site residues (Sine et al., 1994) and pore residues (Revah et al., 1991; Weiland et al., 1996; Kuryatov et al., 1997; Milone et al., 1997).
Structural correlates of AChR desensitization have not been clearly identified. Torpedo AChR have been imaged at 9 Å resolution in both the closed (Unwin, 1993) and open (Unwin, 1995) conformations, but only an 18-Å map of desensitized AChR is currently available (Unwin et al., 1988). In this low resolution map, the extracellular domain of the δ subunit is seen to be tilted tangentially as a consequence of exposure to carbamylcholine for several minutes. Given that desensitization occurs over minute as well as second time scales, it is likely that the electron diffraction patterns of desensitized Torpedo AChR reflect the slower components of inactivation. Fast inactivation of voltage-gated channels has been attributed to a two-gate (“ball and chain”) mechanism (Armstrong et al., 1973; Hoshi et al., 1990), but in AChR it is not known whether the functional distinctions between “closed” and desensitized AChR reflect multiple conformations of a single gate, or different dispositions of multiple gates within the pore.
At the single-channel level, desensitization is manifest as a clustering of channel opening events (Sakmann et al., 1980). Long-lived closed intervals between the clusters reflect times when all AChR in the patch are desensitized. A cluster starts when one AChR recovers from desensitization, and continues with the protein molecule undergoing many cycles of agonist association/dissociation and channel gating. Here, we report desensitization onset and recovery rate constants from the duration and frequencies of single-channel clusters recorded from adult mouse recombinant AChR. The results indicate that the desensitization rate constant is faster when the activation gate is open, and is not a function of the occupancy of the binding sites.
We propose a model in which AChR activation and desensitization reflect the activity of two separate, but interrelated, gates in the ion permeation pathway. In unliganded-closed AChR, the activation gate is usually closed and the desensitization gate is usually open. Binding agonists initiates an allosteric transition (i.e., a global change in structure) in which the binding sites adopt a high-affinity conformation and the activation gate opens. When the activation gate is open, the desensitization gate can close more readily. This configuration (activation gate open and the desensitization gate closed) is very stable. In the two-gate mechanism, the high affinity of a desensitized AChR is simply a consequence of being locked into an activated, but nonconducting, conformation. The recovery process requires agonist dissociation, closing of the main activation gate, and reopening of the desensitization gate. This mechanistic model, which involves only local interactions between the two gates, accounts quantitatively for the phenomenology of AChR desensitization and recovery.
methods
Expression Systems and Electrophysiology
Mouse muscle type nicotinic AChR subunit cDNAs (α, β, δ, ε, or γ) were from the laboratories of Drs. John Merlie and Norman Davidson, and were subcloned into a CMV promoter-based expression vector pcDNAIII (Invitrogen Corp., San Diego, CA). The “wild-type” α subunit differed from the sequence in the GenBank database (accession X03986) and had an alanine, rather than a valine, at position 433 (Zhou et al., 1998).
AChR were expressed in human embryonic kidney (HEK) 293 cells using transient transfection based on calcium phosphate precipitation (Ausubel et al., 1992). For muscle type receptors, a total of 3.5 μg DNA per 35-mm culture dish in the ratio 2:1:1:1 (α:β:δ:ε or γ) was used. The DNA was added to the cells for 12–24 h, after which the medium was changed. Electrophysiological recordings were started 24 h later.
Electrophysiology was performed using the patch clamp technique in the cell-attached configuration (Hamill et al., 1981). The bath was Dulbecco's PBS containing (mM): 137 NaCl, 0.9 CaCl2, 2.7 KCl, 1.5 KH2PO4, 0.5 MgCl2, 6.6 Na2HPO4, pH 7.3. The pipette solution typically contained (mM): 115 NaCl or 142 KCl, 1.8 CaCl2, 1.7 MgCl2, 5.4 NaCl, 10 HEPES, pH 7.4. In some experiments, the concentration of KCl was reduced without replacement. In addition, the pipette solution contained the indicated concentration of ACh or other agonist. All experiments were performed at 22–24°C.
Kinetic Analysis
The details of the kinetic analysis methods are described in Akk et al., 1996. Currents were digitized at 94 kHz (VR-10 and VR-111; Instrutech Corp., Great Neck, NY) and were digitally low-pass filtered (Gaussian) using a cutoff frequency (f c) of 2–7 kHz. Lists of open- and closed-current interval durations were generated via a half amplitude threshold crossing criterion. Clusters were defined as a series of openings separated by closed intervals shorter than some critical duration (τcrit). An interval duration histogram of all closures was compiled and fitted by the sum of two to four exponentials. The values of the time constants varied with the concentration of ACh, the expression level of the receptor, the patch area, and in some cases the analysis bandwidth. Typically, there was a fast component (∼20–50 μs; sensitive to the bandwidth), an intermediate component that predominated (50–0.05 ms; sensitive to the ACh concentration), and a small, variable, slow component (0.5–5 s; sensitive to the number of AChR in the patch). The initial guess of τcrit was more than four times the time constant of the predominant component, or 5 ms, whichever was longer. Clusters were defined accordingly, and an interval duration histogram of all intracluster closures was compiled and fitted by the sum of two to three exponentials. If necessary, the value of τcrit was adjusted and the process repeated. The value of τcrit depended on the type of receptor, the agonist and its concentration, and the expression levels, but was always at least four times longer than the slowest intracluster closed interval duration component. For example, with wild-type, adult AChR, in 1 μM ACh the time constant of the slowest intracluster closed interval component was ∼80 ms and τcrit was 400 ms, while in 100 μM ACh, the slowest component was ∼0.1 ms and τcrit was 5 ms.
The errors associated with cluster definition were not measured for each patch. However, approximate errors can be estimated using typical values for the amplitudes and time constants of the intermediate (a 1 and τ1) and slow (a 2 and τ2) components of the closed interval duration histogram of the entire record. We define r a = a 1/a 2, r τ = τ1/τ2, and x = τcrit /τ1. As shown by Jackson et al. (1983), the fraction of all closed intervals misclassified as being between, rather than within, clusters is a 1 e−x and the fraction of all closed intervals misclassified as being within, rather than between, clusters is
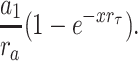 |
These errors will be largest when the agonist concentration is low and the number of AChR in the patch is large. A typical result for a condition with expected large errors (5 μM ACh) was r a = 10, r τ = 0.01, and x = 5. Under these conditions, only ∼0.7% of the intracluster events and 0.5% of the intercluster events would be misclassified. Moreover, the effect of these misclassifications on the cluster duration and open probability would tend to offset, so the net error in these parameters would be even smaller.
The cluster duration was defined as the time between the first opening and last closing transition. To select for clusters with homogeneous amplitude and kinetic properties, as well as to eliminate isolated openings from the accounting, only clusters >100 ms in duration were included in the calculation of the average cluster duration. If cluster durations are exponentially distributed with an inverse time constant λ, the relationship between the mean duration τapp and λ is:
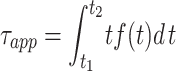 |
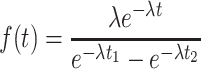 |
where f (t) is the conditional probability density function, t 1 is the minimum cluster duration, and t 2 is the maximum cluster duration included in the average. Assuming no upper limit on the cluster duration, the true cluster duration τc (= λ−1) is:
 |
1 |
Therefore, to correct for the minimum cluster duration requirement, 100 ms was subtracted from the apparent mean cluster duration for each patch.
The probability of being open within a cluster (P o) was calculated from intracluster events as the sum of open interval durations divided by the sum of both open and closed interval durations. To insure that an equal number of open and closed intervals contributed to the P o estimate, the last open interval in the cluster was excluded from this accounting. A mean value of τc and P o was calculated for each patch.
Analysis of Models
Typically, the activation of an AChR requires the association of an agonist molecule to each of the two transmitter binding sites, followed by a concerted channel gating event. These events are encoded in the standard model for AChR activation (del Castillo and Katz, 1957; Magleby and Stevens,  (model i)1972):
(model i)1972):
where A is the agonist concentration, k+1 and k+2 are the agonist association rate constants, k−1 and k−2 are the agonist dissociation rate constants, β is the channel opening rate constant of a diliganded receptor, and α is the channel closing rate constant of a diliganded receptor.
Steady state occupancy probabilities (P) as a function of the agonist concentration calculated according to Model I are:
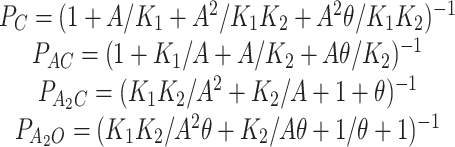 |
2 |
where K 1 and K 2 are the receptor equilibrium dissociation constants, and θ is the gating equilibrium constant (β/α). If the two transmitter binding sites have approximately the same equilibrium dissociation constant (K d), then Eq. 2 can be simplified with K 1 = 0.5 K d and K 2 = 2 K d. Although many studies show that the equilibrium dissociation constants for the two sites are markedly different for some antagonists, analyses of adult mouse AChR indicates that the K ds for ACh are nearly equivalent at the two sites (Akk and Auerbach, 1996; Wang et al., 1997).
With this simplification, the occupancy probabilities can be related to the probability of being open within a cluster:
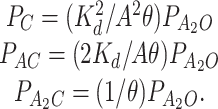 |
3 |
Linear fits were done using Origin (Microcal Software, Northampton, MA). Interval duration histograms and dose-response profiles were fit using NFIT (Island Software, Galveston, TX). The optimization of the rate constants for recovery from desensitization upon washout (see Fig. 9) was carried out by solving the differential equations for the reaction using Scientist (MicroMath, Salt Lake City, UT). Fitted parameters are reported as mean ± SD. Eq. 7 was derived using the symbolic math program Maple (Waterloo Maple, Inc., Waterloo, Ontario, Canada).
Figure 9.

The time course of recovery from desensitization. The points are from Dilger and Liu (1992) and were obtained using a two-pulse protocol. The first pulse of 100 μM ACh desensitized virtually all AChR in the patch. After a delay, the second pulse was applied to test the fraction of AChR that had recovered from desensitization. The abscissa is the interpulse interval (note the logarithmic scale) and the ordinate is the fractional recovery. The solid line is for the optimal values of Model IV (allosteric model). The initial step of recovery, the dissociation of agonist from desensitized AChR, occurs with a rate constant of ∼23 s−1 (per transmitter binding site), and the final, agonist-independent step of recovery occurs with a rate constant of ∼4 s−1.
Mutant AChR
The α subunit mutants shown in Table II were a kind gift from Dr. Steven Sine (Mayo Foundation, Rochester, MN). The ε subunit mutants were made using overlap PCR as described in Higuchi (1990). The final construct was completely sequenced in the region between the ligation sites.
Table II.
Effect of Binding Site Mutations on AChR Desensitization
| AChR | k O +D | n | θ | |||
|---|---|---|---|---|---|---|
| s−1 | ||||||
| Wild-type, adult | 3.5 | 4 | 45.0 | |||
| Wild-type, fetal* ‡ | 4.6 | 5 | 240.0 | |||
| αW149F | 3.0 | 5 | 0.7 | |||
| αY93F | 1.9 | 4 | 0.7 | |||
| αY93W | 3.4 | 5 | 0.4 | |||
| αY198F | 3.4 | 10 | 10.0 | |||
| αN217K§ | 3.7 | 3 | 19.0 | |||
| αG153S‖ | 3.6 | 6 | 51.0 | |||
| αD200N | 4.4 | 3 | 0.1 | |||
| εD175N | 2.2 | 3 | 0.6 | |||
| εE184I | 4.5 | 4 | 0.8 | |||
| εE184A | 2.2 | 4 | 30.0 |
115 mM NaCl in the extracellular solution,
coexpressed with a γ subunit,
activated by carbamylcholine, and
activated by tetramethylammonium. Each of the mutations has been shown to influence the rate constants for agonist association, agonist dissociation, and/or channel opening. k O +D is the diliganded AChR desensitization rate constant, computed from (τc P o)−1, n is the number of patches, and θ is the gating equilibrium constant (opening/closing). The mutations have no significant effect on the desensitization rate constant. (142 mM KCl, −100 mV, adult type (ε subunit) activated by ACh, except where noted.
Drugs
All reagents, including acetylcholine chloride, carbamylcholine chloride, and tetramethylammonium iodide were purchased from Sigma Chemical Co. (St. Louis, MO).
results
Desensitization Versus the Agonist Concentration
Fig. 1 shows example clusters elicited by 20 μM ACh. In this patch, there were 63 clusters >100 ms in duration, and the apparent mean cluster duration was 590 ms. After applying a correction for the minimum cluster duration, the mean cluster duration estimate, τc, was 490 ms. The distribution of cluster durations was fitted by a single exponential function with a characteristic time constant of 513 ± 64 ms. There was a reasonably good agreement between the corrected mean cluster duration and the time constant obtained by fitting the distribution.
Figure 1.

Distribution of cluster durations from one patch. A cluster occurs when a single AChR spontaneously recovers from desensitization, and a cluster ends when that receptor again becomes desensitized. Three example clusters are shown (inward current is down). The distribution of cluster durations is described by a single exponential with a time constant of 513 ms. In this patch, there were 63 clusters longer than 100 ms in duration and the mean probability of being open in a cluster was 0.47. The desensitization rate constant, (τc P o)−1, was 3.9 s−1 (−100 mV; 20 μM ACh; 115 mM NaCl).
Most patches had too few clusters to allow fitting of the cluster duration distribution, thus τc was used as the estimate of the cluster duration time constant. There was substantial scatter in the τc estimates, in part because of the small number of clusters measured in each patch. However, some of the variance may arise from nonstatistical reasons, as almost every quantitative electrophysiological study of AChR desensitization has noted considerable variance in the parameters (see Bufler et al., 1993).
Fig. 2 shows the properties of clusters elicited by 1-500 μM ACh. The probability of being open within a cluster increases with the ACh concentration because the time required to bind agonist decreases, leading to shorter closed interval durations. Over the same concentration range, τc decreases ∼100-fold. However, the product τc P o remains relatively constant, with an average value of 285 ms.
Figure 2.

Cluster properties vs. the concentration of ACh. (top) Example clusters at three different ACh concentrations. (bottom) Between 1 and 100 μM ACh, the probability of being open within a cluster increases and the corrected mean cluster duration (τc) decreases. However, the product of these parameters remains nearly constant at ∼300 ms. The mean desensitization rate constant (τc P o)−1 is 3.5 s−1. Each point is the mean of more than two patches, with 5–63 clusters per patch (mean = 27).
The constancy of the τc P o product with respect to the ACh concentration suggests that receptors desensitize primarily from a diliganded state. To quantify the extent to which states of Model I serve as gateways to desensitized states, the inverse of τc at different ACh concentrations was plotted as a function of the probability a receptor occupies unliganded, monoliganded, and diliganded states (Fig. 3). These probabilities were computed according to Model I (Eq. 2) using the salient equilibrium constants: K d ≅ 100 μM (in 115 mM NaCl) and θ ≅ 50 (at −100 mV).
Figure 3.

The effective desensitization rate as a function of the number of bound ACh molecules. Each symbol is a patch. For each patch, an occupancy probability in unliganded, monoliganded, and diliganded states was calculated from the ACh concentration, the equilibrium dissociation constant (100 μM in 115 mM NaCl and 160 μM in 142 mM KCl), and the gating equilibrium constant according to Model I (see methods). The desensitization rate constant for un- and diliganded AChR was estimated by linear extrapolation to unity occupancy. Diliganded AChR (open plus closed) desensitize at 3.7 s−1 while unliganded AChR desensitize much more slowly, at <0.1 s−1.
In general, the cluster duration distribution will have as many components as states within a cluster. However, if transitions between these states are fast compared with desensitization, the distribution will tend towards a single exponential with:
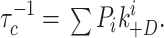 |
4 |
where P i is the steady state probability of occupying state i and ki +D is the desensitization rate constant for that state. Thus, if a state is the predominant outlet to a desensitized state, then the inverse of the cluster duration should increase approximately linearly with occupancy of that state, with a proportionality constant ki +D. Moreover, for each class of AChR, the extrapolated value of τc −1 at P i = 1 is an estimate of the intrinsic desensitization rate constant from that class.
Fig. 3 shows that with ACh as the agonist, the effective desensitization rate is positively correlated with the occupancy of diliganded states, confirming the previously established result that desensitization occurs mainly from diliganded states. From these data, very little can be deduced about desensitization from monoliganded states, as these are occupied with only a small probability. The P values for un- and diliganded AChR span a wide range, allowing estimates of k +D obtained by extrapolation to P = 1 for these classes. For the diliganded points, the fitted straight line has slope of 3.5 ± 0.5 s−1 and an ordinal intercept of 0.24 ± 0.60 s−1, yielding an extrapolated k A2O +D for diliganded receptors of 3.7 ± 1.1 s−1. For the unliganded points, the fitted straight line slope of −3.6 ± 0.9 s−1 and an ordinal intercept of 3.5 ± 0.3 s−1, yielding an extrapolated value of kC +D (unliganded AChR) = −0.1 ± 1.2, which is indistinguishable from zero. The values are scattered and the desensitization rate constants estimated from this analysis are not precise. However, from this analysis we conclude that diliganded AChR desensitize much more rapidly than unliganded receptors.
Desensitization from Open and Closed Diliganded States
Although it is clear that the desensitization of diliganded AChR is relatively fast, the desensitization rate constant of open vs. closed diliganded AChR have not been separately estimated. That is, it is possible that desensitization occurs rapidly from the rarely occupied A2C state, as was assumed by Naranjo and Brehm (1993), or more slowly from the frequently occupied A2O state, as was assumed by Cachelin and Colquhoun (1989), Dilger and Liu (1992), and Franke et al. (1993). Making this distinction is important because it illuminates whether it is the number of bound agonists or the status of the activation gate that influences the desensitization rate constant.
To make this separation, the inverse of τc P o was plotted as a function of the gating equilibrium constant (θ = β/α, from Model I). Because desensitization occurs mainly from diliganded AChR, we combine Eqs. 3 and 4 to produce:
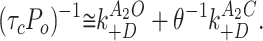 |
5 |
By examining the relationship between the product τc P o (measured on a cluster-by-cluster basis) and θ(estimated from dose-response curves or from single-channel kinetic analysis), the values of k A2O +D and k A2C +D can be separately estimated and a determination can be made whether diliganded closed and/or open states are outlets to desensitized states.
For wild-type, adult mouse AChR activated by ACh, θ ≅ 50 (Sine et al., 1995; Wang et al., 1997); i.e., the fractional occupancy of the diliganded closed state is ∼0.02 that of the open state. Three experimental manipulations were used to change θ: different agonists, different membrane potentials, and mutations (see Table II). With these manipulations, cluster durations and open probabilities could be examined over a wide range of θ values.
Fig. 4 shows that the inverse of the τc P o product is approximately independent of the gating equilibrium constant, θ. The fit of these data by Eq. 5 yields k A2O +D = 3.18 ± 0.33 s−1 and k A2C +D = −0.08 ± 0.21 s−1. The desensitization rate constant for diliganded closed AChR, like that of unliganded closed AChR, is much slower than that of diliganded open AChR and is statistically indistinguishable from zero.
Figure 4.

The desensitization rate constant, (τc P o)−1, for diliganded AChR does not change with the gating equilibrium constant (θ = β/α; see Model I). θ was varied experimentally by using different receptors, agonists, and membrane potentials. The line is the fit by Eq. 5 with k A2O +D = 3.13 s−1 and k A2C +D = −0.08 s−1. Over a ∼1,000-fold range in θ, the slope of the line is indistinguishable from zero, indicating that diliganded-open AChR desensitize much faster than diliganded-closed AChR. This suggests that desensitization is a function of the status of the activation gate rather than the occupancy of the binding sites. Each symbol is the average value for a α2βδε receptor (n patches) (wt [11], αY93F [15], αW149W [4], αG153S [5], εD175N [3], αY198F [10], εE181Q [5], εE184A [4], and I [4]; see Table II) activated under a variety of experimental conditions of agonist (ACh, TMA, CCh), membrane potential (−50, −75, −100, and −130 mV, ) and extracellular salt solution (115 mM NaCl, 140 mM KCl). A total of 61 patches are represented in the plot.
We conclude that the value of (τc P o)−1 is a direct measure of the rate constant of desensitization of diliganded open AChR. To obtain a more global estimate of desensitization and recovery (in the steady presence of agonist) rate constants, (τc P o)−1 and the cluster frequency were measured in 61 patches (115 NaCl or 142 mM KCl in the pipette) activated by 2–1,000 μM ACh (Fig. 5). Although the values are scattered, the main population of patches is centered around values of k A2O +D = 4.6 s−1. There was even greater scatter in the recovery rate for diliganded receptors mainly because this parameter is a linear function of the number of AChR in the patch. Nonetheless, there was a predominant population of cluster frequencies centered around 0.08 s−1. Because we studied cell-attached patches, we could not estimate the number of AChR in each patch. In outside-out patches from embryonic mouse muscle, Franke et al. (1993) found that there were 10–20 AChR per patch. If we assume that in our experiments there is an average of ∼10 AChR in a cell-attached patch, then the recovery rate constant (in the continuous presence of agonist) of a single AChR is ∼0.01 s−1; i.e., that it takes ∼2 min for a diliganded AChR to recover from desensitization.
Figure 5.

Average cluster properties. Solid lines are Gaussians fitted only to the indicated range of bins. (left) The average desensitization rate constant (τc P o)−1 of the main population is 4.6 s−1. (right) The average cluster frequency of the main population is 0.078 s−1. This value divided by the number of channels in the patch (∼10) is an estimate of the rate constant for recovery from desensitization for diliganded AChR (53 patches; 2–1,000 μM ACh).
To summarize, the results presented thus far indicate that desensitization mainly proceeds from a single outlet state, A2O, with a rate constant of ∼4 s−1, and a diliganded AChR recovery rate constant of ∼0.01 s−1.
Effects Of Agonists, Voltage, Ions, and Channel Block
Because desensitization occurs mainly from diliganded, open receptors, k A2O +D (from now on called simply k O +D) can be readily estimated as (τc P o)−1; i.e., without detailed knowledge of the activation rate constants. The value of k O +D was determined for AChR activated by carbamylcholine or tetramethylammonium. AChR activated by these ligands open ∼10× slower than those activated by ACh (Zhang et al., 1995). The results (Table I) indicate that the desensitization rate constant does not vary significantly between these agonists.
Table I.
Effect of Agonists on the Desensitization Rate Constant of Diliganded AChR
| θ | τc P o | k O +D | n | |||||
|---|---|---|---|---|---|---|---|---|
| ms | ||||||||
| Acetylcholine (50 μM) | 45.0 | 222 ± 65 | 4.5 ± 1.0 | 4 | ||||
| Carbamylcholine (200 μM) | 5.1 | 320 ± 98 | 3.1 ± 0.7 | 7 | ||||
| Tetramethylammonium (200 μM) | 5.4 | 312 ± 83 | 3.2 ± 0.6 | 6 |
θ is the gating equilibrium constant (opening/closing), τc P o is the corrected cluster duration multiplied by the probability of being open in the cluster, and k O +D is the inverse of this value, which equals the desensitization rate constant for diliganded AChR. The desensitization rate constant does not vary between these agonists. All experiments in 142 mM KCl, −100 mV.
The properties of the mutant receptor αY93F desensitization were examined at four different voltages. The voltage dependence of the opening and closing rate constants have been determined for this mutant (Auerbach et al., 1996). Fig. 6 shows that for these mutant AChR the product k O +D is approximately constant between −55 and −130 mV. The intrinsic rate constant of AChR desensitization is not sensitive to the membrane potential.
Figure 6.

Voltage dependence of AChR desensitization. Each point is the diliganded AChR desensitization rate constant, (τc P o)−1, for a single patch (αY93F AChR; four patches). The desensitization rate constant of diliganded AChR does not change significantly between −30 and −130 mV.
We next investigated whether the ionic composition of the current influences desensitization. In wild-type AChR (Vm = −100 mV), k O +D was 5.2 ± 0.6 s−1 in 115 NaCl (n = 21), 3.3 ± 0.6 s−1 in 140 KCl (n = 7), and 5.7 ± 1.6 s−1 in 140 mM CsCl (n = 5). With regard to these monovalent species, the extracellular ionic composition does not have a significant effect on AChR desensitization.
At millimolar concentrations, ACh enters the pore region and transiently (<20 μs) occludes the flow of Na+ and K+ until it unbinds (Sine and Steinbach 1984; Ogden and Colquhoun, 1985; Maconichie and Steinbach, 1995). The effects of occupancy of the pore by a channel blocker, ACh, are shown in Fig 7. Under our experimental conditions, channel block by ACh is too fast to be resolved as discrete gaps and is instead manifest as a reduction in the mean open channel current amplitude and an increase in the apparent open channel lifetime. The effect of channel block by ACh was examined in an αY93F AChR. The affinity of the pore of this mutant for ACh was estimated from the single-channel current amplitudes (i) at different ACh concentrations (A): i = i 0/(1 + A/K block), where i 0 is the current amplitude in the absence of blockade and K block is the equilibrium dissociation constant for the ACh-pore interaction. At a membrane potential of −100 mV, K block = 1.9 mM. τc P o was measured at this voltage in αY93F AChR activated by 0.5–8.0 mM ACh. In Fig. 7 (bottom right), the desensitization rate constant is plotted as a function of the fractional occupancy of the pore by ACh (f ): f = (1 + K block/A)−1.
Figure 7.

Channel block by ACh does not influence AChR desensitization. Each symbol is from one patch (αY93F AChR, −100 mV). (top) Example clusters. (bottom left) The single channel amplitude is reduced at high concentrations because of flickery channel block by the agonist. The equilibrium dissociation constant for ACh block is 1.9 mM. (bottom right) The desensitization rate constant plotted as a function of the fractional occupancy of the pore by the blocker. Channel block by ACh does not influence the desensitization rate constant.
Although the data are scattered, over the fractional occupancy range of 0.18–0.77 there is no change in the desensitization rate constant (mean = 2.3 s−1). Thus, the occupancy of the pore by ACh apparently does not affect k O +D. AChR desensitize from either the blocked or unblocked states with essentially the same rate constant.
Effect of Subunit Composition and Mutations
Embryonic AChR contain a γ subunit in place of the ε subunit that is present in adult-type receptors (Mishina et al., 1986). In six patches with embryonic-type AChR (5–100 μM ACh, −100 mV), the mean value of k O +D was 4.6 ± 1.6 s−1 (range = 3.3–8.0 s−1). This value is not significantly different than the desensitization rate constant of adult-type AChR, indicating that the γ vs. ε subunit does not have a significant effect on k O +D.
Several AChR having mutations near the binding site were examined, and the results are shown in Table II. All of the mutations (on the α and ε subunits) lowered the gating equilibrium constant, usually by slowing the channel opening rate constant, and many increased the equilibrium dissociation constant for ACh. However, none of the mutations had a measurable effect on the desensitization rate constant. That k O +D is neither agonist dependent nor sensitive to mutations that otherwise alter binding and gating suggests that conformational changes at the binding sites are not rate limiting to the desensitization of diliganded AChR.
discussion
The most significant experimental finding is that desensitization occurs much faster when the AChR activation gate is open compared with when it is closed. The molecular rate constant for the desensitization of closed AChR is slow for both unliganded and diliganded species, which suggests that in itself the occupancy of the binding sites is essentially irrelevant to the desensitization process. We conclude that experimental manipulations that alter the macroscopic desensitization rate, such as the agonist concentration, membrane potential, temperature, and certain binding site mutations, do so by changing processes that influence the probability that the AChR activation gate is open, rather than the desensitization rate constant per se.
We emphasize that our experiments and analyses only address the molecular mechanism of the component of desensitization that occurs on the second time scale, and that the physical bases of faster and slower components of desensitization may be quite distinct from those we propose.
Phenomenological Model
Under our experimental conditions, un- and monoliganded open states can be ignored because they are occupied with a low probability. In addition, we have found that desensitization rarely occurs from diliganded, closed receptors. For a useful, if phenomenological, model for AChR operation (modified from Katz and Thesleff, 1957; Cachelin and Colquhoun, 1989), see Model II. A is the agonist and C, O, and D represent closed, open, and desensitized AChR, respectively, k
+ and k− are the agonist association/dissociation rate constants for a closed AChR, β and α are the opening/closing rate constants for diliganded AChR, k
O
+D and k
O
−D are the desensitization/recovery rate constants for diliganded-open AChR, j
+ and j− are the agonist association/dissociation rate constants for a desensitized AChR, and k
C
+D and k
C
−D are the desensitization/recovery rate  (model ii)constant for vacant-closed AChR.
(model ii)constant for vacant-closed AChR.
The following equilibrium constants can be defined: K is the equilibrium dissociation constant of the closed conformation (= k −/k +), J is the equilibrium dissociation constant of the desensitized conformation (= j−/j +), θ is the channel gating equilibrium constant (= β/α), D o is the desensitization equilibrium constant of the doubly liganded, open conformation (= k O +D/k O −D), and D c is the desensitization equilibrium constant of the vacant, closed conformation (= k C +D/k C −D).
There are two paths by which AChR can recover from desensitization: a diliganded receptor can directly reopen (A2D ↔ A2O; equilibrium constant D O), or the agonist can dissociate and the protein can return to the closed, resting state (D ↔ C; equilibrium constant D C). From Model II, and assuming detailed balance:
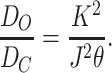 |
6 |
For recombinant mouse adult-type AChR: K = 160 μM (in 142 mM KCl; Akk and Auerbach, 1996) and J = 0.04 μM (Sine et al., 1995; from binding profiles of proadifen-treated receptors). At −100 mV, θ ≅ 50 (Sine et al., 1995; Akk and Auerbach, 1996). Therefore, from Eq. 6, D O/D C = 3.2 × 105.
D O can be estimated from the kinetic parameters. The rate constant for the A2O → A2D transition is 4 s−1. The affinity of desensitized AChR is high and in the continuous presence of agonist the long closed intervals between clusters reflect mainly the A2D → A2O recovery pathway. We estimate that the rate constant for this process is ∼0.01 s−1. From the ratio of these rate constants, D O ≅ 400. In the presence of a high concentration of agonist, an AChR is desensitized ∼99.8% of the time.
With this value for D O, we use Eq. 6 to calculate that D C ≅ 0.0013, which is only approximately four times larger than the estimate obtained from fitting reaction schemes to binding profiles (Sine et al., 1995; D C = 3 × 10−4) and is close to the value obtained from electrophysiological measurements of embryonic mouse muscle AChR (Franke et al., 1993; D C = 10−3). In the absence of ACh, we estimate that only ∼1 receptor in ∼700 is desensitized.
We can convert these equilibrium constants into free energy differences using the relationship ΔG 0 = −RT ln(K ). When the channel is open and diliganded, desensitization produces a net stabilization of the system of −6.0 k B T. When the channel is unliganded, the recovery from desensitization produces a net stabilization of the system of −6.7 k B T. Desensitization has nearly opposite energetic consequences when the activation gate is open compared with when it is closed.
Other workers have used a double-pulse protocol to measure the time course of AChR recovery from desensitization (Katz and Thesleff, 1957; Cachelin and Colquhoun, 1989; Dilger and Liu, 1992; Franke et al., 1993). Upon the removal of acetylcholine, A2D receptors return to the C state with a time constant of ∼300 ms (see Fig. 9). There are no channel opening events during the interpulse interval, indicating that in the absence of agonist, recovery is essentially exclusively via the D → C transition. For the reaction sequence for recovery in the absence of agonist,  (model iii)see Model III.
(model iii)see Model III.
According to this scheme, the recovery time course in the absence of agonist (A2D to C) should be the sum of three exponential components. Under the condition that kC +D << kC −D:
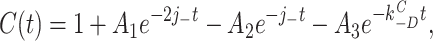 |
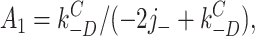 |
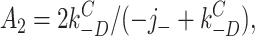 |
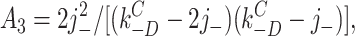 |
7 |
where C(t) is the fraction of AChR in state C at time t. The experimentally determined recovery time course (Dilger and Liu, 1992) was fitted by Eq. 7. The results, shown as the solid line in Fig. 9, were j − = 23.0 ± 4.1 s−1 and kC −D = 4.2 ± 0.4 s−1. Dissociation is only five times faster than the agonist-independent recovery step, and both processes contribute to the recovery time course.
The desensitization and recovery parameters for recombinant AChR are very similar to those for AChR expressed in BC3H1 cells (Dilger and Liu, 1992). This demonstrates that desensitization is determined by factors that are intrinsic to the AChR pentameric complex. Our estimate of the A2O → A2D rate constant (4 s−1) is significantly lower than the estimates of Dilger and Liu (1992) and Franke et al. (1993; embryonic mouse muscle) who measured this rate constant to be ∼20 s−1. This difference cannot be traced to the ε vs. γ subunit difference between the preparations because adult and fetal AChR show similar desensitization kinetics. It is possible that the difference may arise from a difference between outside-out and cell-attached patches, or that posttranslational events can influence AChR desensitization and may differ in native and human embryonic kidney expression systems.
We can estimate the ACh association rate constant to desensitized AChR from the equilibrium dissociation constant (40 nM) and the dissociation rate constant (∼20 s−1). The association rate constant of ACh to desensitized AChR is fast, ∼5 × 108 M−1 s−1. This value is similar to the ACh association rate constant for dansyl-C6-choline to desensitized Torpedo AChR at 0°C (108 M−1 s−1; Heidmann and Changeux, 1979) as well as to nondesensitized (low affinity binding sites) adult mouse AChR (108 M−1 s−1; Akk and Auerbach, 1996). We conclude that desensitization hardly changes the association of ACh to the transmitter binding sites. The increase in affinity that accompanies desensitization is almost completely due to an ∼800-fold decrease in the ACh dissociation rate constant. This indicates that when the binding sites are in their high affinity configuration, each ACh molecule is ∼6.7 k B T more stable than when the binding sites are in their low affinity configuration.
The optimal rate constants of the phenomenological model of AChR activation, desensitization, and recovery rate constants (142 KCl, −100 mV, 22°C, ACh) are shown (model iv) in Model IV.
(model iv) in Model IV.
The reaction free energies according to this scheme are summarized in Fig. 8. The A2D state is −27.3 kT more stable than the resting C state. During recovery, AChR transiently pass through D, which is the only state that is less stable than the resting state. The reaction diagram shows that the desensitization step, A2O → A2D, and the recovery step, D → C, are each accompanied by a net stabilization of the system even though they are functionally inverse processes.
Figure 8.

Energetics of the phenomenological model for AChR activation, desensitization, and recovery. (top) The cyclic reaction (C, closed; O, open; and D, desensitized). For simplicity, the two agonist binding steps have been condensed. The numbers are ΔG O values (k B T ) and were calculated from the ratios of the rate constants shown in Model IV. The sign of the ΔG O value pertains to the clockwise direction. (bottom) Graphical representation of the reaction free energies of the states. Unliganded closed is the ground state and [ACh] = 1 M. The A2D state is the most stable, and only the D state is less stable than the C state. Desensitization has opposite energetic consequences in diliganded (stabilizes) and unliganded (destabilizes) AChR.
Mechanistic Models
The term “desensitization” is a phenomenological one and does not imply any particular physical mechanism for AChR inactivation. In kinetic models of AChR operation, the classification of a state as being ‘C' or ‘O' relates to the conductance status of the pore, which in turn reflects the main allosteric transition of the protein. However, the classification of ‘D' makes no particular physical reference. In this section, we interpret the kinetic results using specific physical models for AChR desensitization.
Activation of AChR is a global change in the structure of the protein that includes rotation of helices at the transmitter binding sites and movement of residues in the pore domain (Unwin, 1995). The functional correlates of this event are a substantial decrease in the dissociation rate of ACh from the transmitter binding sites and a change from a nonconducting to a conducting pore. It is possible that desensitization reflects another such global change in the structure of the protein. Accordingly, the k C ±D and k O ±D rate constants of Model IV describe the rate constants for this additional, global transition.
If there is only one gate, desensitization can be thought of as a change in the coupling between the binding sites and the pore because the solitary gate closes without an accompanying increase in the dissociation rate constant of ACh. If this were so, the kinetic results indicate such interruption occurs readily only when the gate is open (A2O → A2D), but is reestablished readily only when the binding sites are empty (D → C). Moreover, the results indicate that desensitization is an energetically favorable transition when the sites are liganded, but is unfavorable when they are empty.
Certain evidence supports the allosteric hypothesis for AChR desensitization. High resolution electron microscopy reveals only a single structural element in the AChR channel that might serve as a gate (Unwin, 1993), although this barrier has not been detected by cysteine-scanning mutagenesis (Akabas et al., 1994). In addition, low resolution images of profoundly desensitized Torpedo AChR show a change in the tangential tilt of the extracellular domain of the δ subunit, suggesting that a desensitization is accompanied by a large-scale change in the AChR structure (Unwin et al., 1988).
The observation that activated AChR desensitize much faster than resting AChR recalls the two-gate, “ball and chain” inactivation mechanism of some voltage-gated channels (Hoshi et al., 1991), in which an inactivation gate (i.e., a tethered blocking particle, or ball) prevents ion permeation presumably by interacting with residues in the pore that become exposed when the activation gate (which is coupled to the voltage sensor) is open. The simplest form of such a two-gate inactivation mechanism cannot pertain to AChR because neither the relatively rapid recovery from desensitization upon the dissociation of ligand from the binding sites, nor the desensitization of vacant-closed receptors, is accompanied by the formation of an open channel intermediate.
Given this precedent, it is also worth considering a variant of a two-gate mechanism that would account for the phenomenology of AChR desensitization. The observations that the desensitization rate constant is not sensitive to events at the binding sites (occupancy, mutations, agonists), while the recovery rate constant is (being 400× faster when they are empty), motivates us to couple the activity of the inactivation gate with that of the activation gate, which is tightly coupled to the binding sites. Another attraction of the two-gate mechanism is that it could have evolved by the incremental addition of local interactions in the pore, rather than by the imposition of a second global conformational change in the protein. Although an allosteric mechanism for AChR desensitization is a logical possibility, the remainder of this discussion will be devoted to analysis according to a two-gate mechanism.
A Two-Gate Model
A two-gate model for desensitization is outlined in Fig. 10. There are three elements in a two-gate scheme: the occupancy status of the binding sites, the disposition of an activation (A) gate, and the disposition of a desensitization (D) gate. The standard AChR reaction notation must be modified to encode the status of two gates, each of which can be either open or closed. Below, we use C or O to indicate the status of either gate. The two gates are noted in brackets, with the upper letter referring to the A gate and the lower letter to the D gate. For example, in resting, nonconductive receptors, the A gate is closed and the D gate is open, so the notation is [C O]. For a channel to be conducting, both gates must be open (e.g., A2[O O]). In a diliganded, desensitized AChR, the A gate is open and the D gate is closed (A2[O C]).
Figure 10.

A two-gate model for AChR inactivation. The horizontal black bars represent the activation (A) and desensitization (D) gates. The spring represents an interaction between the two gates, which can be ‘relaxed' (compressed) or ‘stressed' (stretched). The square represents the two transmitter binding sites in a low affinity conformation (K d = 160 μM in 140 mM KCl), and the circle represents the binding sites in a high affinity conformation (K d = 40 nM). The agonist is indicated as being positively charged. The pore is permeable only when both gates are open (upper right). The top row is the activation reaction. At part of the main allosteric transition, the binding sites change affinity, the A gate open/closes, the nature of the AD interaction changes. Below each figure is the state notation, where a bound transmitter molecule is indicated by an A and the status of the gates, closed (C ) or open (O), is given in brackets as [A gate D gate]. The large numbers on the outside of the reaction cycle are ΔG 0 values in k B T (sign pertains to the clockwise direction). The smaller numbers on the inside of the reaction cycle break down the ΔG 0 value into more elemental components. Unliganded AChR can adopt three distinct pore conformations, [O C], [C C], or [C O], that interconvert on the millisecond time scale (spontaneous openings, [O O], are extremely rare and have been omitted from the reaction). Unliganded AChR usually reside in the [C O] configuration (top left). Proceeding clockwise, activation consists of binding two molecules of ACh (condensed into a single reaction step for clarity) and opening the channel. In the main gating step, A opens and the AD interaction becomes stressed. AChR desensitize when D closes and the interaction relaxes. Upon washout of agonists, AChR recover via the sequence A2[O C] → [O C] → [C C] → [C O]. After ACh dissociates, A closes and the AD interaction is once again stressed. In the final recovery step, D opens and [C O] returns to its original relaxed condition.
In the two-gate scheme (see Model V), the activation and desensitization steps are identical to those in the one-gate model. Desensitization is a single kinetic step that reflects closing of the D gate. However, in the two-gate model the recovery reaction upon washout requires an explicit accounting of both gate positions. In the presence of ACh, most AChR are diliganded and desensitized; i.e., the binding sites are full, the A gate is open, and the D gate is closed (A2[O C]). When they have completely recovered, the binding sites are empty, the A gate is closed, and the D gate is open ([O C]). Thus, recovery from inactivation upon the removal of the agonist requires the resetting of all three components (opening D, closing A, and agonist dissociation) of the two-gate scheme.
If each of the three components of a two-gate mechanism is binary, there are six possible recovery paths that differ only in the order of events. However, the final step of recovery cannot be the dissociation of agonist because unliganded receptors desensitize. In addition, any reaction that requires the formation of a conducting intermediate (i.e., both gates open) during recovery can be excluded because there are no observed openings during recovery. Thus, the final step of recovery must be the reopening of the D gate. The complete recovery reaction consumes +27.3 k B T.
The first step of recovery could be either the dissociation of the agonist (with A open) or the closing of the A gate (with agonist bound). In the second case the receptor will oscillate between the two diliganded states (A2[O C] ↔ A2[C C]) even in the absence of applied agonist. Number of such oscillations is a function of T 0, the equilibrium constant of the reaction (opening/ closing), and k −2, the agonist dissociation rate constant with two bound agonists.
In the two-gate mechanism, it is assumed that only the status of the A gate determines the agonist dissociation rate constant, and that the status of the D gate per se does not influence the affinity of the binding sites. In a two-gate scheme, the high affinity of desensitized AChR is simply a reflection of the protein being locked in an activated, high-affinity, impermeant condition. If closing A is the first step of recovery, then the subsequent dissociation of two agonist molecules should have the same rate constants (k −2 = 36,000 s−1) and energetic consequences (ΔG 0 = +17.4 k B T, in 142 mM KCl) as during activation.
If the first step of recovery is A gate closure, detailed balance indicates that ΔG 0 for this step would be (−27.3 + 17.6 − 6.7) = −16.4 k B T. T 0 would be large, e +16.4 = 1.3 × 107, as would the ratio T 0/k −2 (>500). The protein would oscillate between the two diliganded, desensitized states for many minutes, and recovery would be hundreds of times slower than what is observed. Given these considerations, we reject the hypothesis that the first step of recovery is A gate closure, and conclude that the most probable recovery sequence is: agonists dissociate, A gate closes, D gate opens.
A reaction sequence that was discounted because it had a conducting intermediate was: agonists dissociate, D gate opens (forming [O O]), A gate closes. The intermediate, open-channel state is the unliganded open state, which is extremely short lived (Jackson, 1986). It is possible that a small amount of current from this specie was not detected either in the single-channel (Franke et al., 1993) or macroscopic (Katz and Thesleff, 1957; Dilger and Liu, 1992) currents, so this reaction path cannot be absolutely excluded.
Model V describes AChR activation, inactivation, and recovery via a two-gate mechanism.  (model v)
(model v)
The recovery path of the two-gate model has one more state than in the phenomenological scheme where two discrete events (closing A and opening D) are condensed into the single D → C transition. In the two-gate model, the step that is open/closing of A when the binding sites are empty and the D gate is closed is related to the main gating event of the phenomenological model; i.e., the open/closing of the A gate when the D gate is open and the binding sites are full (rate constants β and α; equilibrium constant θ). We therefore dub the rate constants for the [C C] ↔ [O C] step a 0 and b 0, and the equilibrium constant T 0 (= b 0/a 0).
Decomposing the Energetics of the Two-Gate Mechanism
To estimate ΔG 0 values for the underlying molecular events that contribute to each step of the reaction scheme shown in Fig. 10, we assume that each transition reflects changes in the status of independent domains. Accordingly, the ΔG 0 of a reaction step can be treated as the sum of free energy changes in each of three “domains”—the A gate (which is coupled to the transmitter binding sites and can be either open or closed), the D gate (open or closed), and the region of interaction between the A and D gates. For example, the transition A2C → A2O entails a change in the A gate (closed to open) plus a change in the interaction between gates ([C O] to [O O]).
The value of a rate constant can be influenced by a change in the transition state energy and/or the starting state energy. To simplify the language of the discussion, we will attribute a difference between rate constants to a difference in the free energies of the starting states. Specifically, we make the assumption that the difference in the desensitization rate constant between diliganded-open and vacant-closed AChR reflects the difference in the free energies of the A2[O O] and [C O] states.
The starting point of this analysis is the energetics of D gate closure. The desensitization rate constant for diliganded-open AChR is ∼4 s−1, while that for unliganded-closed AChR is ∼0.005 s−1. If we assume that this difference is due solely to a difference in the AD interaction energy, then the [C O] configuration of the gates is 6.7 k B T more stable than the [O O] configuration.
The decomposition of the steps of the two-gate model according to changes in discrete domains is given in Fig 10. Consider the step of the reaction that makes the pore conducting: A2C[C O] → A2[O O]. From the ratio of the channel opening and closing rate constants, we estimate that the ΔG 0 for this step is −3.7 k B T. This transition consists of a change in the AD interaction ([C O] → [O O] = +6.7 k B T) plus opening the A gate (for diliganded AChR), which by subtraction must equal −10.4 k B T. This is the intrinsic energy that results from opening A and changing the conformation of the two occupied binding sites.
Continuing clockwise in Model V, from the rate constants we estimate that the next step of the reaction, A2C[O O] → A2[O C], yields −6.0 k B T. This transition consists of two events: closing D and changing the AD interaction from [O O] to [O C] (which alone yields −6.7 k B T ). By subtraction, we estimate that the intrinsic energetic cost of closing D is only +0.7 k B T.
Finally, during recovery upon washout, after ACh dissociates four domain changes occur: the A closes (unliganded AChR), the AD interaction changes from [O C] to [C C], the D gate opens (−0.7 k B T ), and the AD interaction changes from [C C] to [C O]. Together, these events yield −6.7 k B T. By subtraction, we estimate that closing the A gate (unliganded AChR) yields −6.0 k B T. The final step of recovery, in which the D gate closes and the favorable AD interaction is reestablished, yields −7.4 k B T. The breakdown of the rate constants into the elemental free energy changes is shown in Fig. 10.
The equilibrium constants for the pentultimate step of recovery upon washout can be calculated from the elemental ΔG 0 values. In the [O C] ↔ [C C] transition, the A gate closes (unliganded, −6.0 k B T ), but the favorable AD interaction is lost (+6.7 k B T ). We estimate that the net energy of this step is +0.7 k B T, or that T 0 = e +0.7 = 2. In a diliganded, undersensitized AChR, the A gate opens ∼50× faster than it closes. In an unliganded, desensitized AChR, the A gate opens only about twice as fast as it closes.
Before proceeding further, we compare this result with other experimental results regarding spontaneous openings. From our ΔG 0 estimates, we can estimate the equilibrium constant for spontaneous openings. For a vacant AChR to become conducting (i.e., to open spontaneously), the A gate (unliganded AChR) must open (+6.0 k B T ) and the AD interaction must change from [C O] to [O O] (+6.7 k B T ). Thus, from these elemental ΔG 0 values, we predict that an equilibrium constant for spontaneous openings of ∼e −12.7 = 3.0 × 10−6. This value is only about five times higher than the measured value of this equilibrium constant in mouse muscle (6 × 10−7; Jackson, 1986). This is within a range that can be attributed to experimental errors (missed openings and/or an overestimate of the number of AChR per patch) and/or preparation differences.
Returning to the recovery reaction, if D0 = 6.1 × 10−4 and kC +D = 0.005 s−1, then kC −D = 8.2 s−1. The final recovery rate constant is about two times faster than in the one-gate model because in the two-gate scheme after ligand dissociates the protein oscillates between the [O C] and [C C] configurations. Only about half of this time is spent in the [C C] state and as a consequence the effective recovery rate (4 s−1) is about half of the recovery rate constant (8 s−1).
Although we can estimate the equilibrium constant for the [O C] ↔ [C C] transition, we are unable to estimate the rate constants for this step. Fitting the recovery time course (Fig. 9) using an extra reaction step with a fixed equilibrium constant (i.e., one additional free parameter) did not improve the significance of the fit. We speculate that the [O C] ↔ [C C] transitions are rapid and invisible on the time scale of the recovery data, in which the earliest measurement of recovery was at 30 ms. The rate constants a 0 and b 0 need not be very fast (i.e., ∼100 s−1) to have made the additional component absent from the experimental recovery time course.
The rate constants for AChR activation, inactivation, and recovery (142 KCl, −100 mV, 22°C, ACh) according to the two-gate me (model vi)chanism are:
(model vi)chanism are:
Although we have used use the term “favorable” to describe the AD interaction, we emphasize that the gate interaction free energies only relate to the change in the AD interaction when one of the gates opens or closes. We have no information as to whether two open (or closed) gates increase, or one open and one closed gate decreases, the free energy of the system.
Interpreting the Energetics of the Two-Gate Model
Opening and closing the D gate is essentially an energetically neutral event, with only a 0.7 k B T difference between these conditions. In the two-gate model of desensitization, the D gate is essentially oblivious to events at the transmitter binding sites and is mainly driven by local interactions between the gates. The modest difference in the energy of the D gate is in marked contrast to that of the A gate, which strongly (10.4 k B T ) prefers to be open when the binding sites are occupied, or closed (6.0 k B T ) when they are empty.
The difference in the driving force of the A gate with occupancy of the binding sites may be attributed to the energy of the ligand–protein interaction. Accordingly, we estimate that two molecules of ACh contribute ∼16.4 k B T (or 4.8 kcal/mol per molecule) towards the stability of the open A gate conformation. Presumably, this energy arises from more favorable ligand–protein contacts when the binding site assumes its high affinity structure.
A two-gate model of desensitization implies that in the absence of agonists the nonconducting AChR pore can be a dynamic structure. The unliganded AChR is impermeant to ions in three configurations ([C O], [C C], and [O C]) and is permeable only in the [O O] configuration. From the elemental free energies, we estimate that in the absence of agonists the [C O] state clearly predominates (P = 0.998) and has a mean lifetime ∼28 s. The doubly closed and doubly open configurations are occupied only rarely, with equilibrium probabilities of ∼5.4 × 10−4 and 3.4 ×10−6, respectively. Interestingly, the [O C] configuration is occupied a significant fraction of the time, with a probability of 0.0012.
The stability of a desensitized receptor is a function both of increased agonist-receptor interactions in the transmitter binding sites consequent to the change from low-to-high affinity as well as to local interactions between the two gates in the pore. The kinetics of recovery upon washout of agonist are determined by the energy required to disrupt both types of interaction; i.e., the dissociation of the agonists and the disruption of a stable pore configuration. The drive to become conducting (i.e., to open the A gate) and to recover from desensitization (i.e., to open the D gate) both come from the transmitter binding sites. When these sites are fully occupied by ACh, the protein strongly prefers to be in the A-open configuration and the stable AD interaction that kept the D gate open is broken. When the binding sites are empty, the protein strongly prefers to be in the A-closed configuration and the stable AD interaction that kept the D gate closed is broken.
Structural Correlates
High resolution electron microscopy suggests that a structure approximately midway through the M2, pore-lining segment might serve as the main activation gate (Unwin, 1993). In these experiments, Torpedo AChR were imaged only after millisecond exposure to ACh, thus slower changes in the structure of the pore, such as we would expect for the closing of the D gate, would not have been observed. In contrast to these results, cysteine-scanning mutagenesis indicates that, without application of ACh, residues near the cytoplasmic limit of the α subunit M2 segment are covalently labeled (Akabas et al., 1994). The crystallography and labeling results appear to conflict, as the traditional view of the AChR pore holds that residues below the activation gate should not be labeled in the absence of a stimulus that opens the channel.
The two-gate model offers a resolution to this conflict. In the two-gate scheme, an unliganded AChR can exist in one of four conformations: [O C], [C C], [C O], and [O O]. In the [O C] configuration, the A gate is open but the pore is nonconducting. Although this state is occupied with a fairly low probability (∼1%), the cysteine reagents modify covalently and the low occupancy in the absence of agonists would slow the labeling rate, but not the equilibrium labeling pattern of residues that lie between the two gates. Based on the steady state occupancy of the [O C] configuration in the absence of ligand, we estimate that the rate of labeling of these residues should be ∼800× slower in the absence of the agonist compared with when the AChR are conducting.
It is possible that labeling experiments and electron microscopy experiments probe distinct gates within the AChR pore. With a one-gate, allosteric model of desensitization, spontaneous openings could similarly account for the discrepancy between the crystallography and cysteine-labeling results. However, the steady state occupancy in an unliganded, conducting conformation is extremely low (0.0003%), and, all else being equal (i.e., no excess spontaneous openings induced by the cysteine mutation; but see Auerbach et al., 1996), would predict a labeling rate that is 300,000× slower in the absence of agonist than when the channel was conducting.
We propose the following as one possible physical description of AChR desensitization via a two-gate mechanism (Fig. 10). When the A gate is closed, an inactivation particle is held in a position that is out of the ion permeation pathway. The strength of this interaction is ∼6.7 k B T. When the A gate opens as part of the main allosteric transition of the protein, this anchor moves, the interaction is lost, and the inactivation particle becomes free to change its position. After ∼250 ms, the particle moves to a new location where it now prevents ion permeation. The slowness of this movement suggests that significant energy barriers impede the movement of the particle. Once the particle has reached its new location, the highly favorable interaction between the gates is reestablished.
Our experiments do not provide information regarding the residues that form the D gate and other elements of the two-gate mechanism. The main allosteric transition that is the opening and closing of the A gate involves widespread changes in the protein structure (Unwin, 1995) so the AD interaction could take place virtually anywhere. If our hypothesis regarding the cysteine-scanning results is correct, then we speculate that the closed D gate is formed, in part, by α subunit residues below the central leucine; e.g., beyond αT242, which is labeled in cysteine-scanning experiments the absence of applied agonists (Akabas et al., 1994).
Other evidence points to the cytoplasmic limb of M2 as a determinant of AChR desensitization, as mutations to this region influence desensitization onset and recovery. An S-to-F mutation in the human α4 nicotinic receptor subunit at position homologous to α248 (i.e., between L251 and T242) causes a nocturnal epilepsy and alters desensitization onset and recovery kinetics (Weiland et al., 1996; Kuryatov et al., 1997). A V-to-F mutation in human α1 subunit at position 249 causes a congenital myasthenic syndrome and dramatically stabilizes desensitized states (Milone et al., 1997). In our version of a two-gate mechanism, we have speculated that the A and D gates are in close proximity and only make local interactions. Therefore, we speculate that the residues that form the (closed) D gate and the structures that determine the AD interaction may be near the cytoplasmic end of the M2 segment.
Independent of any particular reaction mechanism, our experiments demonstrate that AChR desensitization occurs mainly from the diliganded, open conformation. We speculate that “activation” and desensitization might reflect the operation of two distinct, but interrelated structures within the pore that serve as gates to ion permeation. The two permeation barriers are most stable when they are in opposite functional states (i.e., one open and the other closed).
The one-gate allosteric model and the two-gate model both account quantitatively for the phenomenology of AChR desensitization. Structural information is needed to determine if desensitization reflects a global change in the receptor conformation, or is mediated by local interactions between distinct gates. There are multiple kinetic components to AChR desensitization, and different mechanisms might underlie these different components. Additional mutational, functional, and structural experiments are needed to further define the molecular events that constitute AChR desensitization.
Acknowledgments
This work was supported by National Institutes of Health grant NS-23513.
Abbreviation used in this paper
- AChR
acetylcholine receptors
Footnotes
This paper is dedicated to Professor Bernard Katz. We thank J. Dilger for providing the experimental results shown in Fig. 9, and M. Zhou for the αN217K and αG153S results shown in Table II. Claudio Grosman derived the cluster duration correction. We thank C. Grosman, D. Machonochie, S. Sine, C. Lingle, and F. Qin for comments on the manuscript. We thank Karen Lau for technical assistance.
references
- Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the α subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- Akk G, Auerbach A. Inorganic, monovalent cations compete with agonists for the transmitter binding site of nicotinic acetylcholine receptors. Biophys J. 1996;70:2652–2658. doi: 10.1016/S0006-3495(96)79834-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Sine S, Auerbach A. Binding sites contribute unequally to the gating of mouse nicotinic αD200N acetylcholine receptors. J Physiol (Camb) 1996;496:185–196. doi: 10.1113/jphysiol.1996.sp021676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, Bezanilla F, Rojas E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol. 1973;63:375–391. doi: 10.1085/jgp.62.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A, Sigurdson W, Chen J, Akk G. Different gating charge movements in doubly-, singly-, and unliganded acetylcholine receptors. J Physiol (Camb) 1996;494:155–170. doi: 10.1113/jphysiol.1996.sp021482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1992. Short Protocols in Molecular Biology. John Wiley & Sons, Inc., New York.
- Boyd ND, Cohen JB. Kinetics of binding of [3H]acetylcholine to Torpedo postsynaptic membranes: association and dissociation rate constants by rapid mixing and ultrafiltration. Biochemistry. 1980;19:5353–5358. doi: 10.1021/bi00564a032. [DOI] [PubMed] [Google Scholar]
- Bufler J, Franke C, Witzemann V, Ruppersberg JP, Merlitze S, Dudel J. Desensitization of embryonic nicotinic acetylcholine receptors expressed in Xenopusoocytes. Neurosci Lett. 1993;152:77–80. doi: 10.1016/0304-3940(93)90487-6. [DOI] [PubMed] [Google Scholar]
- Cachelin AB, Colquhoun D. Desensitization of the acetylcholine receptor of frog end-plates measured in a Vaseline-gap voltage clamp. J Physiol (Camb) 1989;415:159–188. doi: 10.1113/jphysiol.1989.sp017717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestnut TJ. Two-component desensitization at the neuromuscular junction of the frog. J Physiol (Camb) 1993;336:229–241. doi: 10.1113/jphysiol.1983.sp014578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol (Camb) 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo J, Katz B. Interaction at end-plate receptors between different choline derivatives. Proc R Soc Lond B Biol Sci. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- Dilger JP, Brett RS. Direct measurement of the concentration- and time-dependent open probability of the nicotinic acetylcholine receptor channel. Biophys J. 1990;57:723–731. doi: 10.1016/S0006-3495(90)82593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilger JP, Liu Y. Desensitization of acetylcholine receptors in BC3H-1 cells. Pflügers Arch. 1992;420:479–485. doi: 10.1007/BF00374622. [DOI] [PubMed] [Google Scholar]
- Feltz A, Trautmann A. Desensitization at the frog neuromuscular junction: a biphasic process. J Physiol (Camb) 1982;322:257–272. doi: 10.1113/jphysiol.1982.sp014036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke C, Parnas H, Hovav G, Dudel J. A molecular scheme for the reaction between acetylcholine and nicotinic channels. Biophys J. 1993;64:339–356. doi: 10.1016/S0006-3495(93)81374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Heidmann T, Changeux JP. Fast kinetic studies on the interaction of a fluorescent agonist with the membrane-bound acetylcholine receptor from . Torpedo marmorata Eur J Biochem. 1979;94:255–279. doi: 10.1111/j.1432-1033.1979.tb12893.x. [DOI] [PubMed] [Google Scholar]
- Higuchi, R. 1990. Recombinant PCR. In PCR Protocols. M. Innis, D. Gelfand, J. Sninsky, and T. White, editors. Academic Press, Inc., San Diego, CA. 177–183.
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shakerpotassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophys J. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB, Wong BS, Morris CE, Lecar H, Christian CN. Successive openings of the same acetylcholine receptor channel are correlated in open time. Biophys J. 1983;42:109–114. doi: 10.1016/S0006-3495(83)84375-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Thesleff S. A study of the ‘desensitization' produced by acetylcholine at the motor end-plate. J Physiol (Camb) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machonichie DJ, Steinbach JH. Block by acetylcholine of mouse muscle nicotinic receptors, stably expressed in fibroblasts. J Gen Physiol. 1995;106:113–147. doi: 10.1085/jgp.106.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL, Palotta BS. A study of desensitization of acetylcholine receptors using nerve-released transmitter in the frog. J Physiol (Camb) 1981;316:225–250. doi: 10.1113/jphysiol.1981.sp013784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL, Stevens CF. A quantitative description of end-plate currents. J Physiol (Camb) 1972;223:173–197. doi: 10.1113/jphysiol.1972.sp009840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor alpha subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature. 1986;321:406–411. doi: 10.1038/321406a0. [DOI] [PubMed] [Google Scholar]
- Naranjo D, Brehm P. Modal shifts in acetylcholine receptor channel gating confer subunit-dependent desensitization. Science. 1993;260:1811–1814. doi: 10.1126/science.8511590. [DOI] [PubMed] [Google Scholar]
- Ochoa EL, Chattopadhyay A, McNamee MG. Desensitization of the nicotinic acetylcholine receptor: molecular mechanisms and effect of modulators. Cell Mol Neurobiol. 1989;9:141–178. doi: 10.1007/BF00713026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden DC, Colquhoun D. Ion channel block by acetylcholine, carbachol and suberyldicholine at the frog neuromuscular junction. Proc R Soc Lond B Biol Sci. 1985;225:329–355. doi: 10.1098/rspb.1985.0065. [DOI] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- Scuka M, Mozrzymas JW. Postsynaptic potentiation and desensitization at the vertebrate end-plate receptors. Prog Neurobiol. 1992;38:19–33. doi: 10.1016/0301-0082(92)90033-b. [DOI] [PubMed] [Google Scholar]
- Sine SM, Steinbach JH. Agonists block currents through acetylcholine receptor channels. Biophys J. 1984;46:277–283. doi: 10.1016/S0006-3495(84)84022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt JN, Engel AG. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- Sine SM, Taylor P. Local anesthetics and histrionicotoxin are allosteric inhibitors of the acetylcholine receptor. J Biol Chem. 1982;257:8106–8114. [PubMed] [Google Scholar]
- Unwin N. Nicotinic acetylcholine receptor at 9 A resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- Unwin N, Toyoshima C, Kubalek E. Arrangement of the acetylcholine receptor subunits in the resting and desensitized states, determined by cryoelectron microscopy of crystallized Torpedo postsynaptic membranes. J Cell Biol. 1988;107:1123–1138. doi: 10.1083/jcb.107.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptors α subunit decreases the rate of agonist dissociation. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, David-Pfeuty T, Changuex JP. Regulation of binding properties of the nicotinic receptor protein by cholinergic ligands in membrane fragments from Torpedo marmorata. Proc Natl Acad Sci USA. 1975;72:3443–3447. doi: 10.1073/pnas.72.9.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett. 1996;398:91–96. doi: 10.1016/s0014-5793(96)01215-x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen J, Auerbach A. Kinetics of wild-type, embryonic nicotinic acetylcholine receptors activated by acetylcholine, carbamylcholine, and tetramethylammonium. J Physiol (Camb) 1995;486:189–206. doi: 10.1113/jphysiol.1995.sp020802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Salamone F, Bouzat C, Sine SM, Auerbach A. Single-channel characterization of a mouse muscle acetylcholine receptor channel with a mutation at position 433 of the M4 segment of the a subunit. Biophys J. 1998;74:A90. [Google Scholar]