

Summary
The X-ray crystal structures of covalent complexes of the Actinomadura R39 DD-peptidase and Escherichia coli penicillin-binding protein 5 with β-lactams bearing peptidoglycan-mimetic side chains have been determined. The structure of the hydrolysis product of an analogous peptide bound non-covalently to the former enzyme has also been obtained. The R39 DD-peptidase structures reveal the presence of a specific binding site for the D-α-aminopimelyl side chain, characteristic of the stem peptide of Actinomadura R39. This binding site features a hydrophobic cleft for the pimelyl methylene groups and strong hydrogen bonding to the polar terminus. Both of these elements of the site are provided by amino acid side chains from two separate domains of the protein. In contrast, no clear electron density corresponding to the terminus of the peptidoglycan-mimetic side chains is present when these β-lactams are covalently bound to penicillin-binding protein 5. There is, therefore, no indication of a specific side chain binding site in this enzyme. These results are in agreement with those from kinetics studies published earlier and support the general prediction made at the time of a direct correlation between the kinetics and structural evidence. The essential high molecular weight penicillin binding proteins have demonstrated, to date, no specific reactivity with peptidoglycan-mimetic peptide substrates and β-lactam inhibitors and thus probably do not possess a specific substrate binding site of the type demonstrated here with the R39 DD-peptidase. This striking deficiency may represent a sophisticated defense mechanism against low molecular weight substrate-analogue inhibitors/antibiotics; its discovery should focus new inhibitor design.
Keywords: penicillin-binding proteins, DD-peptidases, β-lactam, peptidoglycan, peptide
Introduction
Cell wall growth is integral to all bacterial proliferation. The bacterial cell wall is composed of peptidoglycan and grows by incorporation of disaccharide-pentapeptide monomers. This occurs outside the cell membrane and requires two kinds of enzyme active site that separately catalyze transglycosylation and transpeptidation reactions1. The latter reaction serves to stabilize the cell wall by crosslinking the glycan strands. It is catalyzed by a class of enzymes known as D-alanyl-D-alanine transpeptidases, since they catalyze the reaction of Scheme 1, or otherwise simply as DD-peptidases or, inasmuch as they are the targets of β-lactam antibiotics, as penicillin-binding proteins (PBPs).
Scheme 1.

As shown in Scheme 1, the DD-peptidases employ an acyl-(serine)enzyme intermediate that can either be aminolyzed (by R′NH2) to complete the peptidoglycan cross-link, or hydrolyzed in a carboxypeptidase reaction to limit the extent of cross-linking. It seems now quite well established that the essential crosslinking reaction is catalyzed by a high molecular weight (60–100 kDa) group of DD-peptidases, while a lower molecular weight (40–60 kDa) group appears to have, in vivo, only DD-carboxypeptidase and DD-endopeptidase activity.2,3 In vitro, however, several members of the latter group are also able to catalyze transpeptidation reactions with small peptides and esters.3,4 The enzymatic activity of these enzymes has recently been reviewed.5
In view of their in vivo role as DD-peptidases, one would anticipate that these enzymes would exhibit substrate specificity towards peptides analogous in structure to oligopeptide elements of peptidoglycan. In certain instances, this appears to be correct. For example, the peptide 1 is a very specific substrate of the low molecular weight DD-peptidase of Streptomyces R61, which catalyzes both transpeptidase and carboxypeptidase reactions with this substrate as acyl donor.6–8 In view of Streptomyces peptidoglycan structure, 2a, this result is logical, although it is striking that peptide 3a, containing another significant element of 2a, is a poor substrate, comparable to generic D-alanyl-D-alanine-terminating peptides.9 As would be expected on the basis of these results, crystal structures show that 1 binds very snugly to the R61 DD-peptidase active site, which contains what appears to be a specific binding site for the glycyl-L-aminopimelyl moiety.10 β-Lactams with this same side chain, for example 4, are also extremely potent covalent inhibitors of the R61 enzyme, and employ the same specific side chain binding site.11,12

A survey of examples of the major classes of bacterial DD-peptidases, however, showed that the above result is not general, at least with respect to specificity in substrate turnover and β-lactam inhibitory kinetics.9,13 The high molecular weight enzymes, for example, do not show any particular preference for peptidoglycan-mimetic peptides and β-lactams analogous to 1 and 3 or 4. Nor do certain low molecular weight enzymes, for example Escherichia coli PBP5 and Streptococcus pneumoniae PBP3.9,13 On the basis of the R61 DD-peptidase result, we predicted that the crystal structures of enzymes that show kinetic specificity for peptidoglycan-mimetic peptides and β-lactams will contain specific binding sites for the side chains of these molecules, whereas those that show no kinetic specificity will not.13 To support these predictions, we describe in this paper the crystal structures of complexes of two low molecular weight DD-peptidases with peptidoglycan-mimetic ligands, the β-lactams 5 and 6 and the peptide 7 bound to them. Kinetics studies showed that the Actinomadura R39 DD-peptidase does display peptidoglycan-mimetic substrate specificity, analogous to that of the R61 enzyme, but E. coli PBP5 does not.9,13
The Actinomadura R39 DD-peptidase is a low molecular weight, class C enzyme2 with strong amino acid sequence similarity to E. coli PBP4 and to Bacillus subtilis PBP4a.14,15 Crystal structures have confirmed the structural resemblance.16–18 The R39 DD-peptidase is a water-soluble enzyme, loosely associated with the bacterial cell membrane. Although the precise role of the enzyme in vivo is not known, under in vitro conditions it has been shown to catalyze the hydrolysis and aminolysis of small D-alanyl peptides and esters.19,20
PBP5 of E. coli is also categorized as a low molecular weight penicillin-binding protein, but of class A.2 It is not essential to the survival of E. coli but its absence affects cell morphology.21 Under normal conditions, it is apparently responsible for much of the D-alanine carboxypeptidase activity that limits cell wall crosslinking.22 In vivo, PBP5 is attached to the E. coli inner membrane by a C-terminal α-helix23, removal of which affords the solubilized protein used for kinetics studies and crystal structure determination.24 The solubilized construct is inhibited by β-lactams and also catalyzes the hydrolysis and aminolysis of small D-alanyl peptides and esters25, although much less efficiently than the R39 enzyme. The general organization of catalytic functional groups in PBP5 is very similar to that in the R39 DD-peptidase and both resemble that of a class A β-lactamase16; neither, however, contains an analogue of Glu 166 of the β-lactamase, the β-lactam deacylation catalyst. Extensive structural details of the low molecular weight PBPs are provided in a recent review.26
The crystal structures described in this paper strongly support the predictions made above in regard to DD-peptidase substrate specificity: a specific side chain-binding site is revealed in the R39 DD-peptidase but not in PBP5.
Results and Discussion
R39 DD-Peptidase Structures
The asymmetric unit of R39 DD-peptidase crystals contains four protein molecules that are not in the same crystallographic environment.16 The environment is very similar for monomers A and D and for monomers B and C, but different for both pairs.
In the R39-6 complex, all four monomers’ active sites are acylated by the ligand and the conformation of the complex is the same in each monomer. The electron density map calculated in the absence of ligand provides a very clear density showing 6 covalently linked to the enzyme active serine (Figure 1A). The carbonyl oxygen lies in the oxyanion hole, the carboxylate is oriented towards the hydroxyl group of Thr 411 [from the KTG motif], and the amide group of the cephalosporin side chain is wedged between the side chain of Asn 300 (from the SXN motif) and the backbone of the strand that lines the active site. In these features, the structure closely resembles those generally found for complexes of β-lactams with β-lactam-recognizing enzymes, including that of nitrocefin with the R39 DD-peptidase.16
Figure 1.
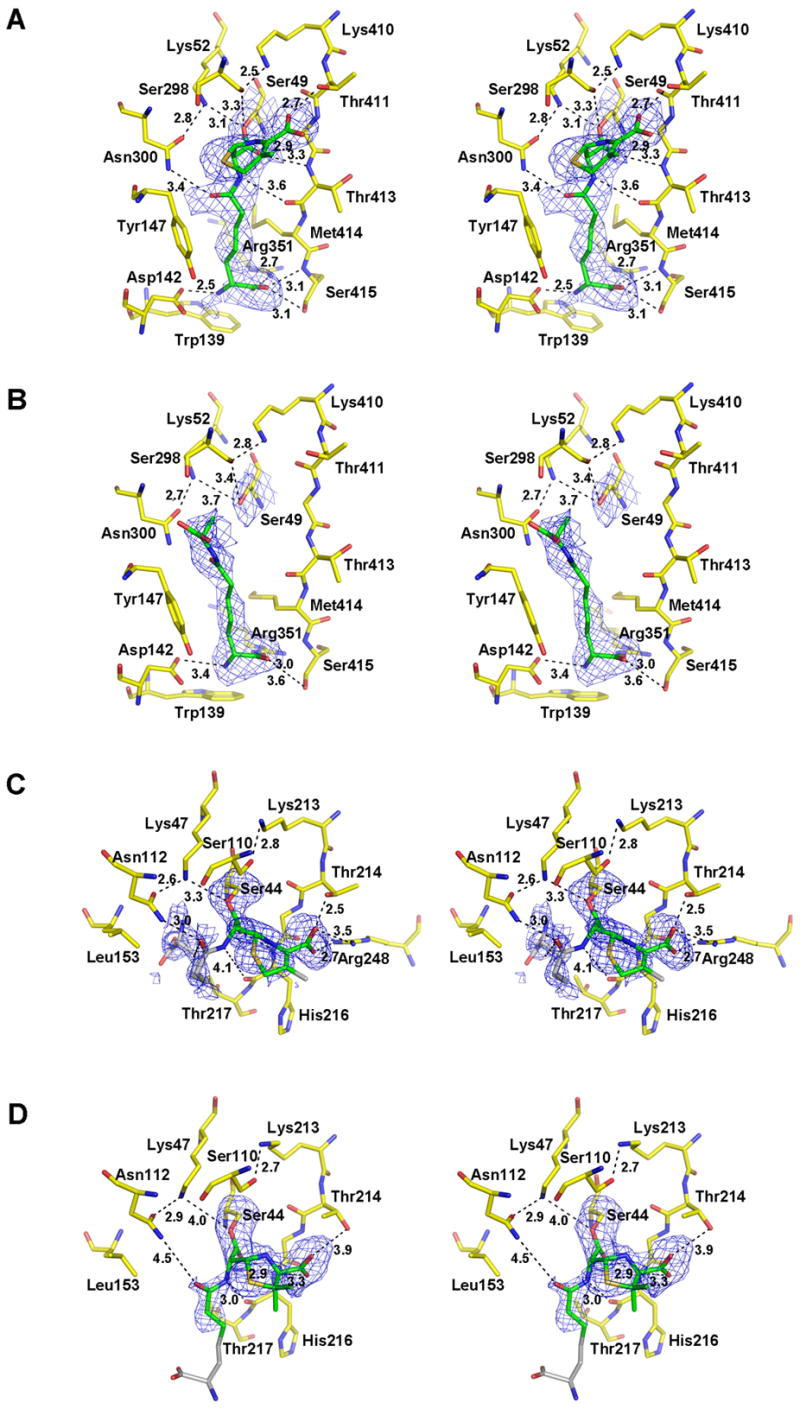
Crystal structures of the R39 DD-peptidase in complex with (A) the cephalosporin 6 and (B) the peptide 7, and of PBP5 from E. coli in complex with (C) the cephalosporin 6 and (D) the penicillin 5. In these stereoviews of the respective active sites, the electron density is a |Fo| − |Fc| difference map calculated from the final coordinates of each model refined in the absence of the ligand. The resulting positive density is shown in blue and is contoured at 2.0 σ Carbon atoms of each ligand that are visible in the electron density and have been included in the final model are colored green. Those that could not be modeled (in PBP 5) due to weak density are colored grey and are included to show the approximate positions of these groups. The carbon atoms of amino acids that form each active site are colored yellow. Oxygen atoms are colored red, nitrogen blue, and sulfur orange. Potential hydrogen bonds are shown as dashed lines and the distances are noted in Å. Some distances beyond hydrogen bonding range are shown in D to compare with the equivalent distances in C. This figure was generated using PYMOL (pymol.sourceforge.net).
Of particular interest to this paper is the disposition of the D-α-aminopimelyl side chain. The methylene groups of the side chain are placed between the side chains of Tyr 157 and Met 414, and the hydrophobic environment is completed by the side chain of Leu 349 in the rear of the active site. The side chain of Tyr 157 essentially occupies the same position as in the free enzyme; in the nitrocefin complex, it moves to provide hydrophobic cover to the thienyl side chain.16 The terminal H3N+-CH-COO−moiety of the aminopimelic acid is inserted in a pocket composed of residues Trp 139 (π-cation), Asp 142, Arg 351 and Ser 415 (hydrogen bonds), with a salt bridge between the aminopimelic acid carboxylate and Arg 351. The aminopimelyl ammonium-terminus hydrogen bonds to the Asp 142 side chain. It is striking that, as with B. subtilis PBP4a 18, the elements of the side chain binding site are derived from two separate domains of the protein16, the β-lactam-binding domain, per se, and domain II, the latter supplying Trp 139, Asp 142 and Tyr 147 (Figure 2A).
Figure 2.

Interaction between Arg 248 and the cephalosporin carboxylate. The backbones of wild-type (green), penicillin 5-bound (purple) and cephalosporin 6-bound (yellow) structures of PBP5 are superimposed showing conformational differences in a loop comprising residues 242–248. In the cephalosporin structure, Arg 248 interacts with the carboxylate of the cephalosporin (colored yellow) whereas in the penicillin-bound structure Arg 248 occupies a position similar to that in wild-type PBP5. There are also differences in the respective positions of Phe 245. Figure prepared using MOLSCRIPT53 and RASTER3D.54
In the complex of the enzyme with the peptide 7, density maps reveal the presence of D-α-amino-ε-pimelyl-D-alanine in monomers A, B and C (Figure 1B). D-α-Amino-ε-pimelyl-D-alanine is one of the products of the carboxypeptidation reaction catalyzed by the R39 DD-peptidase. Electron density that could be attributed to the released D-alanine, the second product of the reaction, may also have been observed but we have attributed this electron density to a sulfate ion as in the apo structure of the R39 enzyme16.
The D-alanine carboxylate of D-α-amino-ε-pimelyl-D-alanine is turned away from the active site serine as if it had rotated around the pimelyl ε-carbon-peptide carbonyl bond after the deacylation. A water molecule is present in the oxyanion hole between the active serine and the alanine carbonyl oxygens. Such a rotation was also observed in the complex of the Streptomyces R61 DD-peptidase with a specific peptide product10, but in that case the carboxylate twisted into the protein, probably by rotation around the D-alanyl α-carbon-peptide N bond, rather than away from the protein as in the present case. Importantly, however, in the present structure, the D-α-aminopimelyl side chain is positioned exactly like the side chain of 6 in the β-lactam complex described above.
The R39 DD-peptidase has close structural homology to the Bacillus subtilis PBP4a, which was previously crystallized and soaked with 7.18 The position of the active site residues of both structures is nearly identical and the side chain of 7 occupies exactly the same position, making identical interactions, particularly with Asp142 and Ser 415 (R39 numbering); the one notable difference is that Arg351 is replaced by a histidine in PBP4a. In the primary structure of most PBPs homologous to R39 (for example, E. coli PBP4 and Neisseiria gonorrhoeae PBP3), this residue is a histidine or an arginine and the other residues forming the meso-diaminopimelate binding pocket are well conserved. The hydrophobic cleft, accommodating the methylene chain is also conserved.
Although monomers A and D have a very similar crystallographic environment, the active site of molecule D is not occupied by the ligand. The Asn 176 side chain of a symmetry mate inserts in the pocket occupied by the aminopimelyl moiety in the other monomers. In molecule A, the Asn 176 side chain (of a symmetric molecule) is clearly shifted and the pocket is occupied by the side chain of 7.
PBP5 Structures
In the complex with cephalosporin 6, the acyl linkage with Ser 44 and most of the dihydrothiazine ring are clearly defined in the electron density, indicating that 6 has bound and reacted covalently with PBP5 (Figure 1C). Weak density is observed in the ring around C2. The C-3′ acetoxy group is lost, as expected.12,27 On moving away from this region, however, less of the molecule is visible. Although some difference peaks are present near residues Thr 217, Asp 41, and Leu 153 that might correspond to the terminal carboxylate and amino groups of the D-α-amino-ε-pimelyl side chain, the side chain could not be modeled without ambiguity and so, extending from the aminopimelyl β-carbon, is excluded from the model. Only a few hydrogen bonding interactions that involve the cephalosporin are present; these hydrogen bonds, none unexpected on the basis of previous structures of cephem complexes, are between the cephem carboxylate and the hydroxyl group of Thr 214 and the side chain terminus of Arg 248 (see below), between the side chain amide carbonyl and a side chain NH of Asn 112 and the backbone NH of Ser 87, and between the acyl-serine carbonyl and the oxyanion hole (NH of His 216 and Ser 44). Neither Lys 47 Nζ nor Ser 110 Oγ is within hydrogen-bonding distance of Ser 44 Oγ. This disposition of the latter functional groups, however, is commonly found in complexes of PBPs with β-lactams and presumably reflects their catalytic incompetence towards deacylation.
A similar picture is observed in the complex of PBP5 with the penicillin 5 (Figure 1D). The acyl-serine linkage and thiazolidine ring are both clearly visible except for weak density corresponding to one of the methyl group substituents on the ring (C16). Some density corresponding to the first part of the side chain is visible, but it disappears from the aminopimelyl Cγ onwards. Consequently, this region is not included in the model. The enzyme appears to participate in only three hydrogen bonds with the penicillin, between the acyl-serine carbonyl in the oxyanion hole (NH of His 216 and Ser 44) and between the side chain amide NH and the backbone carbonyl of His 216. Notably absent are interactions between the penam carboxylate and the protein: the side chains of neither Thr 214 nor Arg 248 appear to make contact, c.f. the cephalosporin structure described above.
The overall paucity of side chain electron density for both ligands is an interesting result and suggests that, for PBP5, at least when organized within a crystal lattice, the β-lactams 5 and 6 can acylate the active site serine but, beyond the region around the covalent bond, these compounds make relatively few contacts with the enzyme. There is certainly no sign of a specific side chain binding site as described above for the R39 DD-peptidase. Indeed, this region is structured quite differently in PBP 5. When compared to the R39 DD-peptidase structure, it is comprised only of residues from the penicillin-binding domain, whereas the R39 DD-peptidase includes residues from its domain II, which has no counterpart in PBP 5, to bind the peptide terminus.
Comparison with wild-type PBP5
Both β-lactam-bound structures were compared with that of the wild-type enzyme.28 All main chain atoms of the 6-bound structure could be superimposed onto the wild-type structure with an RMS deviation of 0.31 Å; the equivalent value for the 5-bound structure was 0.40 Å. Examination of the superimposed backbones showed them to be virtually indistinguishable except for one region comprising residues 242–248, inclusive. In the wild-type enzyme, this region forms a distorted helical region immediately preceding α10, which starts at residue 249 (see ref. 29 for secondary structure assignments). In the 6-bound structure, however, these residues adopt a more canonical helical conformation such that α10 now starts earlier at residue 245 (Figure 2). A possible trigger for this conformational change is the interaction between Arg 248 and the carboxylate of the cephalosporin. To make this interaction, the guanidinium group of the arginine has shifted by approximately 6 Å, when compared to its position in wild-type PBP 5. This change in Arg 248 appears to destabilize Phe 245 because the side chain of this residue now exists in two (and possibly more) conformations. That the carboxylate of the cephalosporin interacts with Arg 248 is a little surprising, as we had postulated previously that this group may interact with Arg 198, which, in wild-type PBP5, appears much better placed. In fact, the side chain of Arg 198 is disordered in the 6-bound structure.
The same region is also slightly altered in the 5-bound structure but in this case the backbone is intermediate in position between wild-type PBP5 and the 6-bound structure (Figure 2). Again, this region is characterized by relatively weak density. Curiously, the side chain of Arg 248 does not interact with penicillin carboxylate and occupies essentially the same position as in the wild-type structure. Thus, there are differences between the interactions with the protein made by the cephalosporin and the penicillin.
Of further interest, a conformational change that occurred in a complex of PBP5 with a boronic acid peptide-mimetic, a shift in residues 152–154 near the active site,30 was not observed in either of the ligand-bound structures presented here. Thus, it appears that different compounds reacting with PBP5 induce different and specific conformational changes in the protein (see below).
Discussion
The structures of complexes of the R39 DD-peptidase with 6 and 7, as described above, show very clearly the presence of a specific binding site for the D-aminopimelyl side chain (Figure 1). This result therefore resembles that obtained for the Streptomyces R61 DD-peptidase where an analogous side chain binding site was observed, appropriate for the glycyl-D-aminopimelyl moiety of Streptomyces peptidoglycan10. An analogous site is likely present on B. subtilis PBP4a.18 In what appears to be sharp contrast, essentially no firm electron density for the side chain, and none particularly for the polar terminus, was observed in the complexes of E. coli PBP5 with 5 and 6 (Figure 1). This suggests the absence of a specific binding site for the N-terminus of the stem peptide in this enzyme. It is possible that crystal packing interactions may have led to this result with PBP5, but there is certainly no overt indication of this problem such as there is, for example, with monomer D of the R39/peptide 7 complex (see above).
These results are strongly supported by kinetics results. The peptide 7 is an excellent substrate of the R39 DD-peptidase (kcat = 7.4 s−1, Km = 1.3 μM, kcat/Km = 5.7 × 106 s−1M−1)10; similarly 5 and 6 are excellent inhibitors.13 These numbers mirror those from the analogous peptidoglycan-mimetic peptide 1 with the R61 DD-peptidase (kcat = 69 s−1, Km = 7.9 μM, kcat/Km = 8.7 × 106 s−1M−1)6 and from analogous β-lactams.11,13 On the other hand, 7 is a very poor substrate of the E. coli PBP5 (kcat/Km < 50 s−1M−1), poorer indeed than the generic peptide N,N′-diacetyl-L-lysyl-D-alanyl-D-alanine.25 The kinetics results, published previously, and the structures presented here are thus in complete agreement. It should also be noted here that neither the R39 DD-peptidase nor E. coli PBP5 catalyze hydrolysis of the peptides 3 at rates suggestive of specific interaction.9 No strong specificity for this element of peptidoglycan structure has yet been demonstrated for any DD-peptidase.13
It seems likely, therefore, that the R39 DD-peptidase has evolved to specifically accept an acyl donor substrate containing the D-α-aminopimelyl side chain. The low Km and therefore high kcat/Km value suggest that the substrate is at low effective concentration in vivo. The R39 DD-peptidase is known to catalyze carboxypeptidation, transpeptidation and endopeptidation reactions in vitro19,20, although its substrate specificity in the latter two reactions has not yet been thoroughly explored. The role of the enzyme in vivo is not known. It may even, as has also been suggested for the Streptomyces R61 DD-peptidase3,31, be a β-lactam scavenger.
It should be noted, however, that other low molecular weight class C enzymes that have structures very similar to that of the R39 enzyme, E. coli PBP4 and B. subtilis PBP4a14,17,18, for example, do appear to have real - if non-essential, in the same sense as E. coli PBP5, perhaps (see below) - roles in bacterial cell wall construction and maintenance.32,33 The latter of these enzymes has also been shown to have high hydrolytic activity against the peptide 7.18
There is no doubt that E. coli PBP5 does participate in bacterial cell wall construction and/or maintenence. Although it is not essential for cell survival and reproduction, its absence does lead to aberrations in cell shape21 and, presumably, a decrease in long-term evolutionary fitness. The structures described above are in agreement with the kinetics results and strongly suggest that no strong binding of peptidoglycan-mimetic peptides occurs, to the solubilized enzyme at least. This seems to be true of the membrane-bound holo-enzyme also, both in vivo and after isolation in membranes.34 It may be that this enzyme recognizes as its specific substrate a much larger segment of peptidoglycan than mimicked by the peptides used in in vitro studies to date. The incorporation of mono and disaccharides in a complete peptidoglycan monomer did not, however, yield enhanced catalysis.35 Alternatively, as has been suggested from time to time, in the two dimensional milieu of the cell membrane, where the substrate may be effectively immobilized, a high affinity for the substrate may not be required, i.e. sufficient affinity in vivo appears as low affinity in three dimensional in vitro solution studies. The ease or otherwise of physical access of these enzymes to their substrates in vivo is, of course, an unknown factor.
Streptococcus pneumoniae PBP3 is another low molecular weight class A enzyme that has been studied to some degree both kinetically and structurally36. It appears to be more active in vitro than E. coli PBP5 [kcat/Km for N,N′-diacetyl-L-lysyl-D-alanyl-D-alanine hydrolysis is 5700 s−1M−1 36] but still has weak affinity for peptide substrates [Km for N,N′-diacetyl-L-lysyl-D-alanyl-D-alanine is 19 mM; the value of this parameter for E. coli PBP5 is at least this high25,37]. This higher activity may reflect the multiple layers of peptidoglycan in Gram positive bacteria. At any event, it is possible that even with a weakly specific binding site and correspondingly high Km values, the kcat values [e.g. for E. coli PBP5 in solution, kcat > 0.2 s−1 25,37 and for S. pneumoniae PBP3, kcat = 110 s−1 36 for N,N′-diacetyl-L-lysyl-D-alanyl-D-alanine] may be large enough for the in vivo role of these enzymes.
Under either of the above scenarios, the lack of structural specificity towards small molecule substrates is striking and may reflect a powerful defense against small molecule peptidoglycan-mimetic inhibitors.34
It is noticeable in the crystal structures of apo-PBP5 and those of its β-lactam complexes reported above that the active site appears in a very open conformation with the active site residues more spread out than expected in an active conformation. Simple measures of this are the Asn 112 or Ser 110 to Gly 215 Cα distances. Relevant data are shown in Table 2. Striking from these data are the smaller distances (narrower active site cleft) in the more reactive S. pneumoniae PBP3 and R39 DD-peptidase structures than in PBP5, both alone and in its complexes with 5 and 6. Striking also are the smaller distances in the PBP5-boronate inhibitor complex where the boronate moiety is hydrogen-bonded to the protein in the manner of a transition state analogue.30 The complexes of PBP5 with 5 and 6 show no such tightening of the structure, which might have been expected to accompany specific recognition. It may be that PBP5 in the crystalline form, in solution, and perhaps when resting in vivo, relaxes to an open inactive conformation unless stimulated by a specific, and perhaps extended, substrate. Such a phenomenon could explain its poor reactivity with small substrates, including small peptidoglycan-mimetics such as 7. S. pneumoniae PBP3 is more reactive but still not specific for elements of peptidoglycan local to the reaction center.13 It may be that the two issues, absolute reactivity and substrate specificity are, partly at least, separable.
Table 2.
Active site distances in DD-peptidases
| Distance (Å) | ||
|---|---|---|
| Enzyme/ligand | Asn Cα-Gly Cαa | Ser Cα-Gly Cαa |
| E. coli PBP5 | 12.7 | 8.9 |
| E. coli PB5/5 | 12.5 | 8.8 |
| E. coli PBP5/6 | 12.4 | 8.7 |
| E. coli PBP5/boronate | 11.8 | 8.1 |
| S. pneumoniae PBP3 | 11.6 | 8.3 |
| R39 DD-peptidase | 11.3 | 8.1 |
| R39 DD-peptidase/6 | 10.8 | 7.8 |
| R39 DD-peptidase/7 | 11.2 | 8.2 |
These refer to the Asn and Ser residues of the SXN motif, and the Gly residue of the KXG motif of the respective enzymes
The situation described above for E. coli PBP5 and other low molecular weight class A enzymes is even more complicated when the essential high molecular weight enzymes are considered, where little, if any, DD-peptidase activity and little affinity for peptidoglycan-mimetic substrates and β-lactams have been demonstrated in vitro.13 The issue of conformational changes necessary for reactivity, both in the crystalline form38,39 and in solution40,41, has been raised for several of the high molecular weight enzymes. As noted above, however, this may relate to absolute reactivity rather than to substrate specificity. With respect to the latter, it would not be surprising if the essential high molecular weight DD-peptidases are even more assiduously protected from small substrate-analogue inhibitors than the low molecular weight enzymes. It might also be noted here that essentially all PBP structures now available, both free and complexed, display active site distances very similar to those of S. pneumoniae PBP3 in Table 2. Since these enzymes are certainly not all equally reactive, with peptides for example, the latter distances may be required for optimal activity but are certainly not sufficient. The exception is PBP2a of Staphylococcus aureus, where the active site gulf in the crystal form is much narrower, a difference that has been interpreted to reflect the very low reactivity of this β-lactam-resistant enzyme.38
It is clear that much more needs to be done, both structurally and kinetically, for us to understand, qualitatively and quantitatively, how these enzymes function in vivo. That they do function there, however, is even clearer - bacteria certainly flourish, for better and for worse.
Experimental Procedures
The β-lactams 5 and 6 and the peptide 7 were obtained as previously described.9,13
R39 DD-Peptidase Crystallography
The R39 DD-peptidase was expressed and purified as described previously.42 The 538 amino-acid protein precursor presents a 49 amino-acid N-terminal signal peptide and, presumably, a 23 amino-acid C-terminal extension that are both cleaved to yield a mature soluble protein of 466 residues. Crystals were grown at 20°C by hanging drop vapor diffusion. The crystals belong to the space group P21 with cell dimensions a = 105.0 Å, b = 93.3 Å, c = 108.2 Å and β = 94.2°. The crystallization medium contained protein [2.5 μl of protein solution (18 mg ml−1), also containing 5 mM MgCl2 and 20 mM Tris, pH 8], 2 μl of well solution (2.0 M ammonium sulfate and 0.1 M MES, pH 6) and 0.5 μl of 0.1 M CoCl2 solution. R39 DD-peptidase crystals were transferred to 2 μl of well solution containing 6 (80 mM) for 48 h to form the R39-6 complex, or 2 μl of well solution containing 7 and D-Ala to final concentrations of 23.4 mM and 900 mM, respectively, for 47 h to form the R39-7 complex. X-ray diffraction experiments were carried out under cryogenic conditions (100K) after transferring the crystals into 100% glycerol. Data were collected with a Rigaku RU-200 rotating anode generator operating at 40 kV and 100 mA and a MarResearch Mar345 Imaging Plate (λ = 1.5418 Å). Intensities were indexed and integrated using XDS.43 Data were scaled with SCALA of the CCP4 program suite44 and all corresponding statistics are given in Table 1.
Table 1.
Data collection and refinement statistics
| Crystal | R39-6 | R39-7 | PBP 5-5 | PBP 5-6 |
|---|---|---|---|---|
| Data Collection: | ||||
| Molarity of soak (mM) | 80.0 | 23.4 | 13.2 | 30 |
| Time of soak (hrs) | 48 | 47 | 1 | 18 |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 0.97934 |
| Resolution range (Å)a | 20–2.4 (2.53–2.4) | 20–2.25 (2.37–2.25) | 47.0–2.0 (2.07–2.00) | 41–1.60 (1.66–1.60) |
| No. of unique reflections | 81,439 | 99,513 | 25,433 | 51,428 |
| Rmerge (%)a,b | 15.0 (59.6) | 14.7 (60.8) | 10.7 (30.5) | 7.4 (41.4) |
| Redundancy a | 3.7 (3.7) | 3.7 (3.5) | 2.5 (2.4) | 3.5 (2.5) |
| Completeness (%)a | 99.8 (100) | 99.1 (94.9) | 94.3 (85.9) | 97.3 (82.7) |
| <I>/<σI> a | 9.6 (2.5) | 9.7 (2.3) | 6.8 (2.0) | 25.7 (1.8) |
| Refinement: | ||||
| Resolution range | 16–2.4 | 15.7–2.25 | 47.0–2.0 | 41–1.6 |
| No. of protein atoms | 13,576 | 13,549 | 2,742 | 2,738 |
| Number of water molecules | 496 | 600 | 188 | 266 |
| R cryst (%) | 20.7 | 23.0 | 23.8 | 19.3 |
| R free (%) | 26.0 | 28.5 | 29.6 | 21.5 |
| RMS deviations from ideal stereochem:- | ||||
| bond lengths (Å) | 0.009 | 0.010 | 0.011 | 0.011 |
| bond angles (°) | 1.22 | 1.28 | 1.40 | 1.29 |
| B factors: | ||||
| Mean B factor (all atoms) (Å2) | 28.9 | 26.4 | 28.0 | 21.3 |
| Mean B factor (main chain) (Å2) | 28.7 | 26.2 | 27.4 | 18.9 |
| RMS deviation in main chain B factors (Å2) | 0.53 | 0.32 | 0.53 | 0.59 |
| Mean B factor (side chains & waters) (Å2) | 29.1 | 26.7 | 28.6 | 23.5 |
| RMS deviation in side chain B factors (Å2) | 1.19 | 0.70 | 1.13 | 1.40 |
| Ramachandran plot: | ||||
| most favoured region (%) | 88.4 | 90.0 | 92.2 | 92.5 |
| additionally allowed regions (%) | 10.8 | 9.4 | 6.9 | 7.2 |
| generously allowed regions (%) | 0.6 | 0.5 | 0.7 | 0.3 |
| disallowed regions (%) | 0.1 | 0.1 | 0.3 | 0.0 |
| RMSD of Cα atom with native structure (Å) | 0.43c | 0.48 | 0.40 | 0.31 |
Statistics for the highest resolution shell are given in parentheses.
Rmerge = Σ | Ii − Im |/ΣIi where Ii is the intensity of the measured reflection and Im is the mean intensity of all symmetry-related reflections. Figures within brackets are for the outer resolution shell.
Monomer A
Using the structure of the apo-enzyme, a first round of rigid-body refinement was carried out with REFMAC5.45 The resulting unbiased electron density maps displayed in Coot46 unequivocally showed 6 covalently bound to the enzyme in all four monomers and 7 in three monomers. The structure of the R39 DD-peptidase bound to 6 was refined to 2.4 Å and the structure with 7 was refined to 2.25 Å. The statistics of refinement are summarized in Table 1.
PBP5 Crystallography
Soluble wild-type PBP 5 protein was purified and crystallized as described previously.28 These crystals belong to the C2 space group and have cell dimensions a = 109.4 Å, b = 50.3 Å, c = 84.5 Å and b = 120.9°.28 Crystal soaking experiments were performed by transferring crystals to stabilizing solutions comprising 8% PEG 400, 50 mM Tris-HCl, pH 8.0 and containing 5 or 6. Various concentrations of 5 and 6 and soak times were tested. The crystals were then cryo-protected by gradually increasing the concentration of glycerol (in the same solution) in 5% increments up to 25% glycerol over a period of about an hour. The degree of incorporation of each compound was assessed by examination of the electron density maps after collection of trial data sets using a rotating anode generator (home X-ray source). The final soaking condition for 5 was 13 mM for 1 h and for 6 was 30 mM for 18 h.
For the complex with 5, data were collected on an R-AXIS IV++ image plate detector mounted on an RU-H3R rotating anode X-ray generator fitted with Osmic Confocal Optics (Rigaku MSC). A total of 180° data were collected and were processed with d*Trek.47
Data for the complex with 6 were collected at the SER-CAT ID22 beam line of the Advanced Photon Source (APS) on a Mar 300 CCD detector at a wavelength of 0.979 Å. The data were processed using HKL2000.48
In both cases, the starting structure for refinement was the structure of wild-type PBP528 but stripped of waters and other ligands. After an initial round of refinement using CNS49 for 6 or REFMAC45 for 5, a difference electron density map (|Fobs| − |Fcalc|) was calculated and displayed using the graphics program O.50 The coordinates for each compound were built in the Monomer Library Sketcher module of REFMAC5 and manually positioned into the unbiased positive electron density in the active site of the enzyme using O. Thereafter, each model was refined using REFMAC5 alternating with rounds of manual revision. A subset (5%) of the reflections were set aside for calculation of the Free R factor.51 Water molecules with reasonable hydrogen bond distances were included in later rounds of refinement and several amino acid side chains were modeled with multiple conformations. The stereochemistries of the final models were evaluated using PROCHECK.52 The structure in complex with the cephalosporin 6 was refined to 1.6 Å resolution with an R factor of 21.2% and Rfree of 23.5% and that with the penicillin 5 was refined at 2.0 Å with an R factor of 23.8 % and Rfree of 29.6%. Both structures have excellent stereochemistry (see Table 1).
Protein Data Bank accession codes
The atomic coordinates for the crystal structures described above have been deposited in the RCSB Protein Data Bank and are available under the accession codes 2VGJ (R39/6 complex), 2VGK (R39/7 complex), 3BEB (PBP5/5), and 3BEC (PBP5/6 complex).
Acknowledgments
Use of the Advanced Photon Source for the PBP5 project (AJP, JH, CD) was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-ENG-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) BM22 beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. The X-ray crystallography facility used for the PBP5 work is supported by the Medical University of South Carolina’s Research Resource Facilities program. The R39 project (ES and PC) was supported in part by the European Commission Sixth Framework Program grants LSMH-CT-COBRA 2003-503335 and LSMH-CT-EUR-INTAFAR 2004-512138, by the Belgian Program on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister’s Office, Science Policy programming (IAP no. P6/19), by the Actions de Recherche Concertées (grant 03/08-297), by the Fonds National de la Recherche Scientifique (IISN 4.4505.00, FRFC 9.45/9.99, FRFC 2.4.508.01.F, FRFC 9.4.538.03.F, FRFC 2.4.524.03) and the University of Liège (Fonds spéciaux, Crédit classique, 1999). This research was also supported by National Institutes of Health Grants AI-17986 (RFP) and GM-66861 (CD).
Abbreviations used
- NAM
N-acetylmuramic acid
- PBP
penicillin-binding protein
- MES
2-(N-morpholino)ethanesulfonic acid
- PEG
polyethylene glycol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Macheboeuf P, Contreras-Martell C, Job V, Dideberg O, Dessen A. Penicillin-binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol Rev. 2006;30:673–691. doi: 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 2.Ghuysen JM. Serine β-lactamases and penicillin-binding proteins. Annu Rev Microbiol. 1991;45:37–67. doi: 10.1146/annurev.mi.45.100191.000345. [DOI] [PubMed] [Google Scholar]
- 3.Goffin C, Ghuysen JM. Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: presence of penicillin-susceptible target proteins versus lack of efficiency of penicillin as a therapeutic agent. Microbiol Mol Biol Rev. 2002;66:702–738. doi: 10.1128/MMBR.66.4.702-738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghuysen JM, Frère JM, Leyh-Bouille M, Coyette J, Dusart J, Nguyen-Distèche M. Use of model enzymes in the determination of the mode of action of penicillins and Δ3-cephalosporins. Annu Rev Biochem. 1979;48:73–101. doi: 10.1146/annurev.bi.48.070179.000445. [DOI] [PubMed] [Google Scholar]
- 5.Pratt RF. Cell Mol Life Sci. 2008. Substrate specificity of bacterial DD-peptidases (penicillin-binding proteins) in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson JW, Pratt RF. Dipeptide binding to the extended active site of the Streptomyces R61 D-alanyl-D-alanine peptidase: the path to a specific substrate. Biochemistry. 2000;39:12200–12209. doi: 10.1021/bi001295w. [DOI] [PubMed] [Google Scholar]
- 7.Kumar I, Pratt RF. Transpeptidation reactions of a specific substrate catalyzed by the Streptomyces R61 DD-peptidase: characterization of a chromogenic substrate and acyl acceptor. Biochemistry. 2005;44:9971–9979. doi: 10.1021/bi050542z. [DOI] [PubMed] [Google Scholar]
- 8.Kumar I, Pratt RF. Transpeptidation reactions of a specific substrate catalyzed by the Streptomyces R61 DD-peptidase: the structural basis of acyl acceptor specificity. Biochemistry. 2005;44:9961–9970. doi: 10.1021/bi0505417. [DOI] [PubMed] [Google Scholar]
- 9.Anderson JW, Adediran SA, Charlier P, Nguyen-Distèche M, Frère JM, Nicholas RA, Pratt RF. On the substrate specificity of bacterial DD-peptidases: evidence from two series of peptidoglycan-mimetic peptides. Biochem J. 2003;373:949–955. doi: 10.1042/BJ20030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonough MA, Anderson JW, Silvaggi NR, Pratt RF, Knox JR, Kelly JA. Structures of two kinetic intermediates reveal species specificity of penicillin-binding proteins. J Mol Biol. 2002;322:111–122. doi: 10.1016/s0022-2836(02)00742-8. [DOI] [PubMed] [Google Scholar]
- 11.Josephine HR, Kumar I, Pratt RF. The perfect penicillin? Inhibition of a bacterial DD-peptidase by peptidoglycan-mimetic β-lactams. J Am Chem Soc. 2004;126:8122–8123. doi: 10.1021/ja048850s. [DOI] [PubMed] [Google Scholar]
- 12.Silvaggi N, Josephine HR, Kuzin AP, Nagarajan R, Pratt RF, Kelly JA. Crystal structures of complexes between the R61 DD-peptidase and peptidoglycan-mimetic β-lactams: a non-covalent complex with a “perfect penicillin”. J Mol Biol. 2005;345:521–533. doi: 10.1016/j.jmb.2004.10.076. [DOI] [PubMed] [Google Scholar]
- 13.Josephine HR, Charlier P, Davies C, Nicholas RA, Pratt RF. Reactivity of penicillin-binding proteins with peptidoglycan-mimetic β-lactams: what’s wrong with these enzymes? Biochemistry. 2006;45:15873–15883. doi: 10.1021/bi061804f. [DOI] [PubMed] [Google Scholar]
- 14.Granier B, Duez C, Lepage S, Englebert S, Dusart J, Dideberg O, Van Beeumen J, Frère JM, Ghuysen JM. Primary and predicted secondary structures of the Actinomadura R39 extracellular DD-peptidase, a penicillin-binding protein (PBP) related to Escherichia coli PBP4. Biochem J. 1992;282:781–788. doi: 10.1042/bj2820781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duez C, Vanhove M, Gallet X, Bouillenne F, Docquier J-D, Brans A, Frère J-M. Purification and characterization of PBP4a, a new low-molecular-weight penicillin-binding protein from Bacillus subtilis. J Bacteriol. 2001;183:1595–1599. doi: 10.1128/JB.183.5.1595-1599.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauvage E, Herman R, Petrella S, Duez C, Bouillenne F, Frère JM, Charlier P. Crystal structure of the Actinomadura R39 DD-peptidase reveals new domains in penicillin-binding proteins. J Biol Chem. 2005;280:31249–31256. doi: 10.1074/jbc.M503271200. [DOI] [PubMed] [Google Scholar]
- 17.Kishida H, Unzai S, Roper DI, Lloyd A, Park S-Y, Tame JRH. Crystal structure of penicillin-binding protein 4 (dacB) from Escherichia coli, both in the native form and covalently linked to various antibiotics. Biochemistry. 2006;45:783–792. doi: 10.1021/bi051533t. [DOI] [PubMed] [Google Scholar]
- 18.Sauvage E, Duez C, Herman R, Kerff F, Perrella S, Anderson JW, Adediran SA, Pratt RF, Frère JM, Charlier P. Crystal structure of the Bacillus subtilis penicillin-binding protein 4a, and its complex with a peptidoglycan-mimetic peptide. J Mol Biol. 2007;371:528–539. doi: 10.1016/j.jmb.2007.05.071. [DOI] [PubMed] [Google Scholar]
- 19.Ghuysen J-M, Leyh-Bouille M, Campbell JN, Moreno R, Frère J-M, Duez C, Nieto M, Perkins HR. Structure of the wall peptidoglycan of Streptomyces R39 and the specificity profile of its exocellular DD-carboxypeptidase-transpeptidase for peptide acceptors. Biochemistry. 1973;12:1243–1251. doi: 10.1021/bi00731a001. [DOI] [PubMed] [Google Scholar]
- 20.Zhao G-H, Duez C, Lepage S, Forceille C, Rhazi N, Klein D, Ghuysen J-M, Frère J-M. Site-directed mutagenesis of the Actinomadura R39 DD-peptidase. Biochem J. 1997;327:377–381. doi: 10.1042/bj3270377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson DE, Young KD. Penicillin-binding protein 5 affects cell diameter, contour and morphology of Escherichia coli. J Bacteriol. 2000;182:1714–1721. doi: 10.1128/jb.182.6.1714-1721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuhashi M, Tamaki S, Curtis SJ, Strominger JL. Mutational evidence for identity of penicillin-binding protein 5 in Escherichia coli with the major D-alanine carboxypeptidase IA activity. J Bacteriol. 1979;137:644–647. doi: 10.1128/jb.137.1.644-647.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris F, Brandenburg K, Seydel U, Phoenix D. Investigations into the mechanisms used by the C-terminal anchors of Escherichia coli penicillin-binding proteins 4, 5,6 and 6b for membrane interactions. Eur J Biochem. 2002;269:5821–5829. doi: 10.1046/j.1432-1033.2002.03295.x. [DOI] [PubMed] [Google Scholar]
- 24.Nicholas RA, Krings S, Tomberg J, Nicola G, Davies C. Crystal structure of wild-type penicillin-binding protein 5 from Escherichia coli. J Biol Chem. 2003;278:644–647. doi: 10.1074/jbc.M310177200. [DOI] [PubMed] [Google Scholar]
- 25.Stefanova ME, Davies C, Nicholas RA, Gutheil WG. pH, inhibitor and substrate specificity studies on Escherichia coli penicillin-binding protein 5. Biochim Biophys Acta. 2002;1597:292–300. doi: 10.1016/s0167-4838(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 26.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 27.Faraci WS, Pratt RF. Interactions of cephalosporins with the Streptomyces R61 DD-transpeptidase/carboxypeptidase. Influence of the 3′-substituent. Biochem J. 1986;238:309–312. doi: 10.1042/bj2380309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicholas RA, Krings S, Tomberg J, Nicola G, Davies C. Crystal structure of wild-type penicillin binding protein 5 from E. coli: Implications for deacylation of the acyl-enzyme complex. J Biol Chem. 2003;278:52826–52833. doi: 10.1074/jbc.M310177200. [DOI] [PubMed] [Google Scholar]
- 29.Davies C, White SW, Nicholas RA. Crystal structure of a deacylation-defective mutant of penicillin-binding protein 5 at 2.3 Å resolution. J Biol Chem. 2001;276:616–623. doi: 10.1074/jbc.M004471200. [DOI] [PubMed] [Google Scholar]
- 30.Nicola G, Peddi S, Stefanova M, Nicholas RA, Gutheil WG, Davies C. Crystal structure of Escherichia coli penicillin-binding protein 5 bound to a tripeptide boronic acid inhibitor: A role for Ser-110 in deacylation. Biochemistry. 2005;44:8207–8217. doi: 10.1021/bi0473004. [DOI] [PubMed] [Google Scholar]
- 31.Kelly JA, Dideberg O, Charlier P, Wery JP, Libert M, Moews PC, Knox JR, Duez C, Fraipont C, Joris B, Dusart J, Frère J-M, Ghuysen J-M. On the origin of bacterial-resistance to penicillin - comparison of a beta-lactamase and a penicillin target. Science. 1986;231:1429–1431. doi: 10.1126/science.3082007. [DOI] [PubMed] [Google Scholar]
- 32.Priyadarshini R, Popham DL, Young KD. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J Bacteriol. 2006;188:5345–5355. doi: 10.1128/JB.00476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheffers DJ, Jones LJ, Errington J. Several distinct localization patterns for penicillin-binding proteins in Bacillus subtilis. Mol Microbiol. 2004;51:749–764. doi: 10.1046/j.1365-2958.2003.03854.x. [DOI] [PubMed] [Google Scholar]
- 34.Kumar I, Josephine HR, Pratt RF. Reactions of peptidoglycan-mimetic β-lactams with penicillin-binding proteins in vivo and in membranes. ACS Chem Biol. 2007;2:620–624. doi: 10.1021/cb7001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hesek D, Suvarov M, Morio K, Lee M, Brown S, Vakulenko SB, Mobashery S. Synthetic peptidoglycan substrates for penicillin-binding proteins of gram-negative bacteria. J Org Chem. 2004;69:778–784. doi: 10.1021/jo035397e. [DOI] [PubMed] [Google Scholar]
- 36.Morlot C, Pernot L, Le Gouellec A, Di Giulmi AM, Vernet T, Dideberg O, Dessen A. Crystal structures of a peptidoglycan synthesis regulatory factor (PBP3) from Streptococcus pneumoniae. J Biol Chem. 2005;280:15984–15991. doi: 10.1074/jbc.M408446200. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, Shi Q, Meroueh SO, Vakulenko SB, Mobashery S. Catalytic mechanism of penicillin-binding protein 5 of Escherichia coli. Biochemistry. 2007;46:10113–10121. doi: 10.1021/bi700777x. [DOI] [PubMed] [Google Scholar]
- 38.Lim D, Strynadka NCJ. Structural basis for the β-lactam resistance of PBP 2a from methicillin-resistant Staphylococcus aureus. Nature Struct Biol. 2002;9:870–876. doi: 10.1038/nsb858. [DOI] [PubMed] [Google Scholar]
- 39.Macheboeuf P, Di Giulmi AM, Job V, Vernet T, Dideberg O, Dessen A. Active site restructuring regulates ligand recognition in class A penicillin-binding proteins. Proc Natl Acad Sci USA. 2005;102:577–582. doi: 10.1073/pnas.0407186102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuda C, Hesek DM, Lee M, Morio K-i, Nowak T, Mobashery S. Activation for catalysis of penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus by bacterial cell wall. J Am Chem Soc. 2005;127:2056–2057. doi: 10.1021/ja0434376. [DOI] [PubMed] [Google Scholar]
- 41.Fuda C, Hesek D, Lee M, Heilmayer W, Novak R, Vakulenko SB, Mobashery S. Mechanistic basis for the action of new cephalosporin antibiotics effective against methicillin-and vancomycin-resistant Staphylococcus aureus. J Biol Chem. 2006;281:10035–10041. doi: 10.1074/jbc.M508846200. [DOI] [PubMed] [Google Scholar]
- 42.Granier B, Duez C, Lepage S, Englebert S, Dusart J, Dideberg O, Van Beeumen J, Frere JM, Ghuysen JM. Primary and predicted secondary structures of the Actinomadura R39 extracellular DD-peptidase, a penicillin-binding protein (PBP) related to the Escherichia coli PBP4. Biochem J. 1992;282:781–788. doi: 10.1042/bj2820781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 44.CCP4. The CCP4 suite: programs for protein crystallography. Acta Cryst D Biol Cryst. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D Biol Cryst. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Cryst D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 50.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein structures in electron-density maps and the location of errors in these models. Acta Cryst A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 51.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 53.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
- 54.Merritt EA, Murphy MEP. Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Cryst D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]