

Terpene synthases often catalyze the committed step in natural products biosynthetic pathways and mediate complex reactions, leading to great interest in their enzymatic mechanisms.1 The resin acid constituents of the oleoresin defensive secretion of grand fir (Abies grandis) are derived from a mixture of abietadiene double bond isomers (1-3) produced by a single diterpene cyclase, abietadiene synthase (AgAS).2 In contrast, the resin acids of Norway spruce (Picea abies) originate from two closely related diterpene cyclases that individually produce either isopimara-7,15-diene (4) (PaPS) or abietadienes (1-3) (PaAS).3 The C13β methyl configuration of 4 is also found in the pimarenyl+ (5) intermediates produced en route to abietadienes in the reaction catalyzed by AgAS,4 and presumably PaAS. This suggests that PaPS terminates its cyclization reaction by deprotonation of a C13ß Me (iso)pimar-15-en-8-yl+ (5a+) intermediate also found in abietadiene formation (Scheme 1). In previous work we found that a specific single residue change (Ile to Thr) was sufficient to ‘short circuit’ the complex cyclization reaction catalyzed by ent-kaurene synthases to instead produce ent-pimara-8(14),15-diene, presumably by deprotonation of a mechanistically relevant ent-pimar-15-en-8-yl+ intermediate.5 Here we demonstrate that AgAS product output can be switched from abietadienes to pimaradienes by a similar single residue change, albeit with subtle differences that have important mechanistic implications.
Scheme 1.
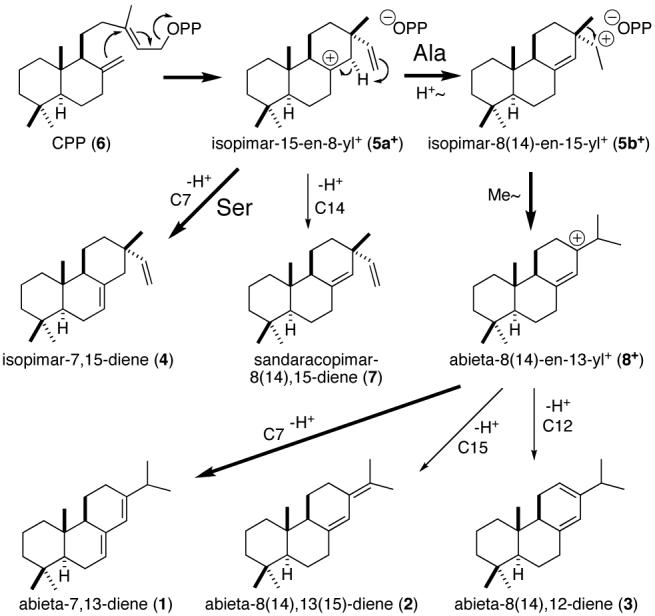
Cyclization to pimaradienes and abietadienes by AgAS.
Early work by Wenkert6 demonstrated the potential biogenetic origin of abietanes from pimaradienes initially suggested by Ruzicka.7 Later mechanistic work with recombinant AgAS established the intermediacy of isopimarenyl+ (5) intermediates in enzymatic production of abietadienes from copalyl diphosphate (CPP, 6).4 In addition, AgAS has been shown to produce sandaracopimara-8(14),15-diene (7) as a minor component (∼2%) of its product output,8 suggesting 7 as a potential stable intermediate. However, labeling studies demonstrated intramolecular proton transfer9 and 7 is not converted to abietadienes by AgAS,2 indicating that if 7 is a true intermediate it is only transiently formed en route to abietadienes in the AgAS active site. The catalyzed intramolecular proton transfer entails a shift from tertiary (5a+) to secondary carbocation that is driven, at least in part, by ion pairing of the resulting isopimar-8(14)-en-15-yl+ (5b+) with the pyrophosphate anion released by initiating ionization of CPP (6).4
Recent work has established the dramatic plasticity of terpene synthases, with small numbers of amino acid changes being sufficient to drive significant changes in enzymatic activity (particularly product outcome).5,10 However, previous alanine scanning mutagenesis of all the polar and charged residues in the relevant modeled active site of rAgAS generally led to very limited changes in product profile. The only exceptions were mutant enzymes that were also severely kinetically compromised (>10,000-fold reductions in catalytic efficiency).11 Note that AgAS, PaPS, and PaAS are all bifunctional diterpene cyclases that catalyze both mechanistically unusual protonation-initiated (i.e. class II) and the more typical diphosphate ionization-initiated (i.e. class I) cyclization reactions in separate active sites.12 The results presented here exclusively pertain to the class I active site analogous to that found in most terpene synthases.
Given the mechanistic and sequence similarity between PaPS and the abietadiene synthases AgAS and PaAS (i.e. the common isopimarenyl+ intermediate and >85% amino acid identity), we hypothesized that there might be a single residue switch responsible for this difference in product outcome similar to that we previously identified for ent-kaurene synthases.5 Alignment of these conifer diterpene cyclases with those from rice (Oryza sativa) utilized in our previous study revealed that there was only a small change in aliphatic side chain size at the corresponding position. In particular, AgAS and PaAS contain a Val at this position, while PaPS has a Leu (Figure 1). However, this type of change in ent-kaurene synthases did not alter product output (e.g. Val substitution for the mechanistically important Ile), indicating that introduction of polarity into the active site is critical. Intriguingly, four residues away PaPS contains a Ser in place of an Ala that is conserved in the abietadienes specific diterpene cyclases. The side chain of this residue forms part of the class I active site in modeled structures of these diterpene cyclases, being one turn of the relevant helix away from the single residue switch found for the ent-kaurene synthases. Considering the difference in configuration of the CPP (6) substrate utilized by the conifer diterpene cyclases and the enantio-stereoisomer utilized by ent-kaurene synthases, we reasoned that this residue might correspond to the hypothetical single residue switch.
Figure 1.
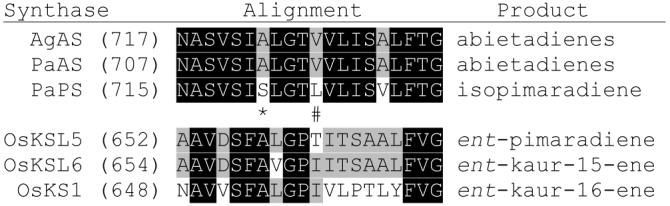
Diterpene synthase alignment. The location of the single residue switch reported here is indicated by * and that previously reported5 by #.
Substitution of this Ala by Ser in AgAS (AgAS:A723S) results in a diterpene cyclase that produces largely pimaradienes. Specifically, 75% isopimara-7,15-diene (4) and 21% sandaracopimara-8(14),15-diene (7), along with minor amounts (∼4%) of an unknown diterpene (Figure 2). Notably, both 4 and 7 have the C13β methyl stereochemistry expected from the known involvement of isopimarenyl+ intermediates in cyclization to abietadienes. In addition, AgAS:A723S retains catalytic activity comparable to the wild type enzyme, actually exhibiting slightly increased specific activity (∼2-fold), consistent with the possibility that this single residue switch might be evolutionarily relevant (i.e. mediate sufficient metabolic flux for resin acid production). While PaPS produces exclusively 4, and other changes would be necessary to convert AgAS to such a selective isopimaradiene synthase, the ability of this single residue change to convert rAgAS from producing >95% abietadienes (1-3) to >95% pimaradienes (4 and 7) is nevertheless quite remarkable.
Figure 2.
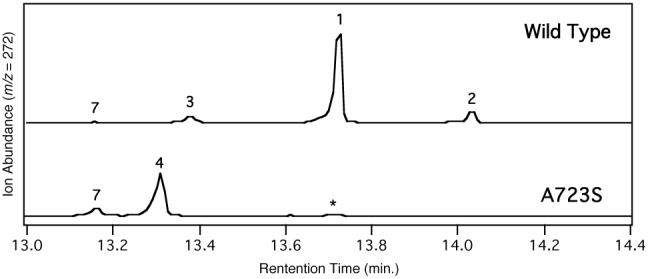
Effect of A723S mutation on AgAS product outcome. Chromatograms from GC-MS analysis of AgAS (either wild type or A723S mutant, as indicated). Numbers correspond to the diterpenes defined in the text and Scheme 1, as identified by comparison of retention time and mass spectra to authentic standards (* unknown diterpene).
The increased active site volume resulting from the Thr for Ile mutation in our previous study5 left open the possibility that the introduced hydroxyl group might coordinate a novel active site water that directly deprotonated the relevant pimarenyl+ intermediate. However, the Ser for Ala mutant reported here reinforces our hypothesis that the introduced hydroxyl group stabilizes the pimar-15-en-8-yl+ intermediate, albeit with the necessary shift in active site position presumably dictated by the difference in CPP configuration. This enables deprotonation, almost certainly by a separate active site general base, as use of the hydroxyl side chain as a general base seems implausible given the generally hydrophobic nature of terpene synthase active sites (including that modeled for AgAS). Indeed, AgAS:A723S seems to use a pre-existing active site general base. In particular, production of isopimar-7,15-diene (4) results from proton removal from C7, which is similarly deprotonated in the production of abieta-7,13-diene (1). This is the only position deprotonated in the final abieta-8(14)-en-13-yl+ (8+) intermediate to form the normally observed abietadienes (1-3) that also will quench the presumably common pimar-15-en-8-yl+ (5a+) intermediate (Scheme 1).
The presence of a pre-existing active site general base specifically for transient production of sandaracopimaradiene (7) might be expected to result in dominant production of 7 by AgAS:A723S. Thus, the relatively small proportion of this 8(14),15-diene actually observed suggests that 7 is not a true intermediate in production of the abietadienes.
In the reaction mechanisms leading to abietadienes and ent-kaurenes, transitions from tertiary pimar-15-en-8-yl carbocations to subsequent secondary carbocation intermediates (pimar-8(14)-en-15-yl+ (5b+) in the case of the abietadienes) are driven by ionic pairing with the pyrophosphate group released upon ionization of the CPP substrate.4,13 In particular, the secondary carbocations are significantly closer to the pyrophosphate than the preceding tertiary carbocations (e.g. Scheme 1). Accordingly, we hypothesize that the introduced hydroxyl group, at the relevant position in either AgAS or the previously examined ent-kaurene synthases, simply acts to stabilize pimar-15-en-8-yl+ intermediates long enough for deprotonation to occur. By contrast, the inert nature of the associated aliphatic residue indicates the more extended reactions leading to production of abietadienes or ent-kaurene are driven by the electrostatic effect of the ionized pyrophosphate group. This provides now two examples of such co-product assisted catalytic specificity. Accordingly, such a role for the ionized pyrophosphate group may be a general feature of terpene synthase catalyzed cyclization/rearrangement reactions.
Regardless of the exact means by which these mutations alter product outcome, their ability to do so is striking. Given the previously demonstrated conversion of an ent-pimaradiene synthase to the production of ent-kaurene by the converse Thr to Ile mutation,5 the similar findings reported here for alternative (normal) CPP stereochemistry offers the possibility of using such single residue switches to engineer product outcome in diterpene synthases more generally.
Supplementary Material
Acknowledgements
We thank Professors Robert M. Coates (Univ. Illinois) and Rodney B. Croteau (Washington State Univ.) for authentic standards, and Meimei Xu and Ke Zhou for technical assistance. This work was funded by a grant from the NIH (GM076324) to R.J.P.
References
- (1).Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- (2).Peters RJ, Flory JE, Jetter R, Ravn MM, Lee H-J, Coates RM, Croteau RB. Biochemistry. 2000;39:15592–15602. doi: 10.1021/bi001997l. [DOI] [PubMed] [Google Scholar]
- (3).Martin DM, Faldt J, Bohlmann J. Plant Physiol. 2004;135:1908–1927. doi: 10.1104/pp.104.042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ravn MM, Peters RJ, Coates RM, Croteau RB. J. Am. Chem. Soc. 2002;124:6998–7006. doi: 10.1021/ja017734b. [DOI] [PubMed] [Google Scholar]
- (5).Xu M, Wilderman PR, Peters RJ. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wenkert E, Chamberlin JW. J. Am. Chem. Soc. 1959;81:688–693. [Google Scholar]
- (7).Ruzicka L, Eschenmoser A, Heusser H. Experentia. 1953;IX:357–367. [Google Scholar]
- (8).LaFever RE, Stofer Vogel B, Croteau R. Arch. Biochem. Biophys. 1994;131:139–149. doi: 10.1006/abbi.1994.1370. [DOI] [PubMed] [Google Scholar]
- (9).Ravn MM, Coates RM, Jetter R, Croteau R. Chem. Commun. 1998:21–22. [Google Scholar]
- (10)(a).Kollner TG, Schnee C, Gershenzon J, Degenhardt J. Plant Cell. 2004;16:1115–1131. doi: 10.1105/tpc.019877. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoshikuni Y, Martin VJJ, Ferrin TE, Keasling JD. Chem. Biol. 2006;13:91–98. doi: 10.1016/j.chembiol.2005.10.016. [DOI] [PubMed] [Google Scholar]; (c) Yoshikuni Y, Ferrin TE, Keasling JD. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]; (d) Greenhagen BT, O’Maille PE, Noel JP, Chappell J. Proc Natl Acad Sci U S A. 2006;103:9826–9831. doi: 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Peters RJ, Croteau RB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:580–584. doi: 10.1073/pnas.022627099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Peters RJ, Ravn MM, Coates RM, Croteau RB. J. Am. Chem. Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]
- (13).Roy A, Roberts F, Wilderman PR, Zhou K, Peters RJ, Coates RM. J. Am. Chem. Soc. 2007;129:12453–12460. doi: 10.1021/ja072447e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.