

Abstract
In experiments designed to characterize the basis of amyloid fibril stability through mutational analysis of the Aβ(1-40) molecule, fibrils exhibit consistent, significant structural malleability. In these results, and in other properties, amyloid fibrils appear to more resemble plastic materials generated from synthetic polymers than they do globular proteins. Thus, like synthetic polymers and plastics, amyloid fibrils exhibit both polymorphism, the ability of one polypeptide to form aggregates of different morphologies, and isomorphism, the ability of different polypeptides to grow into a fibrillar amyloid morphology. This view links amyloid with the prehistorical and 20th Century use of proteins as starting materials to make films, fibers, and plastics, and with the classic protein fiber stretching experiments of the Astbury group. Viewing amyloid from the point of view of the polymer chemist may shed new light on issues such as the role of protofibrils in the mechanism of amyloid formation, the biological potency of fibrils, and the prospects for discovering inhibitors of amyloid fibril formation.
Amyloid fibrils and amyloid-related protein aggregates are associated with a variety of human diseases of the brain and the periphery (1, 2). The past decade has seen significant progress in our understanding of this alternatively folded protein structural motif. At the same time, much remains to be learned about details of amyloid structure, the basis of fibrillogenesis, and the nature of protein aggregate cytotoxicity.
Experimental work over the past 10-15 years has revealed the amyloid folding motif to be a nearly ubiquitous alternative folded state that can be accessed by many polypeptides. For globular proteins, the route to amyloid often depends on initial weakening of the native state; in addition, for all proteins and peptides, amyloid formation also depends on packing constraints within the fibril (3). Reviewed here are mutagenesis experiments designed to focus on understanding the nature of the packing constraints within the amyloid motif.
The results show that amyloid fibrils, like globular proteins, achieve stability through a mixture of hydrophobic and electrostatic forces. At the same time, the response of amyloid to mutagenesis differs in a number of ways from the typical response of globular proteins, and suggests instead a significant resemblance to synthetic polymers and plastics. A relationship to synthetic polymers provides a comfortable context for a number important features of amyloid fibrils, including: (a) the ability of so many naturally occurring proteins to form amyloid, (b) the observation of multiple conformational states of amyloid fibrils from the same polypeptide sequence, (c) the role of protofibrillar intermediates on the amyloid assembly pathway, (d) the biological potency of amyloid prions, and (e) the mixed success of attempts to identify inhibitors of the amyloid formation process.
NATURE OF PACKING INTERACTIONS WITHIN THE AMYLOID FIBRIL
Much of the data and analysis discussed here comes from studies on Aβ(1-40)1 amyloid fibrils, for which a characteristic equilibrium position for fibril elongation has been observed and exploited in the thermodynamic analysis of mutational effects. Thus, Aβ(1-40) fibril elongation tends towards a reproducible equilibrium position characterized by the concentration of monomeric peptide remaining in solution at the end of the fibril formation reaction (4-6). This critical concentration, Cr, is equivalent to the reciprocal of the fibril association equilibrium constant, Ka, which in turn can be used to calculate the ΔG for fibril elongation (6). Although technically the thermodynamics of elongation is not identical to the stability of the fibril, it would seem to be a good approximation of fibril stability, since fibrils are built up by a series of elongation reactions in which, in each cycle, incoming monomer explores the micro-environment at the growth end of the fibril.
Alanine mutagenesis and the fibril elongation equilibrium
Figure 1 shows the effects on the free energy of elongation in response to Ala substitution at different sequence positions in Aβ(1-40) (5). The results show that for many Ala replacements, especially in the C-terminal 2/3 of the molecule that is involved in the formation of the H-bonded amyloid core (Fig. 2a) (7, 8) the elongation equilibrium is destabilizing by ≥ 1 kcal/mol. To check that this degree of destabilization is what might be expected, we compared our data on the Aβ(1-40) parallel, in-register amyloid fibril (Figure 2a) (7, 9) to the stabilities of G(β1) variants in which residues at adjacent positions in parallel β-sheet were mutated in pairwise fashion (10) (Figure 2b). The ΔΔG values for the two studies (Table 1) agree remarkably well, especially considering the differences between the two types of experiment and the expected contributions to the data of unique environments within the protein structures. Thus, although the globular protein data was obtained by extrapolation of denaturation curves (10), while the fibril data was obtained from an equilibrium populated in native conditions (6), it would appear that side chain interactions make very similar contributions to β-sheet stability in fibrils and in globular proteins. This suggests that the analysis of elongation equilibria described here can provide useful data about amyloid fibril structure.
Figure 1.

Elongation thermodynamics of the Aβ(1-40) amyloid fibril in response to alanine replacements. A. The amino acid sequence of Aβ(1-40). B. ΔΔGAla-WT of the elongation equilibrium for Ala mutants of Aβ(1-40). A positive value indicated destabilization of the mutant compared with wild type. The x-axis shows the numbered residue replaced with Ala. The * at position 23 indicates that this mutant peptide could not be dissolved and hence fibrils could not be grown under defined conditions. Adapted from reference (5), with inclusion of additional data (ADW and RW, previously unpublished) obtained using published methods (61).
Figure 2.

(a) A model of a section of the Aβ(1-40) protofibril based on solid state NMR studies, based on pdb coordinates provided by Robert Tycko (7). (b) The G(β1) protein indicating residues 6 and 53 at adjacent positions in parallel β-sheet, from the pdb coordinates 2GB1. Figures rendered in PyMol (DeLano Scientific LLC).
Table 1.
Alanine destabilization of amyloid fibrils and globular proteinsa
| ΔΔG(Ala – residue), kcal/mol | |||||||
|---|---|---|---|---|---|---|---|
| G (β1)b | Aβ(1-40) amyloid fibrils | ||||||
| Mutation | 6 + 53 | 18 | 19 | 20 | 31 | 32 | 36 |
| Val->Ala | 1.25 | 1.3 | 1.0 | ||||
| Phe->Ala | 1.5 | 1.5 | 0.8 | ||||
| Ile->Ala | 1.65 | 2.0 | 1.0 | ||||
The free energy effects of Ala mutations at different sequence positions on the protein folding equilibria of the G(β1) domain and Aβ(1-40) fibrils are listed. Each column contains data for an X->Ala mutation at a particular position (Aβ fibrils) or positions (G(β1)).
Lack of additivity in multiple mutants
With some exceptions, mutations at distal positions in globular proteins tend to act independently of each other and therefore produce additive effects on protein stability and other properties (11, 12). Based on structural models for Aβ(1-40) fibrils derived from solid state NMR (7, 9), hydrogen-deuterium exchange (8), and cysteine mutational (13) studies, we conducted preliminary additivity experiments with Ala mutants of Aβ. We found that when Ala replaces both residues 17 and 34, which are well established to both be on the interior of the Aβ loop and in contact within the amyloid core (7, 13) (Fig. 3), the cumulative destabilization of the two mutations is clearly non-additive (Fig. 4a). Interestingly, when positions 17 and 27 are both replaced with Ala, the cumulative destabilization is also non-additive (Fig. 3a). The lack of additivity between residues 17 and 27 is somewhat surprising, given their remote locations in the structural models (Figs. 2a and 3a). Thus, the structural consequences of these mutations appear to be propagated at a significant distance through the protein structure, a result not usually observed in the analysis of globular proteins.
Figure 3.
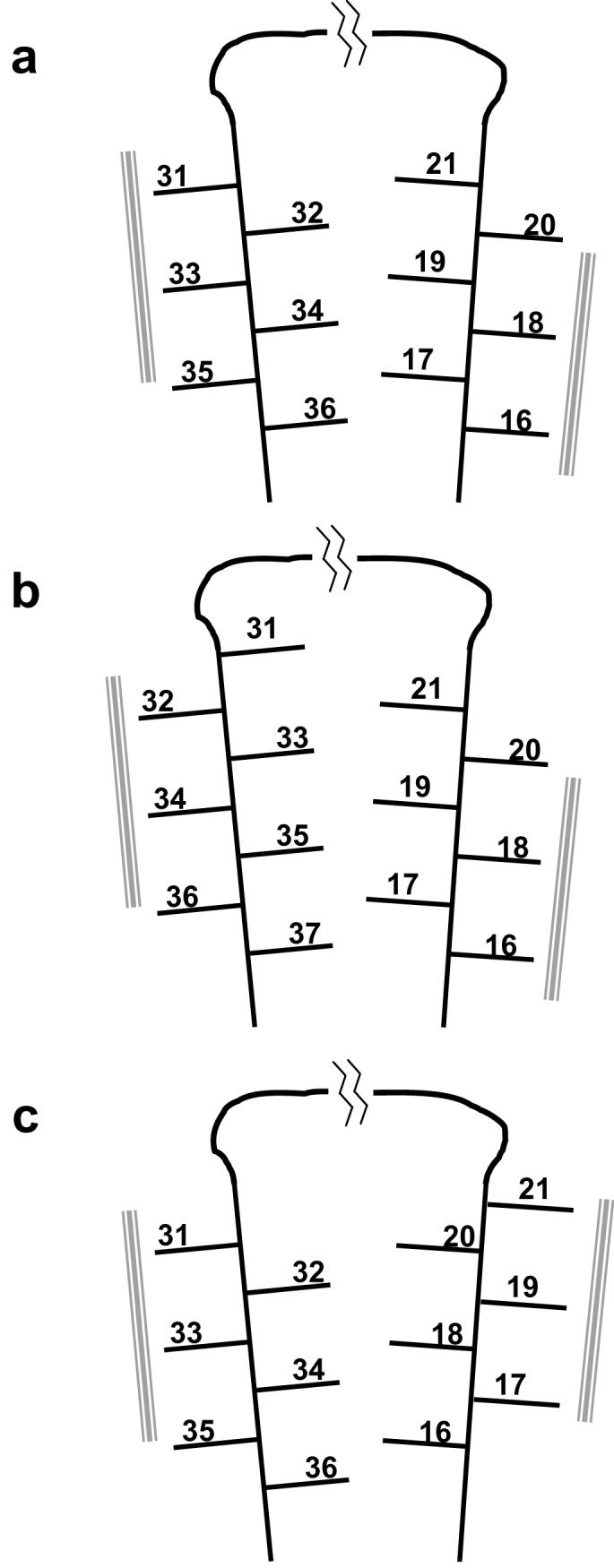
Schematic renderings of different models for the orientation of side chains within the Aβ hairpin in amyloid fibrils. The numbered lines represent the side chains of amino acids involved in β-sheet within the fibril. The gray bars represent apparent intermolecular packing residues. Part a is reproduced from reference (13).
Figure 4.

Elongation thermodynamics of multiple mutants of Aβ(1-40). (a) Additivity experiments of Ala mutants (see text). (b-d) Disulfide crosslinking experiments comparing the elongation equilibria of reduced (cross-hatched bars) and crosslinked, oxidized (open bars) double Cys mutants of Aβ(1-40) at the positions indicated. Part a is reproduced from reference (5) and part b from reference (13). Parts c and d based on previously unpublished data (SS and RW) obtained using published methods (62). Bars labeled “x + y” are mathematical sums of x and y data; bars labeled “x/y” show experimental data on x,y double mutants.
Atypical responses of the Aβ fibril to proline mutations – β-sheet migration within fibrils
We also studied proline mutants to inform on the locations of β-extended chain structure in the Aβ fibril (4, 14). The results (Fig. 5a) indicated two or three contiguous segments of sequence that are highly sensitive to proline replacement, while the N- and C-termini, as well as short elements between predicted β-extended chain segments, are relatively insensitive to replacement (4). One surprising feature of the results, however, is that not a single mutation was found that completely destabilizes fibril structure. This is in sharp contrast to globular proteins like the G(β1) domain (Figure 2b), in which single Pro replacements at β-sheet residues are so destabilizing that no significant fraction of folded protein is populated, leading to an estimated destabilization of ≥4 kcal/mol (15, 16). Figure 5a shows that the highest destabilization found for any Pro mutation in the Aβ fibril is 3 kcal/mol, and many other Pro mutations at residues suspected to be in β-sheet in the fibril (7, 8) are destabilized even less. What is the source of this qualitative difference between an amyloid fibril and a globular protein?
Figure 5.

Proline mutagenesis of Aβ(1-40) amyloid structure and stability. (a) A plot of ΔΔGPro-WT with respect to sequence position of proline mutation. (b) A plot of ΔΔGPro-Ala with respect to sequence position. Diamonds indicate positions not analyzed; * indicates not measurable because of solubility problems. (a) and (b) reprinted from reference (5). (c) Hydrogen-exchange protection of amyloid fibrils from Pro mutants of Aβ(1-40). Higher deuterium exchange (y-axis) indicates less protection and therefore fewer backbone amide hydrogens in β-sheet in the fibrils. Reprinted from reference (4).
One source of the missing folding energy appears to be the additional H-bonds that were surprisingly found to appear in proline-destabilized Aβ fibrils (4). When the number of backbone amide protons protected by the Aβ amyloid structure was assessed for wild type and Pro mutant fibrils, it was found that, in many cases, fibrils that are destabilized by a Pro mutation actually contain a larger number of protected amide protons (and hence, most likely, more H-bonded β-sheet) than the WT fibrils (Fig. 5c). This is especially surprising since it is expected that the effect of the Pro replacement will be a loss of H-bonded structure at the site of mutation. The site of the additional H-bonds must be one or more of the regions of Aβ(1-40) that are not already involved in H-bonded β-sheet in the wild type fibril, such as the N-terminal segment (7, 8, 17, 18). Since each added H-bond contributes stabilization energy, the added folding energy from these additional H-bonds appears to compensate for some of the destabilization caused by the Pro mutation. Many mutations in globular proteins affect the equilibrium by which the native structure is formed but alter the structure of the native folded state modestly (19, 20) or not at all. In contrast, some mutations in the Aβ amyloid fibril appear to produce extensive remodeling of the β-sheet network of the fibril, which has the effect of allowing a fibril to form in spite of a severe mutation.
To further investigate the how proline replacements affect elongation thermodynamics, the difference between the Pro - WT ΔΔG and the Ala – WT ΔΔG was determined and plotted. These values (Fig. 5b), which are effectively the Pro – Ala ΔΔG for each sequence position, should report specifically on the ability of the backbone configuration at a particular site to accept a Pro replacement (5). That is, the component of destabilization due to the loss of a hydrophobic side chain when a residue is replaced by Pro is largely factored out of the data in Figure 5b. The results are similar to those from the Pro scanning experiment (Fig. 5a) but with some significant differences. In particular, some residues that are very likely to be in β-sheet in the fibrils according to structural probes (4, 7, 18), such as residues 31, 34, 35, and 36, exhibit little or no destabilizing due to the main chain disruption effects of Pro (5). This apparent unresponsiveness to Pro is probably explained by compensatory gains from stabilizing interactions, such as additional H-bonds, as discussed above.
Resiliency in fibril structures from disulfide crosslinked monomers
Disulfide engineering has been used to provide important side chain packing information within the Aβ(1-40) fibril (13). In one set of experiments, three double Cys mutants were examined: L17C/L34C, L17C/M35C, and L17C/V36C. Fibrils were grown from each under reducing conditions, then exposed to molecular oxygen. Only one of the three, the 17/34 double, efficiently produced a disulfide-linkage within the monomer (revealed by HPLC after fibril dissolution (13)). These results were interpreted to indicate that (a) the two β-extended chain segments 15-21 and 31-37 containing these residues are oriented within the fibril so that residues 15, 17, 19 and 21 from one segment, and 32, 34 and 36 from the other segment, are directed into the center of the sheet-sheet sandwich, and that (b) within that packed sandwich, residues 17 and 34 are in contact (Figure 3a). (These results are consistent with independent solid-state NMR results (7)). This conclusion was also reached by comparing the elongation thermodynamics of the three double Cys mutants. In this case, oxidized, crosslinked monomers were allowed to form fibrils and their amyloid elongation equilibrium positions assessed. The results are shown in Figure 4b as a series of bar graphs. Confirming the results of the fibril crosslinking experiment described above, the 17/34 crosslinked monomer is the only one of the three to produce a fibril that is significantly more stable than the fibril from the reduced version of the same peptide. Furthermore, only the oxidized 17/34 fibrils exhibited stability very similar to wild type Aβ(1-40) fibrils.
The dramatic surprise in these experimental results was that the other two fibrils from oxidized double Cys mutants are only modestly destabilized compared to fibrils from wild type Aβ or the reduced double Cys mutants. Yet, constrained by the covalent disulfide bond, both of these fibrils must be substantially different in structure compared to the 17/34 crosslinked fibrils or wild type fibrils. Crosslinked 17/36 fibrils must consist of the same faces of the two β-extended chains facing each other within the packed filament (as shown in Fig. 3a), but with the register across the β-sandwich shifted so that now residues 17 and 36 are in contact. In a more extreme case, in crosslinked 17/35 fibrils, the 31-37 β-extended chain element must be engaged in fibril structure in a radically different way, so that the face containing residues 31, 33, 35, and 37 will now be facing the interior of the filament, with the side chains of residues 17 and 35 in contact (as shown in Fig. 3b). In spite of this potentially catastrophic structural change - the flipping of an extended chain element of the β-sheet by 180° - the fibrils so produced are only destabilized compared to wild type fibrils by about 2 kcal/mole. While it is possible that the 17-35 disulfide produces a more local deformation in the β-sheet, rather than a concerted segmental 180° rotation, there appears to be no way that a rearrangement that accommodates the 17-35 disulfide can be subtle or innocuous. Figure 6 shows schematically how segmental rotation of the 31-37 extended chain by 180° dramatically changes the packing arrangements within the β-sheet of a single amyloid filament. In particular, this rotation introduces a potential void in the sheet-sheet packing, due to the altered placement of Gly 33.
Figure 6.

Schematic of the packing of interior residues in Aβ(1-40) fibrils from two different orientations of side chains within the Aβ hairpin. Parts A and D represent wild type Aβ (part a of Figure 3) and parts B and E represent an alternative conformation (part b of Fig. 3) in which the C-terminal extended chain (residues 31-36) is rotated 180°. Part C shows the basic parallel, in-register structure of the Aβ fibril. Green shaded residues indicated residues 16-21; tan/orange residues, residues 31-36. There is no side chain sphere for residue 33, which is Gly. Figure constructed by Sarina Bromberg.
Other disulfide experiments (SS and RW, unpublished results) tell very similar stories. Figure 4c shows a comparison of the elongation ΔΔGs of the oxidized and reduced forms of the three double Cys mutants 17/34, 18/34 and 19/34. Of particular interest is the ΔΔG of the oxidized, crosslinked 18/34 mutant, which is only modestly destabilized compared to wild type Aβ(1-40). For this peptide to form fibrils, it would appear that the 16-21 β-extended strand of the peptide would have to undergo a 180° rotation (Figure 3c). Figure 4d shows a comparison of the elongation ΔΔGs of the oxidized and reduced forms of the three double Cys mutants 19/30, 19/32, and 19/34. Consistent with the model shown in Figure 3a, this experiment shows that only the 19/32 double mutant, when oxidized, produces an amyloid fibril with stability minimally distorted from that of the wild type. Figure 4d also shows that the fibrils formed from oxidized, crosslinked 19/30 and 19/34 mutants are only modestly destabilized, despite the fact that the details of the hydrophobic packing across the loop must be significantly altered from that shown in Figure 3a.
The 17/34 packing interaction implicated in these Cys experiments is consistent with ss-NMR data on Aβ(1-40) fibrils (7). Interestingly, the 17-34 interaction is not part of a structural model for Aβ(1-42) based on hydrogen exchange and other data (21). However, this is likely due to a real difference between Aβ(1-40) and Aβ(1-42) fibril structure and reflects the diversity of packing arrangements within the Aβ peptide capable of generating a stable amyloid fibril structure. Similarly, amyloid fibrils of the 10-40 fragment of Aβ appear to possess an alternative packing arrangement within the peptide as it folds into the amyloid motif (22). The resemblance between these polymorphic forms of Aβ fibrils, and the fibril structures generated by the disulfide experiments described above, is clear.
The packed, cross-β core of an amyloid fibril can be thought of as a three dimensional array in which each dimension consists of a type of stabilizing interaction: (1) covalent bonds (the β-extended peptide chain), (2) electrostatic bonds (the H-bonding direction of the β-sheet), and (3) packing interactions (hydrophobic and other interactions of the side chains). In the experiments described here, interactions in all three dimensions are dramatically altered without destroying the folding stability of the fibril. These experiments reveal a remarkable malleability or plasticity of the Aβ(1-40) fibril structure (4, 5, 13), in which radical changes in individual contacts can occur without a prohibitively large cost to stability. The resiliency of the amyloid fibril appears to be significantly greater than that of globular proteins. Since the folding of globular proteins tends to be highly cooperative, inputs of energy sufficient to disrupt the native state almost always lead to the formation of an essentially completely unfolded molecule, rather than the generation of an alternative structure. In contrast, when fibrils are exposed to mutations that in the globular protein world would be considered catastrophic, amyloid tends to “bend” (that is, structurally adjust), rather than “break” (that is, unfold).
STRUCTURAL PLASTICITY OF AMYLOID FIBRILS
The word “plasticity” is sometimes applied to cases in which globular proteins, in response to point mutations, exhibit local structural polymorphism, lack of additivity, etc (20, 23, 24). The above review of mutational studies of Aβ(1-40) fibrils also uses the word “plasticity” in this sense, to express the apparently very “forgiving” nature of amyloid structure, which appears to have the ability to withstand the stress of potentially highly destabilizing mutations through adjustments in non-covalent packing interactions. Clearly, such examples of structural ambiguity or degeneracy are not formally equivalent to the plastic deformations of semi-crystalline synthetic polymers that are associated with plastic materials (25). At the same time, plastic deformation implies the ability of the material to exist in multiple stable or metastable states that are related to each other by changes in local packing and are capable of interconversion without dissociation. The ability of amyloid fibrils to readily engage alternative packing arrangements in response to mutation, as reviewed in the first half of this article, would seem to reflect a structural degeneracy on a scale that is typically seen in semi-crystalline polymers but almost never observed in globular proteins. The remainder of this article explores the relationship between amyloid and synthetic polymers and plastics.
Amyloid and synthetic polymers – shared features, shared vocabulary
A central conundrum of the amyloid phenomenon is the ability of many different polypeptide sequences to form stable, fibrillar, cross-β structures (26). Understanding how it can be that many different polypeptide sequences, each highly evolved to fold into a particular structure, can also fold into a cross-β amyloid motif has been one of the challenges of the amyloid field. Interestingly, synthetic polymer assemblies often exhibit a similar “isomorphism”. For example, “copolymers can …. show isomorphism if different monomeric units can replace each other without change of lattice structure. Isomorphism of chains is also possible if the two corresponding homopolymers have analogous crystal modifications, similar lattice constraints, and the same helix types.” (27). Might it be that the amazing ability of so many protein sequences to form fibrils is simply due to the polymeric nature of the polypeptide sequence?
With rare exceptions, globular proteins normally exhibit only one stably folded state. In contrast, polymorphism - the ability of a single polypeptide sequence to assemble into two or more structurally distinct and self-propagating amyloid fibrils - has been observed for several proteins, including the sup35 yeast prion (28), Aβ(1-40) (29), and a peptide fragment of β2-microglobulin (30). The structural polymorphism of amyloid is biologically important, since it is likely the molecular basis of both strain phenomena (28) and species barriers (31) in prion biology. Amyloid polymorphism has recently been reviewed, from the point of view of polymer chemistry (32).
Structural polymorphism is commonly observed in plastic materials like the condensed states of synthetic polymers. In fact, a description of this phenomenon, from a basic text on plastics, reads remarkably like a summary of the recent discovery of multiple conformations of amyloid fibrils. “Most polymers show polymorphism: the same constitution and configuration leads to various energetically different crystal modifications. Polymorphism may be caused by different microconformations of polymer chains .. or by different packing of chains with identical chain conformations” (27). Synthetic polymers processed by different crystallization conditions are able to form fibrils, amorphous aggregates, stacks of platelets, and single lamellae (27), all of which are structures previously reported as alternative aggregated states during amyloid fibril formation (33-35). In a particularly striking case, the “Maltese cross” formations sometimes observed under the polarizing microscope in Congo red stained amyloid tissue (36) are also seen in synthetic polymers (27). In both settings, the molecular explanation for the Maltese cross is the formation of “spherulites”, spherical aggregates emanating from a central growth point that appear to form under conditions of limited nucleation. Given the differences in chemical structure between proteins and synthetic polymers such as polypropylene, the resemblance of the spherulites in the EM is startling (Figure 7), suggestive of a deeper physical and structural resemblance of amyloid and synthetic polymers (37).
Figure 7.

Polymer spherulites. “Maltese cross” images in light microscopy. (a) Brain of a mouse model of human Aβ amyloidosis stained with Congo red and visualized by polarizing light microscopy (36); (b) Photomicrograph of a deformed isotactic polypropylene spherulite (courtesy R. Samuels). Reprinted from reference (63). (c) Environmental scanning electron microscopy image of insulin amyloid spherulites; scalebar 30 μm. From reference (64). (d) Phase microscope image of an isotactic polypropylene spherulite (courtesy R. Samuels). Reprinted from reference (63).
In fact, models for the internal structure of semi-crystalline plastics, and how individual polymer chains fold in order to make up those structures (25, 27), bear a remarkable resemblance to models of amyloid fibrils derived from structural studies. Thus, while the structures and stabilities of amyloid fibrils appear to be dictated by the configurational constraints and microscopic interaction energies that also define globular protein structure, the morphological and energetic features, including structural polymorphism, that set them apart from globular proteins are more akin to the world of synthetic polymers and plastics (25, 27).
Amyloid as a synthetic polymer
A resemblance of amyloid fibrils to synthetic polymers and plastics should not be surprising. The first plastic materials used by man were probably the paints used in cave paintings such as those found at Altamira, which were derived from egg white and blood proteins. Cow horn (keratin) was perhaps the first thermoplastic – it was molded into thin sheets to make lantern windows. One of the more well known early commercial synthetic plastics was Galalith, produced by treatment of casein with formaldehyde (27). Over 60 years ago, there was considerable interest in the commercial exploitation of the plastic and textile properties of artificial protein fibers, films, adhesives and plastics (38, 39). Interestingly, Astbury's work on protein fibers can be viewed as plastic deformations induced by applying force to protein fibers. Starting in the 1920s, Astbury and colleagues performed electron diffraction experiments on wool and other protein fibers (40). The designation of “α” and “β” to the two main types of protein secondary structure derive from Astbury's definition of these motifs (long before their underlying structural meaning was clear) in the diffraction patterns of stretched and unstretched wool fibers. The term “cross-β” grew out of similar experiments in which poached eggs produced a cross-β pattern, while stretching of this cross-β form produced fibers with diffraction patterns analogous to those of stretched wool (β-keratin) (39).
Synthetic polymers fall into two broad classes according to their microscopic structures and the properties defined by those structures. Amorphous polymers exhibit little or no long-range order and exist as a glass. In contrast, semi-crystalline polymers exhibit a degree of three dimensional order that is normally thought of as reflecting a mix of highly regular “crystallites” and irregular elements. The degree of order can vary, even for a single polymer, and appears to do so as a result of the nucleated crystallization process. Nucleation can be homogeneous, brought about by alignment within or between polymer molecules; alignment, in turn, can be induced by external forces, such as shear. The requirement for normally rare homogeneous nucleation events can be abrogated by heterogeneous nucleation, in which exogenous factors, such as an added compound or the container wall, provide an amenable surface for initiating aggregation (27). Interestingly, both shear forces (stirring / agitation) (29) and the chemical nature of vessel walls (41) are well known to enhance amyloid growth rates.
In contrast to the simple, two-state crystallization of globular proteins, the crystallization of polymers can involve a number of intermediate states, each with different levels of crystallinity. The transition from melt to solid sometimes only constitutes a “primary stage crystallization”. This is followed by an “after-crystallization” characterized by improved crystallinity, thicker lamellae, and transition to a denser state. Polymorphic forms of polymers can arise as different end states caused by differences in nucleation, cooling rates, etc., or can be stages along the crystallization path toward maximized stability, through a kind of ageing phenomenon (27).
Amyloidogenic proteins exhibit, in their condensed states, two kinds of polymorphism (32). Most of these peptides form spherical oligomers and protofibrils in the early stages of amyloid assembly; as alternative condensed states of these peptides, which in some cases are structurally related to the final amyloid fibrils (8), these states are themselves polymorphic variations. In addition, the final amyloid fibrils also often exist in different polymorphic forms (25, 27); these may be kinetically trapped alternative states, or may be sequential intermediates toward a thermodynamically most stable form. In any case, it is likely that they differ from each other in their degrees of crystallinity, in analogy to semi-crystalline polymers (32).
What is it about amyloid fibril structure that produces properties more like synthetic polymers than globular proteins? Part of the answer likely lies in the malleability of both the H-bonding networks and the hydrophobic packing relationships in the amyloid β-sheets, as discussed above for Aβ fibrils. However, H-bonding networks can't explain the structural plasticity of most synthetic polymeric materials, since the interactions within or between the individual polymeric molecules (such as polypropylene and polyethylene) in many cases do not involve extended H-bonded networks. Instead, the most obvious general similarity between amyloids and synthetic plastics is that both involve condensed states in which individual polymer molecules are associated with each other through non-covalent interactions. That is, while the stability of both individual polymeric molecules and plastics depends on contributions from both intermolecular structural contacts and interactions with the solvent, the structural stability and properties of plastics also depend on the interactions between the individual subunits (25). In amyloid, this means not only the H-bonds and hydrophobic packing between the layers of the basic cross-β filament (Figure 6), but also the less well-characterized interactions between the protofilaments. It seems clear that the more such inter-subunit interactions contribute to overall fibril stability, the less necessary it is that the intramolecular packing contacts within the aggregate be highly optimized. This makes possible the existence of stable structures with only intermediate degrees of crystallinity in the core packing region. This analysis suggests that it will be very important to understanding the structural and energetic aspects of the interactions between the individual protofilaments that make up the mature amyloid fibril.
IMPLICATIONS OF AMYLOID PLASTICITY
Amyloid assembly mechanisms
Due especially to their potential role in human disease (42), there is currently intense interest in the structure of oligomeric pre-amyloid assemblies, and their role in the amyloid assembly mechanism (32). While it may turn out that early intermediates like spherical oligomers and protofibrils are off-pathway, in some cases they may be on-pathway, such that fibril nucleation and/or growth may involve concerted structural transformations within the aggregate as a key step to the formation of a final, amyloid structure (43, 44). Such a mechanism would be supported, for example, by compositional (45) and structural (8) relationships between protofibrils and fibrils. While such large scale structural rearrangements are not within the realm of experience of the protein chemist, they are commonplace among polymer chemists. Transformations can occur by two mechanisms. The first, plastic deformation, requires that the transformation be induced under physical stress, and generally implies that, on relief of stress, the transformation is reversed (25). The second, multi-stage crystallization, implies a conformational search for accessible positions of increased order and stability, in a step-wise transition to higher crystallinities. This latter probably better suits the situations under which oligomeric aggregates are seen to be replaced by mature amyloid fibrils.
Fine structure and specificity co-existent with polymorphism
At first glance, the plastic nature of the amyloid fibril may seem at odds with two previously described properties of amyloid fibrils: self-propagation of polymorphic forms, and well-defined packing interactions. However, while the plasticity of the amyloid folding motif leads to the existence of multiple possible conformers or polymorphs, any one of these polymorphic forms, once produced, exhibits a finite structure of sufficient integrity that it can propagate itself by seeded growth. This is perhaps not so difficult to visualize when the degree of order in the aggregate is high (46). However, even amyloid fibrils with intermediate levels of crystallinity appear to be able to self-replicate with high fidelity (29).
Implications for inhibition of protein aggregation
Significant efforts have been made to discover small molecule inhibitors of amyloid growth, both in industry and academia, for example by screening or design (47, 48). Very often, however, compounds that are discovered to influence the course of an aggregation reaction are required at high concentrations stoichiometric with the amyloidogenic peptide, and/or are found to alter the nature of the aggregation product, rather than completely inhibit aggregate formation (32, 49-51).
A small molecule capable of binding to the growth face of an amyloid fibril might be considered to be the equivalent of a highly destabilizing mutation within the amyloidogenic peptide, such as the Pro replacements described above (Fig. 5). If this is a reasonable analogy, then the expectation would be that, as in the case of the Pro replacements, an aggregate might still be expected to form, but with some alteration of its final structure. Furthermore, compounds that actually stimulate aggregation are well-known in polymer chemistry, where small molecules are sometimes used as heterogeneous nucleating agents (25, 27, 52).
Given the above considerations, it may be a more realistic goal to search for small molecules that can change the course of an amyloid growth reaction so that toxic aggregates are disfavored at the expense of non-toxic aggregate formation (53). This places an emphasis, however, on detailed analysis of the reaction products formed in the presence of candidate inhibitor compounds (51).
Amyloid materials science
As discussed above, thinking of amyloid as a condensed polymer provides insights into the response of fibrils to mutations and to mechanisms of amyloid assembly. In addition, some properties of amyloid as a material appear to play a role in amyloid biology. It is also possible that amyloid will some day be engineered to serve materials science applications. Thus, the strength of a yeast prion phenotype has been shown to be related to the ease with which these fibrils fracture during amyloid elongation (28). Since growth rates depend on the elongation rate constant as well as the concentrations of both monomer and fibril growth ends, multiplication of the latter influences the ability of the prion to propagate in vitro. The strength and mechanical stiffness of insulin amyloid fibrils, assessed by atomic force microscopy, suggest that amyloid may have useful material properties (54). Other investigations of the utility of amyloid and amyloid-like aggregates as materials include surface properties for supporting cell culture (55), use as nanowires (56), and the ability to form liquid crystal phases (57). In contrast to traditional synthetic polymers, polypeptides offer a number of potential advantages as the basis of plastic materials, including uniform lengths, specificity and complexity of sequence, and generally good kinetic solubility allowing slow crystallization.
Implications for amyloid structure
Some amyloid fibrils from short peptides appear to be very highly ordered (46), and micro-crystals of short amyloidogenic peptides have supported high resolution crystal structure determinations that have provided potentially general insights into the amyloid motif (58, 59). In polymer chemistry, however, longer chain lengths normally lead to lower degrees of crystallinity in the condensed state due to the constraints of chain folding (25). Furthermore, as discussed above, one way that polymorphic forms of polymers differ is in their degrees of crystallinity. It may thus be unrealistic to expect that all amyloids will exhibit the same high degrees of order that are found in structures formed by short peptides. Furthermore, while it is generally accepted in protein crystallography that multiple crystal forms may exhibit differences in quality, but not major differences in structure itself, this may not be true for amyloids, for which, like synthetic polymers, polymorphic forms are defined, at least in part, by their degrees of crystallinity. Improving the crystallinity of a particular amyloid polymorph may be commensurate with changing the nature and structure of the polymorph.
CONCLUSIONS
Although the recent concentration of protein-related fields on amyloid fibrils and similar aggregates creates an impression of novelty and freshness, the observation and exploitation of the ability of soluble proteins to undergo a phase change well precedes the scientific enterprise itself, in such prehistoric exercises as the boiling of eggs and the curdling of milk, and also stands at the origins of protein science, in the characterization of the aggregation response of proteins to heat and denaturants (60). In the same way, the results summarized here, while perhaps providing some new insights into fundamental differences between the structural basis of globular proteins and amyloid fibrils, have precedents in the work of Astbury and others on transformations within protein fibers. The idea of the relationship of amyloid fibrils to plastics and synthetic polymers also has precedents in the historical (and pre-historical) use of proteins as precursors for synthetic plastics.
The plastic nature of amyloid appears to involve malleability of both side chain interactions and H-bonded secondary structure, and at its most basic level can be attributed to an apparently significant stability contribution from the non-covalent, multimeric nature of the amyloid fibril, a feature these fibrils share with more traditional plastic materials. At the same time, an amyloid of a particular protein appears to be an unusual plastic in some respects, such as in the homogeneous chain lengths of the monomers, the complexity and homogeneity of the side chain sequences, and the amphipathic nature of many polypeptides. Such unique features, combined with our ever-increasing abilities to dissect amyloid structure and the molecular basis of amyloid stability, may provide a level of control over the final product unprecedented in the synthetic plastics field, and encourage practical uses of amyloid-like fibrils in material science and nanotechnology. In addition, the original impulse for the current focus on amyloid - its connection to human disease - can only be better served by an improved understanding of the biophysical basis of fibril structure, stability, and transformations.
ACKNOWLEDGMENT
We thank Frank Ferrone for helpful discussions.
Footnotes
Supported by NIH grant R01 AG018416 (RW).
ABBREVIATIONS
Aβ, amyloid beta protein; Cr, critical concentration; G(β1), Streptomyces griseus immunoglobulin binding protein, β1 domain.
REFERENCES
- 1.Martin JB. Molecular basis of the neurodegenerative disorders [published erratum appears in N Engl J Med 1999 Oct 28;341(18):1407] N Engl J Med. 1999;340:1970–1980. [Google Scholar]
- 2.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 3.Wetzel R. Mutations and off-pathway aggregation. Trends in Biotechnology. 1994;12:193–198. doi: 10.1016/0167-7799(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 4.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Williams AD, Shivaprasad S, Wetzel R. Alanine scanning mutagenesis of Abeta(1-40) amyloid fibril stability. J Mol Biol. 2006;357:1283–1294. doi: 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 6.O'Nuallain B, Shivaprasad S, Kheterpal I, Wetzel R. Thermodynamics of abeta(1-40) amyloid fibril elongation. Biochemistry. 2005;44:12709–12718. doi: 10.1021/bi050927h. [DOI] [PubMed] [Google Scholar]
- 7.Petkova AT, Yau WM, Tycko R. Experimental Constraints on Quaternary Structure in Alzheimer's beta-Amyloid Fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kheterpal I, Chen M, Cook KD, Wetzel R. Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange--mass spectrometry with on-line proteolytic fragmentation. J Mol Biol. 2006;361:785–795. doi: 10.1016/j.jmb.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 9.Benzinger TL, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer's beta-amyloid(10-35) is parallel beta-sheet with residues in exact register. Proc Natl Acad Sci U S A. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merkel JS, Sturtevant JM, Regan L. Sidechain interactions in parallel beta sheets: the energetics of cross-strand pairings. Structure Fold Des. 1999;7:1333–1343. doi: 10.1016/s0969-2126(00)80023-4. [DOI] [PubMed] [Google Scholar]
- 11.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 12.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1996;1:R121–126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 13.Shivaprasad S, Wetzel R. An intersheet packing interaction in A beta fibrils mapped by disulfide cross-linking. Biochemistry. 2004;43:15310–15317. doi: 10.1021/bi048019s. [DOI] [PubMed] [Google Scholar]
- 14.Wood SJ, Wetzel R, Martin JD, Hurle MR. Prolines and amyloidogenicity in fragments of the Alzheimer's peptide β/A4. Biochem. 1995;34:724–730. doi: 10.1021/bi00003a003. [DOI] [PubMed] [Google Scholar]
- 15.Minor DL, Jr., Kim PS. Measurement of the β-sheet-forming propensities of amino acids. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 16.Minor J, Daniel L, Kim PS. Context is a major determinant of β-sheet propensity. Nature. 1994;371:264–267. doi: 10.1038/371264a0. [DOI] [PubMed] [Google Scholar]
- 17.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural features of the Aβ amyloid fibril elucidated by limited proteolysis. Biochem. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 18.Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and Dynamic Features of Alzheimer's Abeta Peptide in Amyloid Fibrils Studied by Site-directed Spin Labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Baase WA, Baldwin E, Matthews BW. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 1998;7:158–177. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vetter IR, Baase WA, Heinz DW, Xiong JP, Snow S, Matthews BW. Protein structural plasticity exemplified by insertion and deletion mutants in T4 lysozyme. Protein Sci. 1996;5:2399–2415. doi: 10.1002/pro.5560051203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1-42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paravastu AK, Petkova AT, Tycko R. Polymorphic fibril formation by residues 10-40 of the Alzheimer's beta-amyloid peptide. Biophys J. 2006;90:4618–4629. doi: 10.1529/biophysj.105.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D, Petsko GA. Structural basis for conformational plasticity of the Parkinson's disease-associated ubiquitin hydrolase UCH-L1. Proc Natl Acad Sci U S A. 2006;103:4675–4680. doi: 10.1073/pnas.0510403103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Birley AW, Haworth B, Batchelor J. Physics of Plastics. Hanser Verlay; Munich: 1991. [Google Scholar]
- 26.Fandrich M, Fletcher MA, Dobson CM. Amyloid fibrils from muscle myoglobin. Nature. 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 27.Elias H-G. An Introduction to Plastics. 2nd Edition ed. Wiley-VCH; Weinheim: 2003. [Google Scholar]
- 28.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 29.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's {beta}-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi K, Takahashi S, Kawai T, Naiki H, Goto Y. Seeding-dependent propagation and maturation of amyloid fibril conformation. J Mol Biol. 2005;352:952–960. doi: 10.1016/j.jmb.2005.07.061. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission: an infectious conformation compatible with two highly divergent yeast prion proteins. Cell. 2005;121:49–62. doi: 10.1016/j.cell.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol. 2007;17 doi: 10.1016/j.sbi.2007.01.007. In press. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA. 1993;90:3334–3338. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer's peptide Aβ. J. Mol. Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 35.Chen S, Berthelier V, Hamilton JB, O'Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 36.Manuelidis L, Fritch W, Xi YG. Evolution of a strain of CJD that induces BSE-like plaques. Science. 1997;277:94–98. doi: 10.1126/science.277.5322.94. [DOI] [PubMed] [Google Scholar]
- 37.Krebs MR, Macphee CE, Miller AF, Dunlop IE, Dobson CM, Donald AM. The formation of spherulites by amyloid fibrils of bovine insulin. Proc Natl Acad Sci U S A. 2004;101:14420–14424. doi: 10.1073/pnas.0405933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senti FR, Eddy CR, Nutting GC. Conversion of globular to oriented fibrous proteins. I. By heat and mechanical working. J Am Chem Soc. 1943;65:2473. [Google Scholar]
- 39.Astbury WT. Artificial protein fibres: their conception and preparation. Nature. 1945;155:501–503. [Google Scholar]
- 40.Astbury WT, Woods HJ. X-Ray studies of the structure of hair, wool, and related fibres. II. The molecular structure and elastic properties of hair keratin. Philos Trans R Soc Lond B Math Phys. 1934;232:333–394. [Google Scholar]
- 41.Wall J, Murphy CL, Solomon A. In vitro immunoglobulin light chain fibrillogenesis. Methods in Enzymology. 1999:204–217. doi: 10.1016/s0076-6879(99)09016-3. [DOI] [PubMed] [Google Scholar]
- 42.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 43.Harper JD, Lieber CM, Lansbury PT., Jr. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem Biol. 1997;4:951–959. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- 44.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 45.Kheterpal I, Lashuel HA, Hartley DM, Walz T, Lansbury PT, Jr., Wetzel R. Abeta protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry. 2003;42:14092–14098. doi: 10.1021/bi0357816. [DOI] [PubMed] [Google Scholar]
- 46.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Findeis MA. Approaches to discovery and characterization of inhibitors of amyloid beta-peptide polymerization. Biochim Biophys Acta. 2000;1502:76–84. doi: 10.1016/s0925-4439(00)00034-x. [DOI] [PubMed] [Google Scholar]
- 48.Sciarretta KL, Gordon DJ, Meredith SC. Peptide-based inhibitors of amyloid assembly. Methods Enzymol. 2006;413:273–312. doi: 10.1016/S0076-6879(06)13015-3. [DOI] [PubMed] [Google Scholar]
- 49.Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ. New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer's disease. J Biol Chem. 2002;277:42881–42890. doi: 10.1074/jbc.M206593200. [DOI] [PubMed] [Google Scholar]
- 50.Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson's disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- 51.Williams AD, Sega M, Chen M, Kheterpal I, Geva M, Berthelier V, Kaleta DT, Cook KD, Wetzel R. Structural properties of Abeta protofibrils stabilized by a small molecule. Proc Natl Acad Sci U S A. 2005;102:7115–7120. doi: 10.1073/pnas.0408582102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blomenhofer M, Ganzleben S, Hanft D, Schmidt H-W, Kristiansen M, Smith P, Stoll K, Maeder D, Hoffmann K. “Designer” Nucleating Agents for Polypropylene. Macromolecules. 2005;38:3688–3695. [Google Scholar]
- 53.Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc Natl Acad Sci U S A. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith JF, Knowles TP, Dobson CM, Macphee CE, Welland ME. Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci U S A. 2006;103:15806–15811. doi: 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 56.Reches M, Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 57.Scheibel T, Parthasarathy R, Sawicki G, Lin XM, Jaeger H, Lindquist SL. Conducting nanowires built by controlled self-assembly of amyloid fibers and selective metal deposition. Proc Natl Acad Sci U S A. 2003;100:4527–4532. doi: 10.1073/pnas.0431081100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neurath H, Greenstein JP, Putnam FW, Erickson JO. The chemistry of protein denaturation. Chemical Reviews. 1944;34:157–265. [Google Scholar]
- 61.O'Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, Thiagarajan G, Wetzel R. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- 62.Shivaprasad S, Wetzel R. Analysis of amyloid fibril structure by scanning cysteine mutagenesis. Methods Enzymol. 2006;413:182–198. doi: 10.1016/S0076-6879(06)13010-4. [DOI] [PubMed] [Google Scholar]
- 63.Samuels RJ. Structured polymer properties. The Identification, interpretation, and use of polymer structure. Wiley-Interscience; New York: 1974. [Google Scholar]
- 64.Krebs MR, Bromley EH, Rogers SS, Donald AM. The mechanism of amyloid spherulite formation by bovine insulin. Biophys J. 2005;88:2013–2021. doi: 10.1529/biophysj.104.051896. [DOI] [PMC free article] [PubMed] [Google Scholar]