

Abstract
In this study, we establish an MCF-7 xenograft model that mimics the progression of human breast carcinomas typified by loss of p53 integrity, development of centrosome amplification, acquired estrogen receptor (ERα) heterogeneity, overexpression of Mdm2 and metastatic spread from the primary tumor to distant organs. MCF-7 cells with abrogated p53 function (vMCF-7Dnp53) maintained nuclear ERα expression and normal centrosome characteristics in vitro. However, following mitogen stimulation, they developed centrosome amplification and a higher frequency of aberrant mitotic spindles. Centrosome amplification was dependent on cdk2/cyclin activity since treatment with the small molecule inhibitor SU9516 suppressed centriole reduplication. In contrast to the parental MCF-7 cells, when introduced into nude mice as xenografts, tumors derived from the vMCF-7DNp53 cell line developed a strikingly altered phenotype characterized by increased tumor growth, higher tumor histopathology grade, centrosome amplification, loss of nuclear ERα expression, increased expression of Mdm-2 oncoprotein and resistance to the antiestrogen tamoxifen. Importantly, while MCF-7 xenografts did not develop distant metastases, primary tumors derived from vMCF-7DNp53 cells gave rise to lung metastases. Taken together, these observations indicate that abrogation of p53 function and consequent deregulation of the G1/S cell cycle transition leads to centrosome amplification responsible for breast cancer progression.
Keywords: cell cycle, estrogen independence, mitosis, tumor progression
Introduction
The progression of human breast cancer from an estrogen-dependent to an estrogen-independent form and more aggressive phenotype represents a major clinical problem that limits the long-term usefulness of endocrine therapeutic strategies (Ingle, 2004; Glass et al., 2007). Among estrogens, 17-β estradiol is the major promoter of cell proliferation in both normal and neoplastic breast tissue through its binding to the high-affinity estrogen receptor (ERα). ERα functions as an estrogen-activated transcription factor and mediates the expression of target genes involved in the regulation of cell proliferation and inhibition of apoptosis of the breast epithelium (Cicatiello et al., 2004; Helguero et al., 2005). Excessive activation of the ERα pathway due to increased hormonal secretion, prolonged estrogen exposure or increased levels of the receptor may lead to increased risk to develop breast cancer (Calaf, 2006; Lee et al., 2006). Although the ERα mitogen-signaling pathway is implicated in the development of breast cancer, the natural evolution of most breast carcinomas is characterized by loss of ERα expression, antiestrogen resistance and poor outcome (McDonnell and Norris, 2002; Giacinti et al., 2006).
Centrosome duplication plays a critical role in the maintenance of chromosomal stability through the propagation of a diploid karyotype. Centrosome duplication is coordinated with DNA replication, ensuring the formation of a bipolar mitotic spindle and equal chromosome segregation during cytokinesis (Brinkley and Goepfert, 1998; Sluder and Hinchcliffe, 2000). Breast tumor cells, as well as breast cancer cell lines, frequently show centrosome amplification and the consequent formation of aberrant mitotic figures (Lingle et al., 1998; D’Assoro et al., 2002; Daniels et al., 2004). Chromosomal instability and consequent aneuploidy are the hallmarks of breast cancer and represent a mechanism for the evolution of phenotypic diversity in cancer cell populations (Lengauer et al., 1998; Lingle et al., 2002; Kawamura et al., 2004; Fukasawa, 2005;Suizu et al., 2006). Importantly, earlier we established a mechanistic link between estrogen and the development of centrosome amplification and chromosomal instability in the ACI rat model for mammary tumorigenesis (Li et al., 2004).
Centrosome homeostasis is disrupted in cancer cells lacking normal function of the tumor suppressors p53 and Rb or deregulated activity of cdk2 and/or mitotic kinases, leading to centriole reduplication (Fukasawa et al., 1996; D’Assoro et al., 2004; Hernando et al., 2004; Duensing et al., 2006a, b, 2007; Fukasawa, 2007). Furthermore, following treatment with anticancer genotoxic agents, centrosome amplification and consequent chromosomal instability are exacerbated in cancer cells lacking p53 function (Bennett et al., 2004; D’Assoro et al., 2004). Importantly, human breast tumors with abrogated p53 function are also frequently ERα negative and display more aggressive behavior (Angeloni et al., 2004). In a p53-null mouse model for mammary tumorigenesis, cancer cells arising from ERα-positive breast carcinomas develop loss of ERα expression, suggesting that abrogation of p53 function may accelerate the development of phenotypic heterogeneity in breast cancer (Medina et al., 2003). Taken together, these findings suggest that one mechanism by which tumor cell heterogeneity arises in cancer is through abrogation of p53 function, loss of centrosome homeostasis and consequent chromosomal instability.
In this study, we investigate the regulation of centrosome duplication in MCF-7 breast cancer cells with endogenous wild-type p53, or in variant MCF-7 cells (vMCF-7DNp53) engineered to overexpress a dominant negative p53val135 mutant to mask the function of wild-type p53. We show that impaired p53 function leads to centrosome amplification through deregulation of cdk2/cyclin activity following mitogen stimulation in vitro. In addition, to determine if centrosome amplification and aberrant mitoses result in the development of a more aggressive tumor phenotype in vivo, we established MCF-7 and vMCF-7DNP53 xenografts in nude mice. Importantly, in vMCF-7DNp53 xenografts, development of centrosome amplification was linked to acquired ERα phenotypic heterogeneity, antiestrogen resistance and development of metastasis to distant sites. Taken together, these findings suggest a mechanism for the development of subpopulations of ERα-negative cancer cells arising from ERα-positive breast carcinomas through abrogation of p53 function, deregulation of cdk2 activity and consequent centrosome amplification.
Results
Centriole dynamics during cell cycle progression in MCF-7 cells
MCF-7 cells display an early breast cancer phenotype characterized by estrogen dependence for growth, wild-type p53 and normal centrosome phenotype. Therefore, we employed MCF-7 expressing recombinant green fluorescent protein (GFP)-centrin (MCF-7 GFP–cetn2) to directly monitor the timing of centriole duplication (D’Assoro et al., 2001). MCF-7 GFP–cetn2 and the parental MCF-7 cells showed similar proportions of G1, S and G2/M phase cells demonstrating that GFP-centrin expression had no gross effect on cell cycle progression (Figure 1a). To determine the relationship between hormone stimulation, centriole duplication and cell cycle progression, cells were arrested in the G0 phase of the cell cycle by estrogen withdrawal for 48 h followed by stimulation of cell cycle reentry by addition of 17-β estradiol, EGF and IGF-I (Figure 1b). Fluorescence-activated cell sorting analysis showed that following hormone stimulation, cells progressed synchronously through a complete cell cycle, reaching a maximum level of DNA synthesis at 24 h and becoming asynchronous after passing through mitosis by 48 h.
Figure 1.
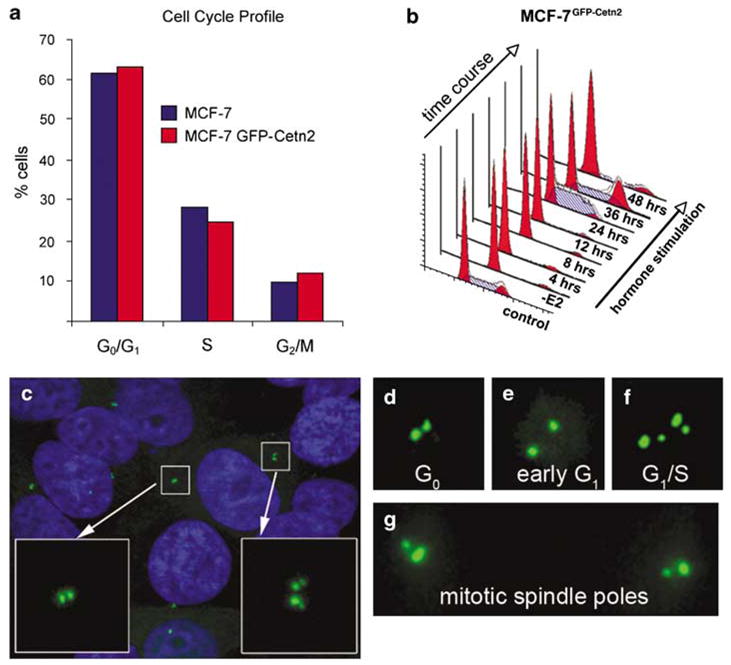
Centriole dynamics during cell cycle progression in MCF-7 cells. (a) Cell cycle analysis (fluorescence-activated cell sorting) for MCF-7 and MCF-7GFP–cetn2 cells showing that green fluorescent protein(GFP)-centrin does not alter cell cycle kinetics. (b) Cell cycle progression following mitogen stimulation in the MCF-7GFP–cetn2 cells. –E2: 48-h withdrawal of 17-β estradiol, EGF and IGF-I. (c) MCF-7GFP–cetn2 cells showing different centriole duplication stages. GFP-centrin (green) and Hoechst stained nuclei (blue). (d) G0 phase with a pair of centrioles adjacent to one another. (e) Early G1 phase with centrioles separated from one another. (f) Late G1 or early S phase characterized by centriole duplication. (g) G2/M phase with a pair of centrioles at each mitotic spindle pole.
Green fluorescent protein-centrin selectively incorporated into the structure of centrioles making them clearly visible in living cells (Figure 1c). MCF-7 cells arrested in the G0 phase of the cycle show a pair of centrioles closely adjacent to one another (Figure 1d). As cells progressed through early G1 following mitogen stimulation, they were characterized by two centrioles, separated a short distance from one another (Figure 1e). G1/S progression was characterized by the formation of new centrioles originating adjacent to the pre-existing ones (Figure 1f). And finally, two pairs of centrioles characterized progression from the S to G2/M phases of the cell cycle, one pair occupying each spindle pole during mitosis (Figure 1g).
Orderly expression of G1/S cyclins during cell cycle progression couples centriole duplication with DNA replication
To determine the timing of centriole duplication during cell cycle progression, we quantified centriole duplication by counting cells with four GFP-centrin2 labeled spots (Figures 2b and d). After 48-h estrogen starvation, only 8% of MCF-7GFP–cetn2 cells showed duplicated centrioles, indicating that centriole duplication was arrested in cells blocked in the G0 phase of the cell cycle. The number of cells with duplicated centrioles increased progressively following mitogen stimulation and by 24 h ~ 56% of cells showed duplicated centrioles (Figures 2b and d).
Figure 2.
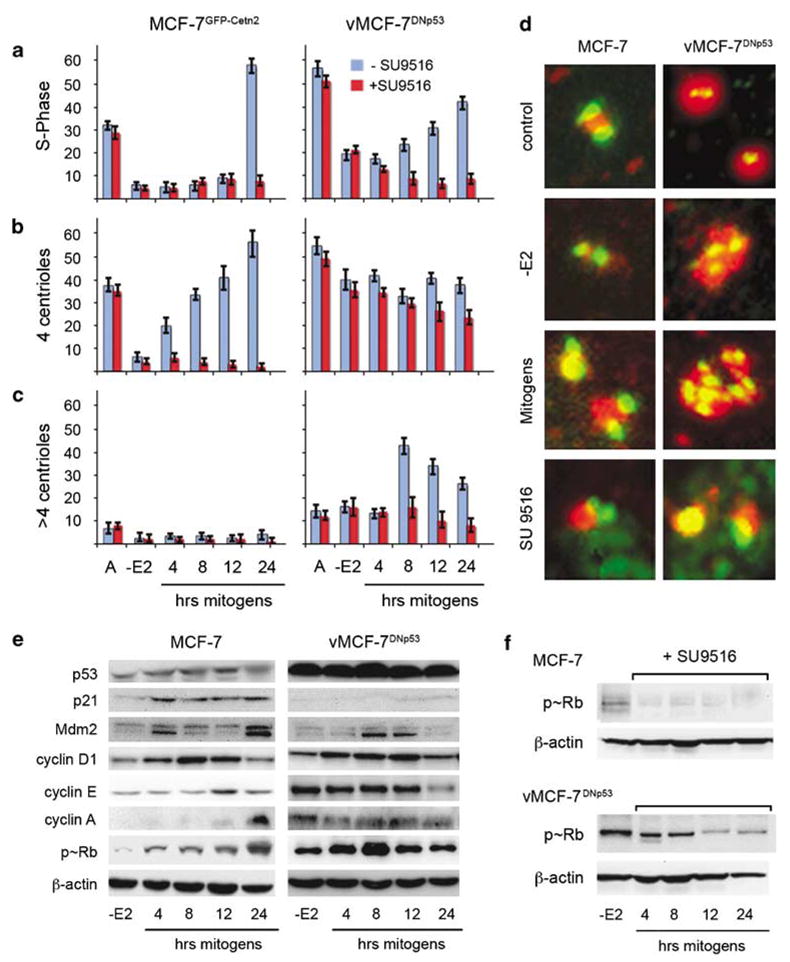
Comparison of cell cycle and centrosome characteristics in MCF-7 and vMCF-7DNp53 cells. Coordination of S-phase entry (a) timing of centriole duplication (b) and centrosome amplification (c) following mitogen stimulation, alone, or in the presence of the small molecule cdk/cyclin inhibitor SU9516. These graphs illustrate the average of percentage of cells from three experiments±s.d. (d) Centrosome characteristics in asynchronous (A) and synchronized cells analyzed before and following 48-h estrogen withdrawal (−E2), and stimulation of cell cycle reentry using 17-β estradiol, EGF and IGF-I, alone, or in combination with the small molecule cdk/cyclin inhibitor, SU9516. Centrioles were labeled for centrin (green), pericentrin (red) and DNA (blue) using Hoechst dye at 1 μg ml−1. (e–f) Western analysis of synchronized cells during cell cycle progression analyzed following 48-h mitogen withdrawal (−E2), and stimulation of cell cycle reentry using 17-β estradiol, EGF and IGF-I (4, 8, 12 and 24 h). Cell lysates (20 μg protein) were loaded in each lane. β-Actin was used as the loading control. (e) Abundance of p53, G1/S cyclins and phospho-Rb. (f) Phospho-Rb following stimulation with mitogens in the presence of the small molecule cdk/cyclin inhibitor, SU9516.
We then performed a time-course immunoblotting analysis to determine at the molecular level the mechanism of coordination of centrosome duplication with DNA replication. The activity of G1/S cyclins was determined by monitoring the phosphorylation status of the tumor suppressor retinoblastoma (Rb) (Figure 2e). After 48-h starvation, cyclin D1, E and A showed low levels of expression and Rb was in its active hypophosphorylated state. Following mitogen stimulation, cyclin D1 expression reached its maximum level by 8 h followed by increase of cyclin E at 12 h and cyclin A at 24 h. The timing of expression of the G1/S cyclins was linked to the progressive increase of Rb phosphorylation, which started after 4 h of hormone stimulation and reached its maximum level by 24 h. Importantly, two downstream transcriptional targets of p53 showed elevated expression following mitogen stimulation, the cdk inhibitor, p21, and the oncoprotein, Mdm2, responsible for p53 degradation (Figure 2e). Taken together, these results demonstrate that the balance of positive and negative regulatory pathways governing the cell cycle coordinates the process of centriole duplication with DNA replication through the orderly expression of G1/S cyclins and consequent Rb inactivation.
Impaired p53 function induces centrosome amplification through deregulation of cdk2 kinase activity and results in mitotic spindle abnormalities
To determine if the coordination of centriole duplication and DNA replication during cell cycle progression was dependent on the normal p53 status of MCF-7 cells, we employed a variant MCF-7 cell line (vMCF-7DNp53) that overexpresses a dominant negative p53val135 mutant. FACS analysis of cycling vMCF-7DNp53 cells showed ~ 55% of cells in the S phase of the cell cycle, indicating a higher proliferative activity compared to the parental cell line (Figure 2a). After 48-h estrogen starvation, vMCF-7DNp53 cells showed a decrease in S phase, indicating that these cells still retained a hormone-dependent phenotype regardless of abrogated p53 function. Importantly, vMCF-7DNp53 cells exhibited a shortened G1/S cell cycle progression following mitogen stimulation, which was characterized by earlier DNA replication compared to the parental MCF-7 cells (Figure 2a). To determine if this accelerated G1/S phase progression was associated with deregulation of centrosome duplication, we performed an immunofluorescence analysis to determine the number of cells with duplicated centrioles. vMCF-7DNp53 cells showed a higher percentage of cells with duplicated centrosomes, compared to the parental cell line, in cycling cultures, as well as in cells arrested for 48 h by estrogen starvation (Figures 2b–d).
As we did for the parental MCF-7 cells, we investigated the expression of p53 downstream targets, G1/S cyclins, and the phosphorylation status of Rb in the vMCF-7DNp53 cells following mitogen stimulation (Figure 2e). As expected the vMCF-7DNp53 cells showed higher level of expression of mutant p53 at all time points. Consequently, due to abrogation of p53 function, p21 and Mdm2 levels were significantly low in the vMCF-7DNp53 cells during cell cycle progression. After 48-h estrogen starvation, cyclin D1 levels were low, while cyclin E, A and phosphorylated Rb were significantly elevated compared to the parental cells. Furthermore, following mitogen stimulation of vMCF-7DNp53 cells, cyclin D1 showed an expression timing similar to that of the parental MCF-7, while in contrast, expression of cyclin E and A and constitutive Rb phosphorylation remained high. To determine if deregulation of cyclin/cdk2 activity was also associated with the development of centrosome amplification in the vMCF-7DNp53 cells, we determined the percent cells with more than four centrioles following hormone stimulation. Figures 2c and d show that centrosome amplification became apparent by 8 h after mitogen stimulation only in the vMCF-7DNp53 cells, and by 24 h this amplification was reduced due to centriole segregation at the time of cell division. These results indicate that impairment of p53-signaling pathway results in the loss of p21 expression and consequent overexpression of cyclin E and A, leading to constitutive Rb inactivation and premature G1/S-phase entry. This deregulated G1/S phase progression is critical for premature centrosome duplication and development of centrosome amplification.
To establish whether suppression of cdk2 activity was sufficient to block premature S-phase entry and centro-some amplification in vMCF-7DNp53 cells, we employed a small molecule inhibitor with high affinity for the cdk2 kinase, SU9516. The inhibitory effect of SU9516 on cdk2 activity was monitored through suppression of Rb phosphorylation (Figure 2f). SU9516 significantly arrested the progression from the G1 to the S phase of the cell cycle in both parental and variant cell lines (Figure 2a), suggesting that suppression of cdk2 activity leads to a reduced level of Rb phosphorylation and its activation regardless of p53 status. Importantly, SU9516 treatment also resulted in a dramatic inhibition of centriole duplication in MCF-7 cells (~4% cells with 4 centrioles) and prevented centriole reduplication in vMCF-7DNp53 cells resulting in a significant reduction of cells with amplified centrosomes (Figures 2c–d). These observations demonstrate that the development of centrosome amplification in vMCF-7DNp53 was dependent on hyperactivity of cdk2 kinase and implicates the cyclin/cdk2 complex as a key player in the deregulation of centrosome homeostasis in cells with abrogated p53 function.
Since centrosome amplification results in the formation of aberrant mitoses, we assessed spindle morphology and compared the number of normal bipolar spindles showing two centrioles at each pole, with abnormal pseudobipolar spindles showing two spindle poles with one or both containing more than two centrioles and multipolar mitotic spindles with more than two poles, each containing two or more centrioles (Figure 3). Following 24-h mitogen stimulation, only the vMCF-7DNp53 cells showed an increase in pseudobipolar and multipolar mitotic spindles, which occasionally gave rise to more than two daughter cells (Figure 3d). Centrosome amplification and aberrant mitoses in breast cancer cells with abrogated p53 function may represent a mechanism for the origin of chromosomal instability and phenotypic heterogeneity among cancer cells.
Figure 3.
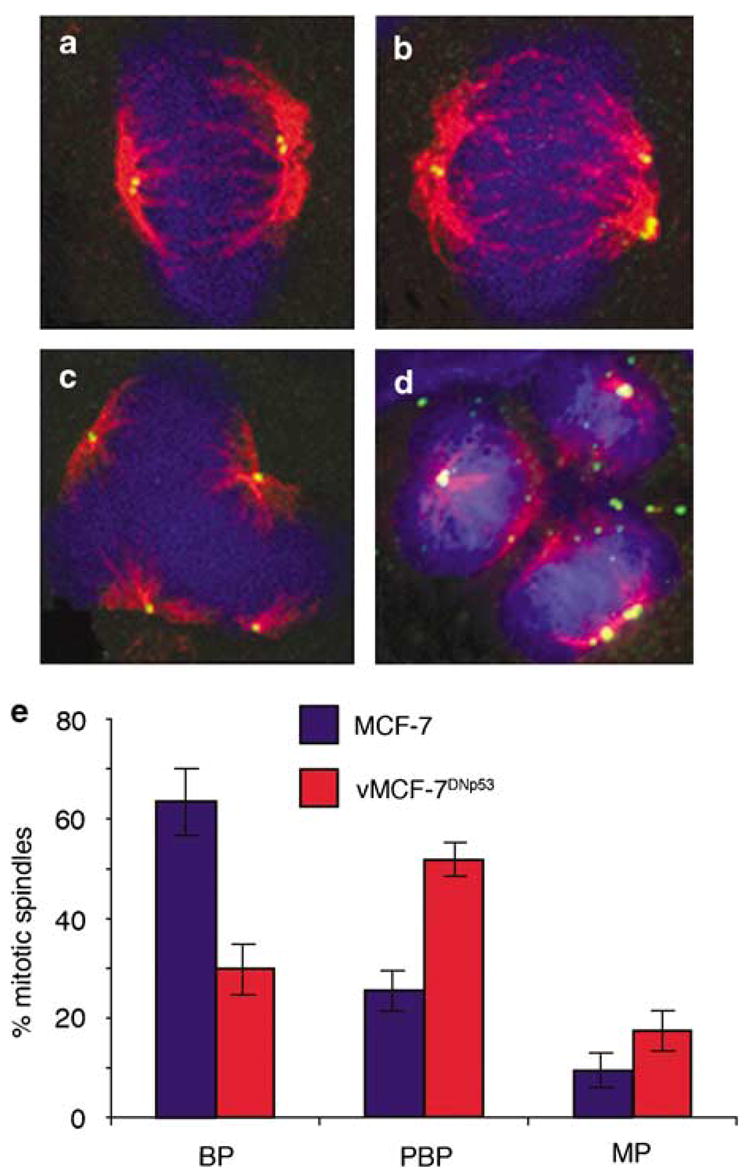
Mitotic spindle morphology in MCF-7 and vMCF-7DNP53 cells. (a–d) Immunofluorescence of bipolar (a) pseudobipolar (b) and multipolar mitotic spindles (c) in the vMCF-7DNP53 cells. (d) An example of a multipolar mitosis resulting in the generation of more than two daughter cells. Centrioles were labeled for centrin (green), mitotic spindles were labeled for the centrosome kinase aurora-A (red) and DNA with Hoechst dye (blue). (e) Graph showing the percentage of mitotic spindles from three experiments±s.d. with bipolar (BP), pseudobipolar (PBP) and multipolar (MP) morphology cells following 24-h mitogen stimulation.
vMCF-7DNP53 xenografts develop high-grade tumors harboring centrosome amplification, ERα phenotypic heterogeneity and distant metastases
To determine if development of centrosome amplification and aberrant mitoses are mechanistically linked to phenotypic heterogeneity and consequent selection of cancer cells with more aggressive properties in vivo, we established MCF-7 and vMCF-7DNp53 xenografts in non-ovariectomized female nude mice. To monitor the xenograft implants and tumor growth in living animals, we established MCF-7 and variant cells expressing firefly luciferase protein (Figure 4a). Xenograft vMCF-7DNp53 tumors showed a significantly higher growth rate than MCF-7 tumors (Figure 4b), consistent with their growth in vitro. After 12 weeks nude mice were sacrificed and primary tumors were assessed for histology and immunohistochemistry (Figure 4c). MCF-7 xenografts developed moderately differentiated tubular tumors and displayed an ERα and a progesterone receptor (PR)-positive phenotype. In contrast, vMCF-7DNp53 xenografts developed poorly differentiated tumors showing a heterogeneous phenotype characterized by the loss of ERα and PR expression. However, when tumor tissues were stained for the expression of HER2/Neu receptor, both MCF-7 and vMCF-7DNp53 xenografts were negative for HER2/Neu receptor. Although loss of ERα expression is generally associated with elevated levels of HER2/Neu, our findings indicate that vMCF-7DNp53 xenografts resemble human breast tumors with a triple negative phenotype (ERα–, PR– and Her2/neu–), which are usually associated with an aggressive phenotype and a poor outcome. Importantly, while cultured vMCF-7DNp53 cells expressed low levels of the onco-protein Mdm2, vMCF-7DNp53 xenografts showed an increased expression of Mdm2 compared to parental MCF-7 xenograft, suggesting that Mdm2 expression during in vivo growth was independent of p53 function. To establish if the heterogeneous phenotype of breast tumors derived from vMCF-7DNp53 xenografts was linked to more aggressive behavior including metastatic spread to distant organs, the presence of cancer cells in tissue samples from the lungs was also examined at the time of nude mice sacrifice (12 weeks). While MCF-7 xenografts did not develop lung metastases, the presence of metastatic cancer cells was observed into the lungs of nude mice carried vMCF-7DNp53 xenografts.
Figure 4.
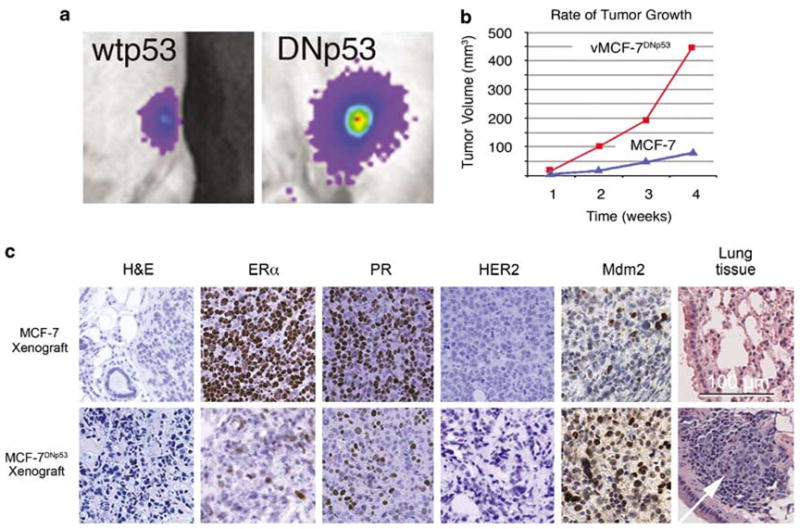
Human breast cancer xenografts in nude mice. (a) Tumor imaging in live animals of MCF-7 (left) and vMCF-7DNp53 (right) xenografts expressing the firefly luciferase reporter 4 weeks after subcutaneous injection. (b) Measurement of weekly tumor growth using digital calipers. (c) Paraffin sections of xenograft tumors (12 weeks) showing: H&E staining of low-grade tubular tumors for MCF-7 and anaplastic vMCF-7DNp53 tumors; loss of estrogen receptor α and progesterone receptor expression in the vMCF-7DNp53 xenografts; negative phenotype for Her2/nue in both xenografts; elevated MDM2 staining and metastatic spread to lungs for the vMCF-7DNp53 xenograft.
To characterize at a cellular level if development of ERα and PR heterogeneity was linked to the development of antiestrogen resistance, cells were recovered from primary tumors at 12 weeks, selected in G418 and recultured as first generation xenograft-derived cell lines (1GX). Importantly, >95% of the parental MCF-7, vMCF-7DNP53 and 1GX MCF-7 cells showed positive ERα expression and nuclear localization when maintained in culture (Figures 5a–c and f). In contrast, the tumor-derived 1GX MCF-7DNp53 cells showed acquired heterogeneity for ERα nuclear localization as was observed in MCF-7DNp53 xenografts (Figures 5d and f). Immunoblotting analysis confirmed the reduced level of ERα expression in 1GX vMCF-7DNp53 cells compared to either the parental MCF-7 or MCF-7DNp53 cells (Figure 5e). To establish if loss of ERα expression led to a hormone independent phenotype, vMCF-7DNp53 and v1GX MCF-7DNp53 cells were cultured for 2 and 8 days in estrogen-depleted medium and the percentage of cells in S phase was determined by FACS analysis (Figure 5g). v1GX MCF-7DNP53 cells showed reduced hormone dependence that was also associated to resistance to the antiestrogen tamoxifen employing a cell viability assay (Figure 5h).
Figure 5.
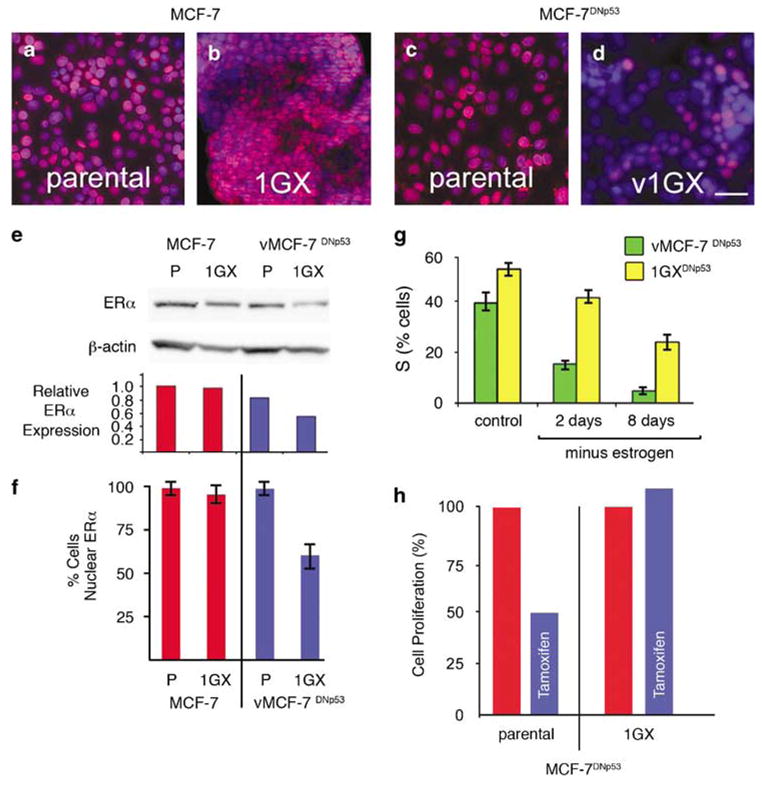
Estrogen receptorα status, estrogen-dependent growth, and tamoxifen sensitivity for MCF-7 and vMCF-7DNp53 and cultures reestablished (1GX) from xenograft tumors. (a–d) ERα staining (red) showing ERα expression and nuclear localization in MCF-7 (a), vMCF-7DNp53 (c), and recultured 1GX MCF-7 (b). Loss of expression and nuclear localization was seen only in the recultured v1GX MCF-7DNp53 cells (d). Nuclei were counterstained for DNA using Hoechst dye (blue). (e) Western analysis of ERα abundance showing reduced expression in the 1GX vMCF-7DNp53 cells. ERα expression relative to β-actin the loading control quantified by densitometry. (f) Quantitative analysis of cells with ERα nuclear localization. (g) FACS analysis of % cells in S-phase before and following estrogen withdrawal showing reduced hormone dependence in v1GX MCF-7DNp53 compared to the parental vMCF-7DNp53 cells. (h) Cell proliferation assay showing loss of tamoxifen sensitivity in the cultures reestablished from v1GX MCF-7DNp53 xenografts.
Furthermore, to determine if the aggressive behavior of tumors derived from vMCF-7DNp53 xenografts was associated with the development of centrosome amplification, we analyzed centrosome phenotype in primary xenograft tumors and in cultures reestablished from them. The tumors derived from vMCF-7DNp53 cells displayed marked centrosome amplification that was not seen in the MCF-7 xenografts (Figures 6a,b). Recultured 1GX MCF-7 cells retained a normal centrosome phenotype and showed normal bipolar mitotic spindles (Figures 6c, e, f and i). In contrast, v1GX MCF-7DNP53 cells retained amplified centrosomes and a high percentage of aberrant mitoses characterized by pseudobipolar and multipolar spindles (Figures 6d, g, h and i). Taken together, these results demonstrate that in vivo growth of MCF-7 cells lacking p53 function leads to centrosome amplification and consequent phenotypic heterogeneity that in turn drives the development of aggressive breast cancer cells with a hormone-independent and metastatic phenotype.
Figure 6.
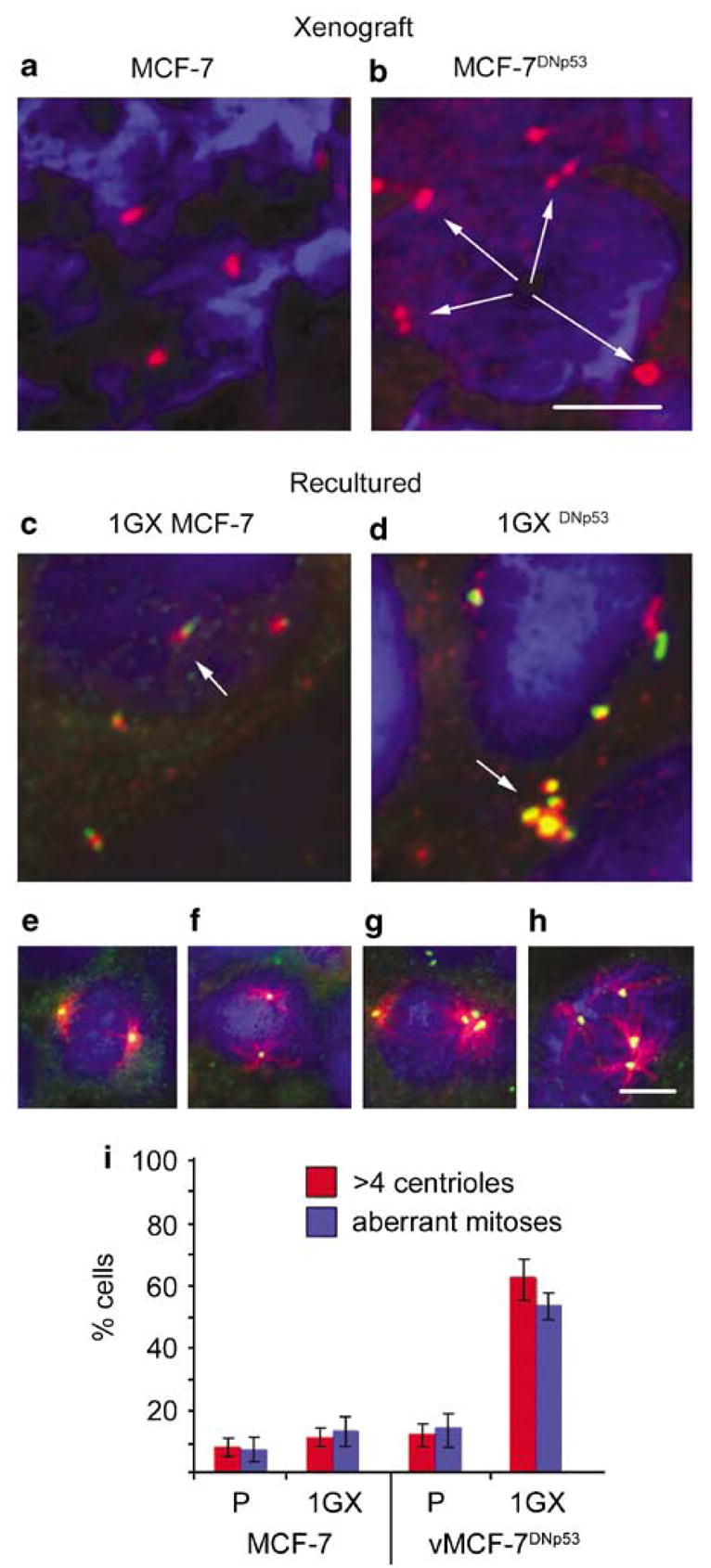
Centrosome phenotype and mitotic spindle morphology in parental and first generation (1GX) cultures reestablished from xenograft tumors. (a, b) g-tubulin staining showing normal centrosomes in MCF-7 xenografts and centrosome amplification in the MCF-7DNp53 xenograft. (c, d) Centrosomes of cells from cultures reestablished from xenografts stained for centrin (green) and pericentrin (red). Normal centrosome staining (c, arrow) was maintained in 1GX cultures reestablished from MCF-7 xenografts (c) and centrosome amplification (d, arrow) was maintained in 1GX cultures reestablished from vMCF-7DNp53 xenografts (d). Bar in (b) ¼5 μm for (a–d). Normal bipolar mitotic spindle morphology in 1GX MCF-7 cells (e, f). Abnormal pseudobipolar and multipolar mitotic spindle morphology in v1GX MCF-7DNp53 cells. (g, h). Bar in (h) = 10 μm for (e–h). Graph showing centrosome characteristics (centrin and pericentrin staining) of parental vs cultures reestablished from xenografts (1GX) showing normal centrosome number and morphology in parental MCF-7, vMCF-7DNp53 and 1GX MCF-7, and centrosome amplification and abnormal mitoses in 1GX vMCF-7DNp53. Bars represent the average percentage of cells with >4 centrioles and those with aberrant mitoses from three experiments±s.d.
Discussion
Reentry into the G1 phase of the cell cycle following mitogen stimulation is governed by cyclin D1/cdk-4 and -6 complex through the phosphorylation and consequent inactivation of the Rb protein (Lewis et al., 2005; Mawson et al., 2005). Likewise, cyclin E/cdk2 and cyclin A/cdk2 complexes are responsible for the G1/S transition and onset of DNA replication, respectively (Hwang and Clurman, 2005; Mailand and Diffley, 2005). Aberrant expression of cyclins is linked to breast cancer development and progression (Bindels et al., 2002; Akli et al., 2004; Caldon et al., 2006). While cyclin D1 overexpression is generally associated with an ERα-positive phenotype and favorable clinical outcome, deregulation of cyclin E and A is linked to induction of centrosome amplification, aneuploidy, loss of ERα and poor outcome (Bindels et al., 2002; Span et al., 2003; Joe et al., 2005).
One mechanism for preservation of genomic stability is through p53-mediated control of centrosome homeostasis (Fukasawa, 2005). Several studies have demonstrated that following DNA damage, centrosome homeostasis is maintained through the G1/S cell cycle checkpoint by p53 transcriptional activation of the cdk inhibitor p21 (Bennett et al., 2004; D’Assoro et al., 2004). However, whether abrogation of p53 function contributes to cancer cell heterogeneity through development of centrosome amplification during breast cancer progression has not been established. To address this issue, we employed ERα-positive MCF-7 cells with wild-type and abrogated p53 function to investigate the molecular mechanism controlling centrosome duplication following mitogen stimulation. We also developed xenograft models using these cell lines to investigate whether impairment of p53 function and development of centrosome amplification in vivo will result in ERα phenotypic heterogeneity and metastatic spread to distant organs characteristic of breast cancer progression.
In this study, we demonstrate that following hormone stimulation in MCF-7 cells, centriole duplication begins in late G1 phase of the cell cycle and is coordinated with DNA replication. At a molecular level, the timing of centriole duplication was correlated with expression of the cdk inhibitor p21, orderly expression of G1/S cyclins and progressive retinoblastoma phosphorylation. While cells overexpressing mutated p53 (vMCF-7DNp53) retained a hormone-dependent phenotype, they nonetheless showed a shortened G1/S phase following mitogen stimulation that was linked to lack of p21 and premature expression of cyclin E and A and consequent Rb hyperphosphorylation. vMCF-7DNp53 cells were also characterized by the development of centrosome amplification following mitogen stimulation. The loss of centrosome homeostasis was dependent on the deregulated cdk2/cyclin complex activity since inhibition of cdk2 activity using the small-molecule inhibitor, SU9516, resulted in the suppression of centrosome reduplication and amplification.
To determine if variant p53 mutant cells develop phenotypes characteristic of tumor progression in vivo, we established MCF-7 and vMCF-7DNp53 xenografts in nude mice. Both parental and vMCF-7DNP53 cells consistently showed estrogen receptor expression and nuclear localization in vitro. Interestingly, tumors derived from MCF-7 xenografts were low grade, retained an ERα and PR positive phenotype, normal centrosomes and bipolar mitotic function. In contrast, vMCF-7DNP53 xenografts developed poorly differentiated tumors showing a heterogeneous phenotype characterized by the loss of ERα and PR expression, centrosome amplification and the ability to give rise to lung metastases. In addition, tumors derived from vMCF-7DNP53 cells resembled a triple negative phenotype seen in subgroup of human breast cancer, since they were also HER2/Neu negative. This suggests the intriguing possibility that triple negative breast carcinomas may derive from early stage ERα+ tumors that development of centrosome amplification due to abrogated of p53 function. In addition, vMCF-7DNP53 xenografts over-express the oncoprotein Mdm2, which has been demonstrated to induce ER degradation and is associated with an aggressive phenotype (Kinyamu and Archer, 2003; Turbin et al., 2006). A recent study by Duong et al. (2007) demonstrated that Mdm2 ubiquitin ligase activity degrades the ERα protein and thus downregulates ERα expression. They show that Mdm2 oncogenic ubiquitin ligase directly interacts with ERα in a ternary complex with p53 and is involved in the regulation of ERα turnover (both in the absence or presence of estrogens). Importantly, in MCF-7 cells, various p53-inducing agents stabilized ERα and abolished its estrogen-dependent turnover. How Mdm2 levels are upregulated in the vMCF-7 DNp53 xenograft cells is not known. There is evidence that increased AKT activity may elevate Mdm2 independent of p53 (Gottlieb et al., 2002), but in the case presented here it is likely that the AKT levels would be increased independent of HER2 signaling, perhaps other growth factor receptors are involved. Therefore, depending on the balance of signals, effective recruitment of AKT may lead to activation of Mdm2 and inactivation of ERα.
Loss of ERα expression and nuclear localization persisted in first generation v1GX MCF-7DNP53 reestablished from xenograft tumors. Importantly, v1GX MCF-7DNP53 cells showed reduced hormone dependence and tamoxifen resistance compared to the vMCF-7DNP53 cells that had only been maintained in culture, suggesting that loss of ERα was dependent on the selection of subclonal populations in the context of in vivo tumor progression. Development of tamoxifen resistance has been associated to overexpression of HER2/Neu (Kurokawa et al., 2000; Kurokawa and Arteaga, 2001, 2003). Our findings suggest an alternative mechanism responsible for the development of antiestrogen resistance based on the development of centrosome amplification, chromosomal instability, Mdm-2 overexpression and loss of ERα.
In conclusion, we establish an experimental model that mimics the progression of human breast carcinomas typified by loss of p53 integrity, centrosome amplification, development of high-grade tumors, acquired ERα heterogeneity and distant metastases. Development of centrosome amplification in breast cancers lacking p53 function could result from deregulation of G1/S phase progression during in vivo tumor growth. Precisely how aneuploidy affects tumor progression is not well understood. Recent studies in yeast (Torres et al., 2007) suggest that allelic imbalance resulting from aneuploidy may provoke additional acquired epigenetic and physiological changes. If a similar mechanistic process operates in cancer, this could help to explain the diverse forms of genomic instability seen during tumor progression. Taken together, these observations suggest that chromosomal instability due to centrosome and other mitotic defects, coupled with frank mutations and acquired epigenetic changes, operate in concert to give the persistent generation of new genetic variations and an increasingly diverse pool of tumor cells, from which ultimately selection in vivo leads to more aggressive phenotypes with increased metastatic potential and chemoresistance during tumor progression. Importantly, these studies also suggest that centrosome amplification may accelerate the natural evolution of ERα breast carcinomas from a hormone-dependent to a hormone-independent and metastatic phenotype. Therefore, we propose that novel molecular therapeutic strategies targeted to centrosome function may be useful in the delay of chemo-endocrine resistance that characterize tumor progression and poor outcome in breast cancer patients.
Materials and methods
Human breast cancer cell lines
The human breast cancer cell line, MCF-7 (ATCC, Manassas, VA, USA), was modified to express a recombinant GFP-centrin2 chimera (MCF-7GFP–cetn2) and a temperature sensitive p53 construct mutated at residue 135 of valine (vMCF-7DNp53) as described previously (Knippschild et al., 1996; D’Assoro et al., 2001, 2004). For imaging of tumor localization and growth in living animals, cells were engineered to express a firefly luciferase protein (Hasegawa et al., 2006). Cultures were maintained in EMEM medium containing 5 mM glutamine, 1% penicillin/streptomycin, 20 μg ml−1 insulin and 10% FBS at 37°C in 5% CO2, and the engineered clones were maintained under 500 μg ml−1 G418 selection.
Synchronization and cell cycle progression
Cultures were allowed to proliferate for 2 days in complete medium, washed twice in PBS and synchronized in G0 of the cell cycle in defined phenol red-free EMEM medium supplemented with 5% charcoal-stripped FBS serum and 2 mM L-glutamine. This medium was changed once daily for 2 days. After 48 h of starvation, cells were washed in 1 × PBS and stimulated to reenter active proliferation by incubation with 10 nM 17-β estradiol, 10 ng ml−1 EGF and 10 ng ml−1 IGF-I. The cdk2 inhibitor SU9516 (Sigma, St Louis, MO, USA) was diluted in DMSO and 5 μM was added to the defined EMEM medium containing the mitogens indicated above.
Cell cycle profile
For fluorescence-activated cell sorting analysis, cells were washed with cold PBS, fixed in 95% ethanol, stained with propidium iodide overnight and analyzed by flow cytometry using Facscan by Becton Dickinson (Franklin Lakes, NJ, USA). Profiles based on 20 000 events, were analyzed using the ModFit program by Verity Software House (Topsham, ME, USA). Experiments were performed in duplicate with similar results.
Cell proliferation assay
For cell proliferation assay, MCF-7 and vMCF-7DNp53 cells were cultured in a 96-well plate at a density of 2000 cells in each plate and treated with 100 nM of tamoxifen (Sigma). Following 7 days incubation, cell viability was determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA), performed according to the manufacturer’s protocol.
Fluorescence microscopy
Cells were fixed in absolute methanol at –20 °C for 10 min, blocked in 5% normal goat serum, 1% glycerol, 0.1% BSA, 0.1% fish skin gelatin, 0.04% sodium azide and incubated with primary antibodies. Primary antibodies against the proteins centrin (20H5 or hCetn-2.4 produced in our laboratory), γ-tubulin (Sigma), pericentrin (Sigma) and Aurora-A (Santa Cruz, Santa Cruz, CA, USA) were followed by secondary antibodies conjugated with Alexa 488 or Alexa 568 (Molecular Probes, Eugene, OR, USA). GFP-labeled centrioles were counted in cells fixed in 4% formaldehyde, incubated in Hoescht dye at 1 μg ml−1 to stain DNA and mounted using ProLong antifade (Molecular Probes). Images were digitally recorded at multiple focal planes using a Zeiss Axiovert 200M fluorescence microscope and analyzed as maximum projections. Values reported represent the average of 100 cells in each of two independent experiments.
Immunoblotting
Cell lysates (20 μg protein) were run in 12% SDS–PAGE, transferred to PVDF membrane, fixed in 0.25% glutaraldehyde, blocked in 5% nonfat dry milk and incubated with primary antibodies against the following proteins: p53 (D07 DAKO), p21 (oncogene), Mdm-2 (Santa Cruz), phosphoretinoblastoma (Sigma), cyclins D1, E, A (Santa Cruz) and β-Actin (Sigma) as loading control, followed by HRP secondary antibodies (Amersham, Piscataway, NJ, USA), and detected using the ECL-plus reagent (Amersham) and an UVP BioImaging system.
Xenograft models
Procedures established by the Institutional Animal Care and Use Committee based on US NIH guidelines for the care and use of laboratory animals were followed for all experiments. Four-week-old non-ovariectomized female NCR/Nu/Nu nude mice were anesthetized by exposure to 3% isoflurane and injected subcutaneously with 2 ×106 cells suspended in 50 μl of 50% Matrigel (BD Bioscience, Bedford, MA, USA). Tumor localization and growth was monitored using the IVS imaging system from the ventral view 10 min after luciferin injection. Tumor volume was also monitored weekly using digital calipers. After 12 weeks, mice were killed and xenograft tumors were processed for histology, immunohistochemistry and immunofluorescence analyses. To reculture 1GX explants, tumors were excised from killed animals, minced using sterile scissors, transferred to complete culture medium and fibroblast-free tumor cell lines were reestablished by serial passages in culture.
Acknowledgments
This work was supported by NCI CA72836 to JLS, USAMRMC BC022276 to ABD and the Mayo Clinic School of Medicine.
Abbreviations
- 1GX
cultures reestablished from xenografts
- ACI
August/ Copenhagen/Irish
- EGF
epidermal growth factor
- ERα
estrogen receptor α
- IGF
insulin-like growth factor
- PVDF
polyvinylidene difluoride
- vMCF-7DNp53
a variant cell line derived from MCF-7 that expresses a recombinant dominant negative p53 mutation (Val134)
References
- Akli S, Zheng PJ, Multani AS, Wingate HF, Pathak S, Zhang N, et al. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res. 2004;64:3198–3208. doi: 10.1158/0008-5472.can-03-3672. [DOI] [PubMed] [Google Scholar]
- Angeloni SV, Martin MB, Garcia-Morales P, Castro-Galache MD, Ferragut JA, Saceda M. Regulation of estrogen receptor-alpha expression by the tumor suppressor gene p53 in MCF-7 cells. J Endocrinol. 2004;180:497–504. doi: 10.1677/joe.0.1800497. [DOI] [PubMed] [Google Scholar]
- Bennett RA, Izumi H, Fukasawa K. Induction of centrosome amplification and chromosome instability in p53-null cells by transient exposure to subtoxic levels of S-phase-targeting anticancer drugs. Oncogene. 2004;23:6823–6829. doi: 10.1038/sj.onc.1207561. [DOI] [PubMed] [Google Scholar]
- Bindels EM, Lallemand F, Balkenende A, Verwoerd D, Michalides R. Involvement of G1/S cyclins in estrogen-independent proliferation of estrogen receptor-positive breast cancer cells. Oncogene. 2002;21:8158–8165. doi: 10.1038/sj.onc.1206012. [DOI] [PubMed] [Google Scholar]
- Brinkley BR, Goepfert TM. Supernumerary centrosomes and cancer: Boveri’s hypothesis resurrected. Cell Motil Cytoskeleton. 1998;41:281–288. doi: 10.1002/(SICI)1097-0169(1998)41:4<281::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Calaf GM. Susceptibility of human breast epithelial cells in vitro to hormones and drugs. Int J Oncol. 2006;28:285–295. [PubMed] [Google Scholar]
- Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem. 2006;97:261–274. doi: 10.1002/jcb.20690. [DOI] [PubMed] [Google Scholar]
- Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, et al. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol. 2004;24:7260–7274. doi: 10.1128/MCB.24.16.7260-7274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Assoro AB, Busby R, Suino K, Delva E, Almodovar-Mercado GJ, Johnson H, et al. Genotoxic stress leads to centrosome amplification in breast cancer cell lines that have an inactive G1/S cell cycle checkpoint. Oncogene. 2004;23:4068–4075. doi: 10.1038/sj.onc.1207568. [DOI] [PubMed] [Google Scholar]
- D’Assoro AB, Lingle WL, Salisbury JL. Centrosome amplifica-tion and the development of cancer. Oncogene. 2002;21:6146–6153. doi: 10.1038/sj.onc.1205772. [DOI] [PubMed] [Google Scholar]
- D’Assoro AB, Stivala F, Barrett S, Ferrigno G, Salisbury JL. GFP-centrin as a marker for centriole dynamics in the human breast cancer cell line MCF-7. Ital J Anat Embryol. 2001;106:103–110. [PubMed] [Google Scholar]
- Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–879. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- Duensing A, Ghanem L, Steinman RA, Liu Y, Duensing S. p21(Waf1/Cip1) deficiency stimulates centriole overduplication. Cell Cycle. 2006a;5:2899–2902. doi: 10.4161/cc.5.24.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26:6280–6288. doi: 10.1038/sj.onc.1210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Tseng M, Malumbres M, Barbacid M, Duensing S. Cyclin-dependent kinase 2 is dispensable for normal centrosome duplication but required for oncogene-induced centro-some overduplication. Oncogene. 2006b;25:2943–2949. doi: 10.1038/sj.onc.1209310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, et al. Differential regulation of estrogen receptor alpha turnover and transactivation by Mdm2 and stress-inducing agents. Cancer Res. 2007;67:5513–5521. doi: 10.1158/0008-5472.CAN-07-0967. [DOI] [PubMed] [Google Scholar]
- Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911–924. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- Giacinti L, Claudio PP, Lopez M, Giordano A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist. 2006;11:1–8. doi: 10.1634/theoncologist.11-1-1. [DOI] [PubMed] [Google Scholar]
- Glass AG, Lacey JV, Jr, Carreon JD, Hoover RN. Breast cancer incidence, 1980–2006: combined roles of menopausal hormone therapy, screening mammography, and estrogen receptor status. J Natl Cancer Inst. 2007;99:1152–1161. doi: 10.1093/jnci/djm059. [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–1303. doi: 10.1038/sj.onc.1205181. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Pham L, O’Connor MK, Federspiel MJ, Russell SJ, Peng KW. Dual therapy of ovarian cancer using measles viruses expressing carcinoembryonic antigen and sodium iodide symporter. Clin Cancer Res. 2006;12:1868–1875. doi: 10.1158/1078-0432.CCR-05-1803. [DOI] [PubMed] [Google Scholar]
- Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA. Estrogen receptors alfa (ERalpha) and beta (ERbeta) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24:6605–6616. doi: 10.1038/sj.onc.1208807. [DOI] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- Ingle JN. Sequencing of endocrine therapy in postmenopausal women with advanced breast cancer. Clin Cancer Res. 2004;10:362S–367S. doi: 10.1158/1078-0432.ccr-031200. [DOI] [PubMed] [Google Scholar]
- Joe AK, Memeo L, McKoy J, Mansukhani M, Liu H, Avila-Bront A, et al. Cyclin D1 overexpression is associated with estrogen receptor expression in Caucasian but not African–American breast cancer. Anticancer Res. 2005;25:273–281. [PubMed] [Google Scholar]
- Kawamura K, Izumi H, Ma Z, Ikeda R, Moriyama M, Tanaka T, et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res. 2004;64:4800–4809. doi: 10.1158/0008-5472.CAN-03-3908. [DOI] [PubMed] [Google Scholar]
- Kinyamu HK, Archer TK. Estrogen receptor-dependent proteasomal degradation of the glucocorticoid receptor is coupled to an increase in mdm2 protein expression. Mol Cell Biol. 2003;23:5867–5881. doi: 10.1128/MCB.23.16.5867-5881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knippschild U, Oren M, Deppert W. Abrogation of wild-type p53 mediated growth-inhibition by nuclear exclusion. Oncogene. 1996;12:1755–1765. [PubMed] [Google Scholar]
- Kurokawa H, Arteaga CL. Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer. Clin Cancer Res. 2001;7:4436s–4442s. [PubMed] [Google Scholar]
- Kurokawa H, Arteaga CL. ErbB (HER) receptors can abrogate antiestrogen action in human breast cancer by multiple signaling mechanisms. Clin Cancer Res. 2003;9:511S–515S. [PubMed] [Google Scholar]
- Kurokawa H, Lenferink AE, Simpson JF, Pisacane PI, Sliwkowski MX, Forbes JT, et al. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000;60:5887–5894. [PubMed] [Google Scholar]
- Lee S, Mohsin SK, Mao S, Hilsenbeck SG, Medina D, Allred DC. Hormones, receptors, and growth in hyperplastic enlarged lobular units: early potential precursors of breast cancer. Breast Cancer Res. 2006;8:R6. doi: 10.1186/bcr1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- Lewis JS, Vijayanathan V, Thomas TJ, Pestell RG, Albanese C, Gallo MA, et al. Activation of cyclin D1 by estradiol and spermine in MCF-7 breast cancer cells: a mechanism involving the p38 MAP kinase and phosphorylation of ATF-2. Oncol Res. 2005;15:113–128. doi: 10.3727/096504005776367924. [DOI] [PubMed] [Google Scholar]
- Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates Aurora-A overexpression, centrosome amplifica-tion, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci USA. 2004;101:18123–18128. doi: 10.1073/pnas.0408273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D’Assoro AB, Boeneman K, Liu W, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci USA. 1998;95:2950–2955. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122:915–926. doi: 10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Mawson A, Lai A, Carroll JS, Sergio CM, Mitchell CJ, Sarcevic B. Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer cells through differential regulation of c-Myc and cyclin D1. Mol Cell Endocrinol. 2005;229:161–173. doi: 10.1016/j.mce.2004.08.002. [DOI] [PubMed] [Google Scholar]
- McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- Medina D, Kittrell FS, Shepard A, Contreras A, Rosen JM, Lydon J. Hormone dependence in premalignant mammary progression. Cancer Res. 2003;63:1067–1072. [PubMed] [Google Scholar]
- Sluder G, Hinchcliffe EH. Centrosome in Cell Replication and Early Development, Current Topics in Developmental Biology. Academic press; San Diego: 2000. The coordination of centrosome reproduction with nuclear events during the cell cycle; pp. 267–289. [DOI] [PubMed] [Google Scholar]
- Span PN, Tjan-Heijnen VC, Manders P, Beex LV, Sweep CG. Cyclin-E is a strong predictor of endocrine therapy failure in human breast cancer. Oncogene. 2003;22:4898–4904. doi: 10.1038/sj.onc.1206818. [DOI] [PubMed] [Google Scholar]
- Suizu F, Ryo A, Wulf G, Lim J, Lu KP. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol Cell Biol. 2006;26:1463–1479. doi: 10.1128/MCB.26.4.1463-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Turbin DA, Cheang MC, Bajdik CD, Gelmon KA, Yorida E, De Luca A, et al. MDM2 protein expression is a negative prognostic marker in breast carcinoma. Mod Pathol. 2006;19:69–74. doi: 10.1038/modpathol.3800484. [DOI] [PubMed] [Google Scholar]