

Abstract
This study describes the antibacterial properties of synthetically-produced mixed aryl alkyl disulfide compounds as a means to control the growth of Staphylococcus aureus and Bacillus anthracis. Some of these compounds exerted strong in vitro bioactivity. Our results indicate that among the twelve different aryl substituents examined, nitrophenyl derivatives provide the strongest antibiotic activities. This may be the result of electronic activation of the arylthio moiety as a leaving group for nucleophilic attack on the disulfide bond. Small alkyl residues on the other sulfur provide the best activity as well, which for different bacteria appears to be somewhat dependent on the nature of the alkyl moiety. The mechanism of action of these lipophilic disulfides is likely similar to that of previously reported N-thiolated β-lactams, which have been shown to produce alkyl-CoA disulfides through a thiol-disulfide exchange within the cytoplasm, ultimately inhibiting type II fatty acid synthesis. However, the mixed alkyl-CoA disulfides themselves show no antibacterial activity, presumably due to the inability of the highly polar compounds to cross the bacterial cell membrane. These structurally simple disulfides have been found to inhibit β-ketoacyl-acyl carrier protein synthase III, or FabH, a key enzyme in type II fatty acid biosynthesis, and thus may serve as new leads to the development of effective antibacterials for MRSA and anthrax infections.
Keywords: Disulfides, MRSA, Bacillus anthracis, lipid biosynthesis, FabH protein
1. Introduction
The design of new antimicrobial agents has reached unprecedented urgency as drug resistance in certain medically-relevant bacteria continues to climb. Our laboratory has recently been investigating the microbiological properties of N-alkylthio β-lactams1–9 and N-alkylthio-2-oxazolidinones10 (Figure 1) as potential antibiotics for treatment of infections caused by methicillin-resistant Staphylococcus aureus (MRSA) and Bacillus anthracis (the causative agent of anthrax).
Figure 1.

Antibacterially-active sulfenylating agents.
These compounds exert selective bacteriostatic properties against Staphylococcus and Bacillus microbes over most other common bacterial genera, and demonstrate unique structure-activity profiles never seen before for β-lactam and 2-oxazolidinone antibiotics. One of the structural requirements of these compounds is that the residues on the heterocyclic ring and sulfur side chain must be highly lipophilic in order to bestow the most potent antibacterial activities. Most recently our studies have determined that N-methylthio β-lactams block type II fatty acid biosynthesis in S. aureus through the initial transfer of the N-alkylthio moiety from the ring nitrogen onto a cellular target. 9 We have since identified this target as being coenzyme A (CoASH), which is present in large quantities in the cytoplasm and comprises the redox buffer system of these bacteria. It is postulated that sulfenylation of coenzyme A produces an alkyl-CoA disulfide (CoASSR) responsible for inhibiting lipid biosynthesis (Scheme 1).
Scheme 1.

Sulfenylation of coenzyme A by N-thiolated β-lactams.
Recent work in the Reynolds laboratory has demonstrated that these mixed alkyl-CoA disulfides, CoASSR, covalently modify the fatty acid biosynthesis protein, FabH, and that the inhibitory effects are highly dependent on the nature of the alkyl side chain R.11 In our initial investigations, we examined the possibility that the CoASSR disulfides could directly inhibit microbial growth if fed to bacteria growing in culture media. However, incubation of S. aureus cells with CoASSMe does not lead to growth inhibition, as judged by both Kirby-Bauer disk diffusion studies and minimum inhibitory concentration assays in broth. Although this seemed surprising at first, given that this compound is likely responsible for bioactivity in the cell, this suggests that the multiply charged CoA moiety may not easily traverse the cell membrane to be able to exert an inhibitory effect within the cytoplasm.12 This led to the realization that the N-thiolated β-lactam, and presumably the N-thiolated oxazolidinone as well, may have the requisite cell permeability and sulfenylation capabilities for antibacterial activity, and could generate the CoASSR adduct intracellularly. Since it was apparent that the lactam or the oxazolidinone rings did not have any special structural features needed for activity, other than being relatively lipophilic and able to serve as a leaving group during nucleophilic attack on sulfur, we thought we could utilize lipophilic disulfides in place of the N-thiolated β-lactams as a means to delivery electrophilic sulfur species more effectively into the bacterial cell. In fact, nature has long used disulfides and trisulfides as natural sulfenylation agents, as evidenced by the large number of naturally-occurring compounds that have anti-infective properties.13 Highly notable examples include diallyl disulfide, diallyl trisulfide, ajoene, S-allylmercaptocysteine, and allicin, found only in freshly crushed garlic (Figure 2).14–17 The biological targets and mode of action of most of these compounds are not precisely known, but in the case of allicin, there are distinct similarities in the antibiotic properties to the N-thiolated β-lactams (and N-thiolated oxazolidinones). In addition to exhibiting activity against a wide range of bacteria18–21, allicin possesses antifungal22–24, antiviral25,26, antiparisitic27,28, and anticancer properties. Both the lactams and allicin strongly inhibit fatty acid synthesis29–32, but also partially inhibit protein and nucleic acid biosynthesis.33 Both compounds exert their strongest antibacterial activities against microbes such as S. aureus and B. anthracis that express unusually low levels of glutathione and relatively high levels of coenzyme A. Allicin reacts with sulfhydryl residues of various proteins such as RNA polymerase, thioredoxin reductase, and alcohol dehydrogenase, and reversibly inhibits acetyl-CoA synthetases.34,35 This certainly suggests similar modes of action in causing intracellular sulfenylation of their biological targets. A survey of the patent literature also uncovered two recent U.S. patent applications describing the effects of mixed alkyl disulfides as inhibitors of redox buffers in mammalian cells as a means to restore normal cellular function, a feature attributed to their high reactivity towards and inhibition of thioredoxin.36,37
Figure 2.

Naturally-occurring disulfide and trisulfide antibacterials from garlic.
2. Results and Discussion
To initiate these investigations, a selection of unsymmetrical aryl methyl disulfides was synthesized from commercially-available arylthiols as shown in Scheme 2. We selected a group of electronically-diverse arylthiols as coupling partners in this reaction to obtain aryl methyl disulfides 1–8 for evaluation. The synthesis was done simply by combining a methanolic solution of the aryl thiol (100 mL of 1.0 M) with a slight excess of the alkyl methanethiolsulfonate (105 mL of 1.0 M methanolic solution) and stirring under an inert atmosphere for 1 hour. The solutions were then treated with a small amount (about 0.3 eq.) of cysteine hydrochloride to consume the excess methanethiolsulfonate. The solution was concentrated and taken up in dichloromethane (1 mL), filtered to remove any cysteine products, treated with Amberlyst 21resin (weakly basic), then flushed through a small silica plug to remove impurities. In most instances, this yielded products >90% pure by 1H NMR. Yields for these reactions were in most cases high and the disulfides were used in microbiological screening assays without further purification. We carried out initial antibacterial assays on these eight aryl methyl disulfides against methicillin-susceptible Staphylococcus aureus by Kirby-Bauer disk diffusion on agar plates, according to National Committee for Clinical Laboratory Standards guidelines.38 In each case, 20 µg of the test compound in DMSO was applied to 6-mm wide wells bored directly into the agar, prior to inoculation and incubation. The average zones of growth inhibition (from three trials) produced by the compounds after 24 h of incubation at 37 °C are presented in Table 1. The Kirby-Bauer data indicates that most of the disulfides have weak or no antimicrobial activity, which suggests that the disulfide bond itself is not enough of a prerequisite to induce antibacterial activity. Of these eight compounds, the p-nitrophenyl analogue 8 showed the strongest in vitro activity against S. aureus, with zones sizes comparable to some of our previously reported N-thiolated β-lactam compounds.3–8
Scheme 2.

Synthesis of aryl methyl disulfides 1–8.
Table 1.
Zones of bacterial growth inhibition data from Kirby-Bauer testing of disulfides 1–8 against S.aureus (ATCC 25923) on agar plates
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| Diameter of zone (mm) | 11 | 10 | 9 | 14 | 23 | 20 | 0 | 35 |
We then expanded on the structure-activity properties of these compounds by making structural modifications to the most potent one, aryl disulfide 8. These include changing the location of the electron-withdrawing nitro group on the phenyl ring, as well as the length and branching of the S-alkyl substituent. The same procedure shown in Scheme 1 was used to prepare meta-nitrophenyl disulfides 9 and ortho-nitrophenyl disulfides 10, employing the corresponding alkylthio sulfonates as sulfenylating agents.
Antimicrobial screening was done by Kirby-Bauer testing as well as by determining the minimum inhibitory concentration (MIC) values of compounds 8–10 by broth serial dilution. The data shown in Table 2 indicates that all three nitrophenyl disulfide systems were active against S. aureus and B. anthracis, but it appeared that p-nitrophenyl disulfides 8 and m-nitro isomers 9 were both considerably more active than the o-nitro analogues 10. The enhanced bioactivity of these nitrophenyl disulfides compared to other aryl-substituted analogues suggests that nucleophilic scission of the disulfide linkage is enhanced by strong electron-deficiency on the sulfur centers.
Table 2.
Zones of Inhibition (on agar plates) and MIC values (in broth) for aryl disulfides 8a–f, 9a–d, and 10a–d against S. aureus, MRSA, and B. anthracis.
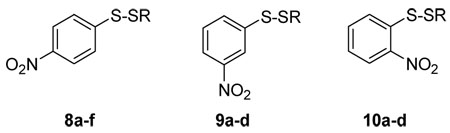 | ||||
|---|---|---|---|---|
| Cpd | Alkyl R |
S. aureus (ATCC 25923) |
MRSA (ATCC 43300) |
B.anthracis |
| 8a | methyl | 35 mm | 23 mm | 54 mm |
| 16 ug/ml | 32 ug/ml | 1 ug/ml | ||
| 8b | ethyl | 54 mm | 38 mm | 67 mm |
| 1 ug/ml | 16 ug/ml | 0.25ug/ml | ||
| 8c | isopropyl | 85 mm | 75 mm | 71 mm |
| <0.125 ug/ml | <0.125 ug/ml | 0.125ug/ml | ||
| 8d | sec-butyl | 68 mm | 54 mm | 62 mm |
| 0.8 ug/ml | 1.0 ug/ml | 0.5 ug/ml | ||
| 8e | n-propyl | 43 mm | 38 mm | 57 mm |
| 16 ug/ml | 16 ug/ml | 1 ug/ml | ||
| 8f | n-butyl | 53 mm | 28 mm | 52 mm |
| 1 ug/ml | 32 ug/ml | 1 ug/ml | ||
| 9a | methyl | 36 mm | 15 mm | 43 mm |
| 16 ug/ml | 64 ug/ml | 8 ug/ml | ||
| 9b | ethyl | 45 mm | 12 mm | 40 mm |
| 8 ug/ml | 64 ug/ml | 8 ug/ml | ||
| 9c | isopropyl | 85 mm | 38 mm | 54 mm |
| <0.125 ug/ml | 16 ug/ml | 1 ug/ml | ||
| 9d | sec-butyl | 45 mm | 22 mm | 41 mm |
| 8 ug/ml | 32 ug/ml | 8 ug/ml | ||
| 10a | methyl | 37 mm | 26 mm | 31 mm |
| 16 ug/ml | 32 ug/ml | 16 ug/ml | ||
| 10b | ethyl | 40 mm | 28 mm | 35 mm |
| 16 ug/ml | 32 ug/ml | 16 ug/ml | ||
| 10c | isopropyl | 31 mm | 20 mm | 27 mm |
| 16 ug/ml | 32 ug/ml | 32 ug/ml | ||
| 10d | sec-butyl | 24 mm | 20 mm | 23 mm |
| 32 ug/ml | 32 ug/ml | 32 ug/ml | ||
| Penicillin G | 33 mm | 14 mm | nt | |
| 0.03 ug/ml | 64 ug/ml | nt | ||
| Vancomycin | 27 mm | 22 mm | nt | |
| 0.25 ug/ml | 0.5 ug/ml | nt | ||
The top value for each compound and microbe is the average diameter of growth inhibition (3 trials) produced after 24 h of incubation using 20 ug of test compound (in DMSO). The lower value is the minimum inhibitory concentration (MIC) of drug (in µg/mL), the lowest concentration of compound in the agar where bacterial growth is completely inhibited. MIC values were determined by serial dilution in 24-well plates according to NCCLS protocols.38 nt =not tested.
These data revealed that increasing the alkyl chain length of the aryl alkyl disulfide from methyl to ethyl to propyl to butyl increased bioactivity somewhat, but not as much as introducing branching in the chain (Table 2). Thus, the isopropyl disulfides and sec-butyl, 8c and 8d, respectively, were considerably more active than the unbranched side chain analogues. Additionally, all of the compounds appeared to be somewhat more active against S. aureus than MRSA, which is opposite to what we observed previously for the N-thiolated β-lactams.7 We do not believe this difference has anything to do with penicillinase production in MRSA, since repeating the testing of S. aureus in the presence of penicillinase protein does not cause a reduction in bioactivity.
The most active series of compounds, p-nitrophenyl disulfides 8a–f, was also tested against E. coli (K12 strain, ATCC 23590) (Table 3). With the lowest MICs being around 16 ug/ml, none of these derivatives were appreciably active against E. coli. However, it is interesting that the methyl and ethyl disulfides possess the strongest activity, which drops off dramatically as the alkyl chain is elongated or branched. This is opposite to that observed for S. aureus and B. anthracis.
Table 3.
Comparison of the minimum inhibitory concentration (MIC) values of p-nitrophenyl alkyl disulfides 8a–f against E. coli in broth.
| Compound |
E. coli (ATCC 23590) |
|---|---|
| 8a | 16 ug/ml |
| 8b | 16 ug/ml |
| 8c | 96 ug/ml |
| 8d | 128 ug/ml |
| 8e | 64 ug/ml |
| 8f | 96 ug/ml |
The need for an aryl moiety directly on one of the sulfurs was made apparent when we tested p-nitrobenzyl- and p-nitrophenethyl disulfides, 11 and 12 (Figure 3). Neither of these compounds showed antibacterial activity against S. aureus, but had very weak activities against B. anthracis (MICs of 32 and 64 ug/mL, respectively). Thus, insertion of a methylene or ethylene group between the aryl ring and sulfur center dramatically reduces antibacterial activity, probably due to significantly lower reactivity of the disulfide toward nucleophilic displacement.
Figure 3.

p-Nitrobenzyl and p-nitrophenethyl disulfides 11 and 12.
Likewise, the corresponding sulfide 13 and Ellman’s reagent (14) were both found to be effectively inactive against S. aureus, illustrating the need for both a disulfide linkage (in the case of inactive compound 13) and high lipophilicity (in the case of inactive disulfide 14) for antimicrobial activity (Figure 4).
Figure 4.

Methyl p-nitrophenyl sulfide (13) and Ellman’s reagent (14).
In addition to the in vitro properties, we were interested to determine if the antibacterial activity of the disulfides could be due to inhibition of a key enzymatic step in lipid biosynthesis in bacteria. Given our recent finding that N-thiolated lactams react with coenzyme A to create a mixed CoA disulfide and inhibits fatty acid biosynthesis, we decided to investigate whether the disulfides could directly inhibit β-ketoacyl-acyl carrier protein synthase III, or FabH, a key enzyme in type II fatty acid biosynthesis. Thus, additional experiments using compounds 8a–f to determine inhibition capabilities against purified E. coli FabH verify that the disulfides are highly active against this enzyme. This was carried out as described previously.39 Each compound in this series showed >90% inhibition of E. coli FabH at a disulfide concentration of 5 µM (Table 4).
Table 4.
Percent inhibitiona of FabH derived from E. coli by disulfides 8a–f.
| Compound | ecFabH (%) |
|---|---|
| 8a | 94 |
| 8b | 98 |
| 8c | 98.5 |
| 8d | 96 |
| 8e | 91 |
| 8f | 93 |
Measured as the percent decrease in activity of FabH in the presence of 5 µM of disulfides 8a–f, as compared to the same enzyme in the absence of the disulfide.
For comparison, N-alkylthio β-lactams 15a and 15b were also tested, and both of these thiolating lactams exhibited inhibitory activity against purified E. coli FabH (Figure 5). At 1 µM, methylthio lactam 15a inhibits FabH catalytic activity by 89%, while sec-butylthio lactam 15b induces a 30% decrease in activity. These assays were done in the absence of coenzyme A, thus showing that the compounds directly act on FabH protein.
Figure 5.


N-Alkylthio β-lactams (15a and 15b).
FabH inhibitor disulfide 16.
These results provide the first evidence that the disulfides and the N-alkylthio β-lactams can inhibit bacterial growth in the same manner, directly through FabH inactivation. It should be mentioned, however, that once inside the bacterial cell, the disulfides and N-alkylthio β-lactams can exert indirectly inhibit FabH catalysis by readily forming alkyl-CoA mixed disulfide adducts, which in turn can impede FabH function. Indeed, Reynolds has previously reported that methyl-CoA mixed disulfide effectively inhibits E. coli-expressed FabH (IC50 = 57 µM).11 Data is not yet available for inhibition studies on FabH from S. aureus.
It is noteworthy that the trends in activity observed for the nitrophenyl disulfides 8–10 coincide with previous observations on the relative dimensions of the active site in each of the microbial species studied. The active site of the E. coli FabH has been reported to preferentially bind S-acetyl CoA over larger or branched acyl CoA analogs.40 By contrast, the FabH from both Staphylococcus41 and Bacillus42 have larger active site pockets, which favor reactions with branched acyl-CoA species such as isobutyryl-CoA.43 These trends are followed precisely in the bioactivities of the disulfide series, providing strong indirect evidence that these compounds may selectively bind to FabH in a mechanism-based manner. One possibility is that the aryl alkyl disulfides may inhibit FabH by reversibly “capping” the active site cysteine through a thiol-disulfide exchange. The reported X-ray crystal structures44,45 of FabH from S. aureus indicates a fairly deep, lipophilic “pocket” into which acyl-CoA or acyl-ACP are able to stretch their lipophilic arm during binding. A proximal cysteine residue is located within the active site, and it is this available thiol that we speculate may be sulfenylated by entry of the aryl alkyl disulfides into the enzyme. Reynolds has recently reported that the asymmetric disulfide 16 inhibits the activity of M. tuberculosis and E. coli FabH enzymes, by capping an active site cysteine thiol by sulfenylation, and that this capping destroys catalytic functioning of the enzyme (Figure 5). We presume that a similar event could be occurring intracellularly in the FabH protein of S. aureus in the presence of the disulfides.46
3. Conclusions
The aryl alkyl disulfides examined in this initial study possess strong in vitro antimicrobial properties against S. aureus (including MRSA) and B. anthracis, and only weak activity against E. coli. Of the structural variants included in our study, the nitrophenyl alkyl disulfides exhibit the greatest in vitro potency. Perhaps even more interestingly, it seems that the efficacy of these compounds may be “tuned” to select for a particular bacterial species: for E. coli, the methyl aryl disulfides are optimal, while for Staphylococcus and Bacillus, the isopropyl- and sec-butyl aryl disulfides appear to be more efficacious. The fact that the in vitro microbiological properties of these compounds, determined from agar diffusion and agar MIC measurements, coincides with the FabH inhibition capabilities certainly points to the fact that this key enzyme is associated with the biological properties of the compounds. These results also highlight the fact that the N-thiolated β-lactams (and presumably N-thiolated oxazolidinones), reported previously as selective bacteriostatic agents for Staphylococcus and Bacillus, may act similarly by directly inhibiting FabH protein in MRSA and B. anthracis.
The structural effects on bioactivity of these compounds raise several key questions regarding the role of the aryl nitro substituent, whether it can be satisfactorily substituted for other functionalities (such as water solubilizing groups), and if the aryl ring substituent in some way aids in specific binding of the molecule to the FabH active site prior to sulfenylation. Understanding the ability of alkyl-CoA mixed disulfides to covalently deactivate bacterial fatty acid synthesis represents a major advance in the quest for novel antibacterials. However, due to the demonstrated inability of CoA mixed disulfides to traverse the cell membrane, the alkyl-CoA disulfides are not directly useful as therapeutics. This research shows that prodrugs such as the described disulfides and the previously reported N-alkylthio β-lactams can be used to produce the CoA mixed disulfides within the bacterial target.
The activity of methyl-CoA disulfide against ecFabH provides strong evidence that in prokaryotic cells which contain CoA, the alkyl-aryl disulfides and N-alkylthio β-lactams may function as prodrugs to produce alkyl-CoA disulfides, and that these mixed disulfides can then inhibit the fatty acid cycle through inhibition of FabH. This mechanism is unique in that it involves a thiol-disulfide exchange from the alkyl-CoA disulfide to the active-site cysteine of FabH, a suggestion that is supported by protein crystallographic studies. Reynolds reported an X-ray crystal structure of E. coli FabH showing the methylthio group from methyl-CoA disulfide being covalently bound to the active site cysteine (112Cys). While the catalytic activity of the methylthiolated (deactivated) FabH protein could not be regenerated simply by dialysis, treatment with dithiothreitol (DTT) quickly restores full catalytic function. This suggests that formation of the cysteine-alkyl FabH disulfide (either via the alkyl-CoA disulfide, the alkyl-aryl disulfide, or N-alkylthio β-lactam) is irreversible under buffered aqueous conditions, but that addition of a thiol can regenerate the catalytic form through thiol-disulfide exchange.
The finding that these simple alkyl-aryl disulfides are generally less active against E. coli than S. aureus or B. anthracis may also reflect differences in the thiol-redox buffers of these three microbes.13,47 Staphylococcus and Bacillus utilize a coenzyme-A based thiol-redox buffer, while E. coli uses a glutathione buffer. Consequently, the effects of the aryl alkyl disulfides in glutathionebased eukaryotic cells would likely also be dampened, which is a question we are now exploring.
For the past decade, there has been considerable interest in the development of FabH inhibitors as potential antibacterial agents.48,49 The key role of FabH in Type II fatty acid synthesis, as well as the unique differences between bacterial and mammalian FAS pathways, make this protein a desirable target for the selective inhibition of bacterial growth. We are exploring potential applications of these disulfide inhibitors of FabH as antibacterial agents for life-threatening bacterial infections.
4. Experimental
4.1. General methods
Reagents were purchased from Sigma-Aldrich Chemical Company or Acros Chemical Company. Methanethiolsulfonates were purchased from Toronto Research Chemicals. Reagents were used without further purification. Solvents were obtained from Fischer Scientific Company. Thin-layer chromatography (TLC) was carried out using EM Reagent plates with fluorescence indicator (SiO2-60, F-254). Products were purified by flash chromatography using J.T. Baker flash chromatography silica gel (40 µm). NMR spectra were recorded in CDCl3 unless otherwise noted. 13C NMR spectra were proton broad-band decoupled. Methylene chloride and THF were distilled prior to use. Prior to the preparation of the disulfides, methanol was purged of oxygen by bubbling nitrogen an inert gas through it for several minutes.
4.2. m-Nitrobenzenethiol.50
To a solution of m-nitrobenzene disulfide (300 mg, 0.97 mmol) in anhydrous THF (2 mL) was added solid NaBH4 (140 mg, 3.4 mmol) in small portions. The resulting mixture was stirred at rt under an inert atmosphere. After 2 hr, the reaction mixture was cooled in an ice bath, then about 5 mL of icewater was added to the mixture. The resulting mixture was acidified with HCl (1 M), then extracted with CH2Cl2 (10 mL). The organic layer was then washed with water (10 mL), then brine (10 mL), dried over MgSO4, and concentrated in vacuo to give the thiol (248 mg, 83%) as a pale yellow oil. 1H NMR (CDCl3, 400 MHz): δ 8.10 (d, J=2.0 Hz, 1H); 7.97 (dd, J=8.0, 2.0 Hz, 1H); 7.54 (d, J=8.0 Hz, 1H); 7.38 (t, J=8.0 Hz, 1H); 3.68 (s, 1H); 13C NMR (CDCl3, 100 MHz): δ 134.9, 130.0, 123.9, 120.7.
4.3. o-Nitrobenzenethiol
Prepared exactly as described above, except that the reaction was allowed to run for 3 h, and the products required chromatography (silica with hexanes: CH2Cl2) to give the thiol (160 mg, 61%) as a pale yellow flocculent solid. Mp: 44–45 °C. 1H NMR (CDCl3, 400 MHz): δ 8.24 (d, J=8.0 Hz, 1H); 7.42 (d, J=4.0 Hz, 1H); 7.29-7.24 (m, 1H); 4.00 (s, 1H). 13C NMR (CDCl3, 100 MHz): δ 133.9, 132.2, 126.5, 126.0.
4.4 p-Nitrobenzyl thiol
Thioacetic acid (386 µl, 5.40 mmol) was added to a mixture of 4-nitrobenzyl bromide (600 mg, 2.79 mmol) and anhydrous K2CO3 (380 mg, 2.75 mmol) in acetone (3 ml), and the resulting mixture was stirred at rt for 2 h under an inert atmosphere. The mix was then partitioned between CH2Cl2 (10 ml) and water (10 ml). The organic layer was separated, then washed quickly with saturated aqueous NaHCO3 (10 ml) and brine (10 ml). The organic solution was concentrated in vacuo, then taken up in methanol (5 ml). Aqueous H2SO4 (50%, 1 ml) was added, and the solution was heated to reflux for 3h, under N2. The solution was partitioned between ether (30 ml) and water (30 ml). The ether layer was washed with water (2×30 ml), then brine (10 ml). The solution was dried over Na2SO4, then concentrated in vacuo to give the thiol (317 mg, 67%) as a yellow solid. mp: 49–52 °C. 1H NMR (200 MHz,CDCl3): δ 8.19 (d, J=8.8 Hz, 2H); 7.02 (d, J=8.6 Hz, 2H); 3.82 (d, J=7.8 Hz, 2H); 1.84 (t, J=8.1 Hz, 1H).
4.5. p-Nitrophenethyl thiol
Prepared according to the general procedure described above, except that the reaction time was 45 min. Isolated 170 mg (69%) as a yellow liquid. 1H NMR (200 MHz, CDCl3): δ 8.17 (d, J=8.8 Hz, 2H); 7.35 (d, J=8.8 Hz, 2H); 3.04 (t, J=7.7 Hz, 2H); 2.84 (dt, J=5.9 Hz, 7.7 Hz, 2H); 1.38 (t, J=5.9 Hz, 1H).
4.6. Synthesis of alkyl aryl disulfides: General procedure
To a 1.0 M solution of aryl thiol in MeOH (100 µl) was added a solution of alkyl methanethiolsulfonate in MeOH (105 µl of a 1.0 M solution, 1.05 eq) After stirring at rt under an inert atmosphere for 1 h, the solutions were treated with a small amount (about 0.3 eq) of cysteine hydrochloride in order to consume the excess thiosulfonate. The mixture was then concentrated under reduced pressure. The solid was taken up in CH2Cl2 (1 mL), and the insoluble material was filtered off. The solution was stirred with Amberlyst-21 resin (weakly basic), (approx. 0.2 eq, pre-swelled in CH2Cl2) for 3 min. The solution was drawn off of the resin, quickly passed through a small silica plug using dichloromethane, and concentrated in vacuo to give the desired product.
4.6.1. 3,4-Difluorophenyl methyl disulfide (1)
9.6 mg (50%) as a colorless oil. 1H NMR (250 MHz, CDCl3): δ 7.37-7.29 (m, 1H); 7.18-7.00 (m, 2H); 2.34 (s, 3H).
4.6.2 3,4-Dimethylphenyl methyl disulfide (2)
Isolated 9.7 mg (53%) as a colorless oil. 1H NMR (250 MHz, CDCl3): δ 7.22 (d, J=8.4 Hz, 1H); 7.18 (s, 1H); 7.02 (d, J=7.7 Hz, 1H); 2.70 (s, 3H); 2.19 (s, 3H); 2.18 (s, 3H).
4.6.3. m-Ethoxyphenyl methyl disulfide (3)
Isolated 5.6 mg (28%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.21 (t, J=8.0 Hz, 1H); 7.09-7.05 (m, 2H); 6.74 (dd, J=8.0, 1.2 Hz, 1H); 4.04 (q, J=7.1 Hz, 2H); 2.43 (s, 3H); 1.41 (t, J=7.4 Hz, 3H).
4.6.4 o-(Hydroxymethyl)phenyl methyl disulfide (4)
1H NMR (400 MHz, CDCl3): δ 7.73 (d, J=8.8 Hz, 1H); 7.44 (dd, J=6.6, 2.2 Hz, 1H); 7.32-7.24 (m, 2H); 4.83 (d, J=5.2 Hz, 2H); 2.42 (s, 3H); 2.02 (broad t, J=6.0 Hz, 1H).
4.6.5. Methyl 2-pyridinyl disulfide (5)
1H NMR indicates about 10% of material is unreacted thiol. 1H NMR (400 MHz, CDCl3): δ 8.46 (d, J=4.4 Hz, 1H); 7.68-7.61 (m, 2H); 7.07 (t, J=6.6 Hz, 1H); 2.49 (s, 3H).
4.6.6. p-Acetamidophenyl methyl disulfide (6)
To a solution of p-acetamidobenzenethiol (50 mg, 0.3 mmol) in dry CH2Cl2 (3 ml) was added triethylamine (127 µL, 0.9 mmol, 3 eq.), followed by acetyl chloride (32 µL, 0.45 mmol). The reaction was stirred at RT for 1 hr, then was diluted with dichloromethane, washed with water, then 5% aq. HCl, then 5% aq. NaHCO3. Dried over MgSO4, then concentrated in vacuo to give a yellow oil which solidified on standing. Chromotography (1:1 petroleum ether:ethyl acetate) gave 18 mg (29%) as a light brown solid. 1H NMR (400 MHz, CDCl3): δ 7.47 (s, 4H); 2.41 (s, 3H); 2.16 (s, 3H).
4.6.7. p-Aminophenyl methyl disulfide (7)
To a solution of p-aminobenzenethiol (125 mg, 1 mmol) in methanol (1 ml) was added methylthio methanesulfonate (94 µl, 1 mmol) in a single portion. The reaction was stirred at RT under N2. After 2 hr, the solution was loaded onto an SCX column and eluted with methanol, followed by 1M ammonia in methanol. The basic eluents were concentrated in vacuo to a yellow oil. Chromatography (1:1 hexanes:CH2Cl2 with 1% isopropyl amine) yielded 8 (140 mg, 82%) as a pale yellow oil. 1H NMR (250 MHz, CDCl3): δ 7.28 (d, J=8.6 Hz, 2H); 6.54 (d, J=8.6 Hz, 2H); 3.60 (broad s, 2H); 2.34 (s, 3H). Treatment with HCl/ether gave 5 mg of the HCl salt. Biological testing was performed on the acid salt. 1H NMR (400 MHz, d6-DMSO): δ 7.46 (broad s, 2H); 7.05 (broad s, 2H); 2.47 (s, 3H); 2.43 (s, 3H).
4.6.8. p-Nitrophenyl methyl disulfide (8a)
Isolated 15.3 mg (76%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J=8.8 Hz, 2H); 7.63 (d, J=8.8 Hz, 2H); 2.46 (s, 3H). Resynthesis using 10x scale yielded 184 mg (92%) as a slightly oily, bright yellow solid. Mp: 29–32 °C. 13C NMR (100 MHz, CDCl3): δ 126.6, 126.0, 124.7, 124.4, 22.9.
4.6.9. Ethyl p-nitrophenyl disulfide (8b)
1H NMR (400 MHz, CDCl3): δ 8.14 (d, J=8.8 Hz, 2H); 7.64 (d, J=8.8 Hz, 2H); 2.77 (q, J=7.0 Hz, 2H); 1.31 (t, J=7.2 Hz, 3H).
4.6.10. Isopropyl p-nitrophenyl disulfide (8c)
1H NMR (400 MHz, CDCl3): δ 8.14 (d, J=8.8 Hz, 2H); 7.64 (d, J=8.8 Hz, 2H); 3.10 (septuplet, J= 6.5 Hz, 1H); 1.30 (d, J=6.8 Hz, 6H).
4.6.11. sec-Butyl p-nitrophenyl disulfide (8d)
1H NMR (400 MHz, CDCl3): δ 8.14 (d, J=8.8 Hz, 2H); 7.64 (d, J=8.8 Hz, 2H); 2.88-2.83 (m, 1H); 1.73-1.64 (m, 1H); 1.57-1.50 (m, 1H); 1.27 (d, J=6.8 Hz, 3H); 0.97 (t, J=7.2 Hz, 3H).
4.6.12. p-Nitrophenyl n-propyl disulfide (8e)
1H NMR (400 MHz, CDCl3): δ 8.14 (d, J=8.8 Hz, 2H); 7.63 (d, J=8.8 Hz, 2H); 2.73 (t, J=7.2 Hz, 2H); 1.68 (tq, J=7.2, 7.6 Hz, 2H); 0.98 (t, J=7.6 Hz, 3H).
4.6.13. n-Butyl p-nitrophenyl disulfide (8f)
1H NMR (400 MHz, CDCl3): δ 8.15 (d, J=8.0 Hz, 2H); δ 7.64 (d, J=8.8 Hz, 2H); δ 2.95 (t, J=7.4 Hz, 2H); δ 1.65-1.61 (m, 2H); δ 1.42-1.37 (m, 2H); δ 0.88 (t, J=7.2 Hz, 3H).
4.6.14. Methyl o-nitrophenyl disulfide (9a)
Isolated 13.9 mg (69%) as an oily yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.27-8.23 (m, 2H); 7.69 (t, J=7.4 Hz, 1H); 7.35 (d, J=7.2 Hz, 1H); 2.41 (s, 3H).
4.6.15. Ethyl o-nitrophenyl disulfide (9b)
Isolated 13.2 mg (62%) as an oily yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J=8.0 Hz, 1H); 8.24 (d, J=8.0 Hz, 1H); 7.66 (t, J=7.4 Hz, 1H); 7.33 (t, J=7.6 Hz, 1H); 2.74 (q, J=7.3 Hz, 2H); 1.31 (t, J=7.2 Hz, 3H).
4.6.16. Isopropyl o-nitrophenyl disulfide (9c)
Chromatographed on silica with 3% dichloromethane in hexanes as eluent. Isolated 13.7 mg (60%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J=8.8 Hz, 1H); 8.23 (d, J=8.8 Hz, 1H); 7.64 (t, J=7.9 Hz, 1H); 7.31 (t, J=7.6 Hz, 1H); 3.05 (s, J= 6.8 Hz, 1H); 1.31 (d, J=6.8 Hz, 6H).
4.6.17. Butyl o-nitrophenyl disulfide (9d)
Chromatographed on silica with 3% dichloromethane in hexanes as eluent. Isolated 12.9 mg (54%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 8.29 (d, J=8.0 Hz, 1H); 8.22 (d, J=8.8 Hz, 1H); 7.64 (t, J=7.8 Hz, 1H); 7.31 (t, J=8.0 Hz, 1H); 2.86-2.79 (m, 1H); 1.75-1.60 (m, 1H); 1.58-1.50 (m, 1H); 1.28 (d, J=7.2 Hz, 3H); 0.98 (t, J=7.8 Hz, 3H).
4.6.18. Methyl m-nitrophenyl disulfide (10a)
Isolated 7.6 mg (38%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.39 (s, 1H); 8.04 (d, J=6.4Hz, 1H); 7.79 (d, J=8.0 Hz, 1H); 7.49 (t, J=8.0 Hz, 1H); 2.47 (s, 3H).
4.6.19. Ethyl m-nitrophenyl disulfide (10b)
Isolated 7.7 mg (36%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.41 (s, 1H); 8.02 (d, J=7.2 Hz, 1H); 7.80 (d, J=7.2 Hz, 1H); 7.47 (t, J=7.2 Hz, 1H); 2.78 (q, J=7.3 Hz, 2H); 1.32 (t, J=7.2 Hz, 3H).
4.6.20. Isopropyl m-nitrophenyl disulfide (10c)
Isolated 17.0 mg (75%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.41 (s, 1H); 8.01 (d, J=8.0 Hz, 1H); 7.80 (d, J=7.2 Hz, 1H); 7.46 (t, J=7.2 Hz, 1H); 3.09 (septet, J=6.8 Hz, 1H); 1.30 (d, J=6.8 Hz, 6H).
4.6.21. Butyl m-nitrophenyl disulfide (10d)
Isolated 12.6 mg (52%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.42 (s, 1H) 8.00 (d, J=8.0 Hz, 1H); 7.80 (d, J=8.0 Hz, 1H); 7.45 (t, J=8.0 Hz, 1H); 2.88-2.83 (m, 1H); 1.73-1.68 (m, 1H); 1.57-1.50 (m, 1H); 1.28 (d, J=7.2 Hz, 3H); 0.97 (t, J=7.2 Hz, 3H).
4.6.22. Methyl 4-nitrobenzyl disulfide (11)
Prepared according to the general procedure outlined above for the aryl alkyl disulfides. Following chromatography (3:1 hexanes:CH2Cl2), 3.1 mg (5%) was obtained as a pale yellow solid. 1H NMR (200 MHz, CDCl3): δ 8.20 (d, J=8.7 Hz , 2H); 7.51 (d, J=8.6 Hz, 2H); 3.94 (s, 2H); 2.15 (s, 3H).
4.6.23. Methyl 2-(p-nitrophenethyl) disulfide (12)
Obtained 9.7 mg (15%) as a pale yellow oil. 1H NMR (200 MHz, CDCl3): δ 8.17 (d, J=8.0 Hz, 2H); 7.37 (d, J=8.0 Hz, 2H);. 3.19-2.85 (m, 4H); 2.43 (s, 3H). MS (EI) m/z: 229 (M+).
4.7. General microbiological methods
S. aureus (ATCC 25923) and MRSA (ATCC 43300) were purchased from ATCC sources. Bacillus bacteria from Sterne spore vaccine was purchased from Colorado Serum Co., Denver. CO.
4.7.1. Culture preparation
From a freezer stock in tryptic soy broth (Difco Laboratories, Detroit, MI) and 20 % glycerol, a culture of each microorganism was transferred with a sterile Dacron swab to Trypticase® Soy Agar (TSA) plates (Becton-Dickinson Laboratories, Cockeysville, MD), streaked for isolation, and incubated at 37 °C for 24 h. A 108 standardized cell count suspension was then made in sterile phosphate-buffered saline (pH 7.2) and swabbed across fresh TSA plates.
4.7.2. Antimicrobial testing: Kirby-Bauer method
Sterile saline (5 mL) was inoculated with a swab of bacteria, then the concentration was adjusted to 0.5 McFarland standard. Bacterial solution was then streaked across a TSA plate to give an even lawn of bacteria. 1–30 µL sterile pipet tips were used to drill 6 mm wells into the agar plate, then 20 µL of 1 mg/mL drug in DMSO was added to the well. Plates were inoculated overnight at 37°C.
4.7.3. Agar dilution minimal inhibitory concentration (MIC) assay
A stock solution of 1 mg/mL of each disulfide was prepared in DMSO. 1.5 mL of freshly-prepared Mueller-Hinton agar was added to each well of a 24-well plate, and to each well was then added a specified amount of the disulfide test solution in order to a give a final drug concentration of 256 mg/mL down to 0.125 ug/mL. One well within each series received 0.1 mL of DMSO as a blank. The contents of each well were thoroughly stirred to evenly distribute the antimicrobial solution within the agar. Once the agar completely solidified, 10 µL of sterile saline containing 0.5 McFarland standard of the test bacteria was pipeted on top of each well and the plates were then incubated for 24 hours at 37° C. Bacterial growth was assessed by visual observation of growth.
4.7.4 FabH enzyme assay
FabH assays were carried out using a standard coupled trichloroacetic acid precipitation assay which determines the rate of formation of radiolabeled 3-ketoacyl ACP from malonyl ACP and radiolabeled acetyl CoA. In this coupled assay Streptomyces glauscescens FabD is used to generate the malonyl ACP substrate from malonyl CoA and Streptomyces glauscescens ACP. For inhibition studies, A standard 20 µL reaction mixture of ecFabH (2 pmol of monomer, 100 nM) and test compound (0.02 nmol, 1µM for compounds 15a and 15b; 0.1 nmol, 5 µM for compounds 8a–f) in 50 mM sodium phosphate buffer (pH 7.4) was incubated at room temperature for 15 min prior to addition to a solution of [1-14C]acetylCoA (0.8 nmole, 40 µM) and MACP (0.2 nmole, 10 µM). The inhibition was measures using standard FabH assay.51 The % inhibition was determined my measuring FabH activity in the presence of test compound relative to a negative control that has no inhibitor (100% activity). The assay was run in duplicate.
Figure 6.

Methyl-coenzyme A mixed disulfide.
Acknowledgements
We thank the NIH for supporting these studies through research grant R01 AI51351.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Turos E, Konaklieva MI, Ren RX-F, Shi H, Gonzalez J, Dickey S, Lim D. Tetrahedron. 2000;56:5571. [Google Scholar]
- 2.Long TE, Turos E. Curr. Med. Chem.- Anti-infective Agents. 2002;1:251. [Google Scholar]
- 3.Turos E, Long TE, Konaklieva MI, Coates C, Shim J-Y, Dickey S, Lim DV, Cannons A. Bioorg. Med. Chem. Lett. 2002;12:2229. doi: 10.1016/s0960-894x(02)00343-8. [DOI] [PubMed] [Google Scholar]
- 4.Coates C, Long TE, Turos E, Dickey S, Lim DV. Bioorg. Med. Chem. 2003;11:193. doi: 10.1016/s0968-0896(02)00383-8. [DOI] [PubMed] [Google Scholar]
- 5.Long TE, Turos E, Konaklieva MI, Blum AL, Amry A, Baker EA, Suwandi LS, McCain MD, Rahman MF, Dickey S, Lim DV. Bioorg. Med. Chem. 2003;11:1859. doi: 10.1016/s0968-0896(03)00037-3. [DOI] [PubMed] [Google Scholar]
- 6.Turos E, Coates C, Shim J-Y, Wang Y, Leslie JM, Long TE, Reddy GSK, Ortiz A, Culbreath M, Dickey S, Lim DV, Alonso E, Gonzalez J. Bioorg. Med. Chem. 2005;13:6289. doi: 10.1016/j.bmc.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 7.Heldreth B, Long TE, Jang S, Reddy GSK, Turos E, Dickey S, Lim DV. Bioorg. Med. Chem. 2006;14:3775. doi: 10.1016/j.bmc.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 8.Turos E, Long TE, Heldreth B, Leslie JM, Reddy GSK, Wang Y, Coates C, Konaklieva M, Dickey S, Lim DV, Alonso E, Gonzalez J. Bioorg. Med. Chem. Lett. 2006;16:2084. doi: 10.1016/j.bmcl.2006.01.070. [DOI] [PubMed] [Google Scholar]
- 9.Revell KD, Heldreth B, Long TE, Jang S, Turos E. Bioorg. Med. Chem. 2007;15:2453–2467. doi: 10.1016/j.bmc.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mishra RK, Revell KD, Coates CM, Turos E, Dickey S, Lim DV. Bioorg. Med. Chem. Lett. 2006;16:2081–2083. doi: 10.1016/j.bmcl.2006.01.058. [DOI] [PubMed] [Google Scholar]
- 11.Wright HT, Reynolds KA. Curr. Opin. Microbiol. 2007;10:447. doi: 10.1016/j.mib.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke KM, Mercer AC, La Clair JJ, Burkart MD. J. Am. Chem. Soc. 2005;237:11234. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]
- 13.Heldreth B, Turos E. Curr. Med. Chem.- Anti-infective Agents. 2005;4:295–315. [Google Scholar]
- 14.Cavallito C, Bailey JH. J. Am. Chem. Soc. 1944;66:1944. [Google Scholar]
- 15.Ankri S, Mirelman D. Microbes and Infection. 1999;2:125. doi: 10.1016/s1286-4579(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 16.Block E. Scient. Am. 1985;252:94. doi: 10.1038/scientificamerican0385-114. [DOI] [PubMed] [Google Scholar]
- 17.Koch HP, Lawson LD. Garlic: The Science and Therapeutic Application of Allium sativum L. Baltimore: Williams and Wilkins; 1996. pp. 1–233. [Google Scholar]
- 18.Uchida Y, Takahashi T, Sato N. Jpn. J. Antibiotics. 1975;28:638. [PubMed] [Google Scholar]
- 19.Cellini L, Campli E, Masulli M, Di Bartolomeo S, Allocati N. FEMS Immunol. Med. Microbiol. 1996;13:273. doi: 10.1111/j.1574-695X.1996.tb00251.x. [DOI] [PubMed] [Google Scholar]
- 20.Gonzales-Fandos E, Garcia-Lopez ML, Sierrra ML, Otera A. J. Appl. Bacteriol. 1994;77:549. doi: 10.1111/j.1365-2672.1994.tb04400.x. [DOI] [PubMed] [Google Scholar]
- 21.Gimenez MA, Solanes RE, Gimenez DF. Rev. Argent. Microbiol. 1988;20:17. [PubMed] [Google Scholar]
- 22.Davis LE, Shen J, Royer RE. Planta Med. 1994;60:546. doi: 10.1055/s-2006-959568. [DOI] [PubMed] [Google Scholar]
- 23.Hughes BG, Lawson LD. Photother. Res. 1991;5:154. [Google Scholar]
- 24.Yamada Y, Azuma K. Antimicrob. Agents Chemother. 1997;11:743. doi: 10.1128/aac.11.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai Y, Cole LL, Dav Lockwood SJ, Simmons V, Wild GC. Planta Med. 1985;5:460. doi: 10.1055/s-2007-969553. [DOI] [PubMed] [Google Scholar]
- 26.Tatarintsev AV, Vrzhets PV, Ershov DE, Turgiev AS, Karamov EV, Kornilaeva GV, Makarova TV, Federov NA, Varfolomeev SD. Vestn. Ross. Akad. Med. Nauk. 1992;11:6. [PubMed] [Google Scholar]
- 27.Mirelman D, Monheit D, Varon S. J. Infect. Dis. 1987;156:243. doi: 10.1093/infdis/156.1.243. [DOI] [PubMed] [Google Scholar]
- 28.Ankri S, Miron T, Rabinkov A, Wilchek M, Mirelman D. Antimicrob. Agents Chemother. 1997;10:2286. doi: 10.1128/aac.41.10.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adetumbi M, Javor GT, Lau BH. Antimicrob. Agents Chemother. 1986;30:499. doi: 10.1128/aac.30.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghannoum MA. J. Gen. Microbiol. 1988;134:2917. doi: 10.1099/00221287-134-11-2917. [DOI] [PubMed] [Google Scholar]
- 31.Neuwirth Z, Sundstrom DC, Thompson NH. Antimicrob. Agents Chemother. 1988;32:1763. doi: 10.1128/aac.32.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Augusti KT, Mathew PT. Experentia. 1974;30:468. doi: 10.1007/BF01926297. [DOI] [PubMed] [Google Scholar]
- 33.Feldberg RS, Chang SC, Kotik AN, Nadler M, Neuwirth Z, Sundstrom DC, Thompson NH. Antimicrob. Agents Chemother. 1988;32:1763. doi: 10.1128/aac.32.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Focke M, Feld A, Lichtenthaler HK. FEBS Lett. 1990;261:106. doi: 10.1016/0014-5793(90)80647-2. [DOI] [PubMed] [Google Scholar]
- 35.Ozolin ON, Uteshev TA, Kim IA, Deev AA, Kamzolova SG. Mol. Biol. (Mosk.) 1990;24:1057. [PubMed] [Google Scholar]
- 36.Kirkpatrick DL. Asymmetric disulfides and methods of using same. 20030176512. US Patent application. 2003 September 18;
- 37.Kirkpatrick DL, Powis G. Asymmetric disulfides and methods of using same. 20040116496. US Patent application. 2004 June 17;
- 38.NCCLS (National Committee for Clinical Laboratory Standards) Methods for Dilution of Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. NCCLS Document M7-A4. 1997;Vol. 17(2) [Google Scholar]
- 39.Alhamadsheh MM, Musayev F, Komissarov AA, Sachdeva S, Wright HT, Scarsdale N, Florova G, Reynolds KA. Chem. and Biol. 2007;14:513. doi: 10.1016/j.chembiol.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Kang SL, Rybak ML. Antimicrob Agents Chemother. 1995;39:1505. doi: 10.1128/aac.39.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He X, Reynolds KA. Antimicrob. Agents and Chemotherapy. 2002;46:1310. doi: 10.1128/AAC.46.5.1310-1318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi K, Heath RJ, Rock CO. Journal of Bacteriology. 2000;182:365. doi: 10.1128/jb.182.2.365-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi K-H, Heath RJ, Rock CO. J. Bacteriology. 2000;182:365. doi: 10.1128/jb.182.2.365-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daines RA, Pendrak I, Sham K, Van Aller GS, Konstantinidis AK, Lonsdale JT, Janson CA, Qiu X, Brandt M, Khandekar SS, Silverman C, Head MS. J. Med. Chem. 2003;46:5. doi: 10.1021/jm025571b. [DOI] [PubMed] [Google Scholar]
- 45.Qui X, Choudhry AE, Janson CA, Grooms M, Daines RA, Lonsdale JT, Khandekar SS. Protein Science. 2005;14:2087. doi: 10.1110/ps.051501605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sachdeva S, Musayev FN, Alhamadsheh MM, Scarsdale JN, Wright HT, Reynolds KA. Chem. and Biol. 2008;15:402. doi: 10.1016/j.chembiol.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 47.Poole LB, Karplus PA, Claiborne A. Ann. Rev. Pharmacol. Toxicol. 2004;44:325. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 48.Marrakchi H, Zhang Y-M, Rock CO. Biochem. Soc. Trans. 2002;30:1050. doi: 10.1042/bst0301050. [DOI] [PubMed] [Google Scholar]
- 49.Campbell JW, Cronan JE. Ann. Rev. Microbiol. 2001;55:305. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- 50.Cashman JR, Olsen LD, Young G, Bern H. Chem. Res. Toxicol. 1989;2:392. doi: 10.1021/tx00012a007. [DOI] [PubMed] [Google Scholar]
- 51.Lobo S, Florova G, Reynolds KA. Biochemistry. 2001;40:11955. doi: 10.1021/bi011325a. [DOI] [PubMed] [Google Scholar]