

Abstract
Leptin signaling in the hypothalamus is critical for normal food intake and body weight regulation. While hyperleptinemia in obese people suggests a state of leptin resistance, the mechanism is not clearly understood. In a rat model of central leptin infusion in which animals develop resistance to the satiety action of leptin, orexigenic peptide producing neuropeptide Y neurons in the hypothalamus develop leptin resistance. However, it is still unknown if increased hypothalamic leptin tone caused by central leptin infusion results in the development of leptin resistance in anorexigenic peptide producing proopiomelanocortin (POMC) and neurotensin (NT) neurons. To this end, male rats were infused chronically with leptin (160 ng/hr) or vehicle into the lateral cerebroventricle for 16 days. On day 4 of leptin infusion when food intake was decreased, POMC and NT mRNA levels, as determined by RNAse protection assay, were significantly increased as compared to control. By contrast, on day 16 of leptin infusion, when food intake was mostly normalized, both POMC and NT mRNA levels remained unchanged compared with control. These findings suggest the development of leptin resistance in the POMC and NT neurons following chronic elevation of hypothalamic leptin tone, which may be involved in the development of resistance to the satiety action of leptin following central infusion of this peptide hormone.
Keywords: leptin, POMC, NT, hypothalamus, leptin resistance
Introduction
A large body of evidence suggests that leptin signals nutritional status to key regulatory centers in the hypothalamus and it has emerged as an important signal regulating energy homeostasis [11,35,40]. Leptin administration centrally or peripherally decreases food intake and body weight in a variety of animals [11]. The evidence that serum leptin levels are significantly higher in obese humans relative to non-obese humans [8,11], and that leptin administration shows very limited response in obese people [13], suggests that a state of leptin–resistance is present in obese individuals. However, the mechanisms behind the development of leptin resistance are not clearly understood. It has been known that human or rodents, made obese by dietary manipulation, have elevated levels of circulating leptin but maintain a normal food intake [8,11,44]. Although a defective leptin transport is thought to be one of the many factors behind the development of leptin resistance [2,6,39], available data from diet-induced obese (DIO) rodents, which may represent the form of obesity seen in most humans, strongly suggests that central leptin resistance also contributes to the development of obesity [20,22,47]. Notably, rats fed with high-fat diet (HF) shows elevated leptin levels within 1 day of HF feeding [49]. Thus, it is possible that an extended period of exposure of the brain, especially the hypothalamus, to a high level of leptin may result in the development of central leptin resistance.
We have previously demonstrated that rats develop resistance to the satiety action of leptin following chronic central leptin infusion in association with the development of leptin resistance in neuropeptide Y (NPY) neurons within two weeks of leptin infusion [34]. However, the question still remains whether other leptin-sensitive neurons develop leptin resistance following chronic increase in hypothalamic leptin tone caused by central leptin infusion. In this regard, proopiomelanocortin (POMC) producing neurons are known to play a significant role in energy homeostasis and in transducing leptin action in the hypothalamus [7,9,31,35,40]. Furthermore, several reports have shown that the melanocortin system is itself intact during states of leptin resistance, with normal or enhanced responsiveness to melanotan II (MT-II) seen when this agonist is injected either peripherally [5] or centrally [38] in rodents. Recently, Enriori et al. [10] have shown that the MC4-R receptor is up-regulated in rats in the leptin-resistance states, perhaps explaining the enhanced responsiveness to experimentally administered agonists. However, the later group also showed a resistance to the effects of leptin on alpha-MSH secretion in DIO rats. Interestingly, there was no effect of leptin on POMC mRNA levels not only in DIO rats but also in control animals [10]. Several studies have reported neurotensin (NT) as an important centrally acting anorectic signal [21,23,41], which acts partially through histamine 1 receptor [29]. NT neurons are localized in the hypothalamus [17] and they are also the targets of leptin signaling [31]. NT antagonist or antibody reverses the anorectic effect of leptin [33]. These results suggest that NT may play a role in mediating leptin action in the hypothalamus. In the present study, we sought to examine whether POMC as well as NT neurons, like NPY, develop leptin resistance following chronic central leptin infusion, because if they do, then it would provide additional evidence in support of the development of leptin resistance in the hypothalamic neuropeptidergic circuitry, involved in regulation of food intake and bodyweight, following chronic central leptin infusion.
Materials and methods
Adult male Sprague-Dawley rats, weighing ~250 g, obtained from Taconic Farms (Germantown, NY) were housed individually in a light (lights on 0500 h to 1900 h) and temperature (22 °C)-controlled room with food (pelleted Purina rat chow) and water available ad libitum. After 7 days of acclimatization, rats were subjected to the following experiments according to an approved Institutional Animal Care and Use Committee protocol.
Rats were implanted stereotaxically with 22-gauge osmotic pump connector cannulae (Plastic One, Roanoke, VA, USA) into the lateral cerebroventricle under pentobarbital anesthesia as described previously [34]. The lateral ventricle was then connected via Medical Vinyl tubing to an artificial cerebrospinal fluid (aCSF, pH 7.4, Ref.12)-filled Alzet osmotic pump (model#2002, DURECT Corp., Cupertino, CA) implanted sc in the back. Seven days later, aCSF pumps were replaced with new pumps to infuse with either recombinant murine leptin (Dr. A.F. Parlow, NHPP, Torrance, CA, USA) at a dose of 160ng/0.5µl/hr or aCSF vehicle for 16 days. A group of aCSF infused rats was fed the amount of food that was consumed on the day before by the leptin-infused group, and served as pair-fed (PF) group. Food intake and body weight were measured daily for 16 days. Rats were killed by decapitation between 0900 and 1200 h on day 4 or 16 of infusion. Brains were removed immediately and the medial basal hypothalamus (MBH) were dissected out [30,34], frozen in liquid nitrogen and kept at −80 C until processed for RNA extraction. Trunk blood was collected for glucose, insulin and leptin determination. Epididymal fat (WAT) was dissected out and weighed.
POMC and NT mRNA levels were measured by ribonuclease protection assay (RPA) [32]. Total RNA was isolated from MBH, using RNAzol (RNA STAT 60) followed by precipitation with isopropanol and ethanol washes according to the manufacturer’s instructions (TEL-TEST, Inc., Friendswood, TX). Rat POMC [16] and NT [18] cDNAs were kindly provided by Dr. J. L. Roberts (Mount Sinai School of Medicine, New York, NY) and P. R. Dobner (University of Massachusetts, Amherst, MA), respectively. A riboprobe generated from a plasmid containing a rat-specific β-actin cDNA fragment (Ambion Inc., Austin, TX) served as an internal control in all RPA. [α-32P]UTP-labeled antisense cRNA probes were synthesized using T7 RNA polymerases using a transcription kit (Ambion Inc., Austin, TX). Six µg of MBH RNA, 32P-labeled POMC and NT (200,000 cpm) and β-actin (20,000 cpm) cRNA probes, and 12 µg yeast tRNA (Boehringer Mannheim, Indianapolis, IN, USA) were allowed to hybridize in solution at 45 °C overnight, followed by combined RNAse A and T1 digestion of non-hybridized probe at 32 °C for 1 hour. Stable hybrids were extracted with phenol-chloroform followed by ethanol precipitation and then separated on 6% polyacrylamide-8M urea gels. The dried gels were exposed in a Bio-Rad Molecular Imaging Screen-K for 6 to 40 hours, and the image of each gel was acquired using a Molecular Imager FX (Bio-Rad Laboratories, Hercules, CA, USA). The volume analysis of each band was performed using Quantity One Software (Bio-Rad). POMC and NT mRNA values were first normalized with β-actin mRNA levels and then the values were expressed in relation to aCSF control.
Plasma leptin was determined with a rat leptin radioimmunoassay (RIA) kit (Linco Research, St. Charles, MO). Plasma insulin was measured with a rat insulin RIA kit (Linco). All samples were assayed in the same assay. Plasma glucose was determined by the Trinder method [43] using a kit (Sigma).
All values are expressed as means ± standard error (SE). Statistical significance of differences in food intake and body weight were analyzed using repeated measures one or two-way analysis of variance (ANOVA) with post-hoc testing using Student-Newman-Keuls multiple range test. All other data were analyzed by randomized one-way ANOVA followed by Student-Newman-Keuls multiple range test. Comparisons with p < 0.05 were considered to be significant.
Results
The changes in food intake and body weight during the 16-day infusion period in aCSF, pair-fed and leptin-treated rats were essentially similar as described previously [34]. In control aCSF infused rats, body weight (F 14,240 = 221.70; p < 0.0001) and food intake (F 14,240 = 3.30; p < 0.0001) were progressively increased throughout the infusion period. In leptin infused rats, body weight gradually decreased to a nadir at day 10 and remained stabilized at lower level thereafter (F 14,240 = 25.08; p < 0.0001). Food intake showed an initial dramatic decrease followed by a recovery by day 16, although it remained significantly (p < 0.01) lower than that of the aCSF control (Fig. 1). In pair-fed rats, body weight also changed significantly during the 16-day period (F 14,240 = 56.69; p < 0.0001). Additionally, during the later period of infusion, the pair-fed rats showed significant increase in body weight as compared to that in leptin group.
Fig. 1.

Changes in food intake and body weight during central recombinant murine leptin (160 ng/0.5 µl/hr) infusion for 16 days. Rats were infused into the lateral cerebroventricle with artificial cerebrospinal fluid (aCSF) via Alzet osmotic minipump (0.5µl/hr) for 7 days before infusion with leptin or aCSF. One group of aCSF infused rats was paired-fed to that of leptin-infused group. Values represent the mean ± SEM for n = 13, 11 and 15 in aCSF, pair-fed and leptin groups, respectively.
As reported previously [34], plasma leptin levels were significantly increased (Day 4, aCSF: 3.01 ± 0.28, pair-fed: 0.99 ± 0.10, leptin: 18.29 ± 2.59, p < 0.0001, n = 12–14/group; Day 16, aCSF: 4.02 ± 0.25, pair-fed: 3.58 ± 0.24, leptin: 14.14 ± 2.21, ng/ml, p < 0.0001, n = 10–14/group; mean ± SEM) and plasma insulin levels were significantly decreased in the leptin-infused rats (Day 4, aCSF: 2.58 ± 0.27, pair-fed: 1.29 ± 0.23, leptin: 0.71 ± 0.15, p < 0.0001, n = 12–14/group; Day 16, aCSF: 2.64 ± 0.23, pair-fed: 2.15 ± 0.26, leptin: 0.29 ± 0.04; ng/ml, p < 0.0001, n = 10–15/group; mean ± SEM), compared with that of aCSF infused rats. Plasma glucose levels remained unchanged between the groups. On day 16 of infusion, epididymal fat weight was decreased in leptin treated group by 87% (p < 0.0001) and in pair-fed group by 24% (p < 0.01) as compared to that of aCSF control animals (aCSF: 5.51 ± 0.35g; pair-fed: 4.41± 0.37g; leptin: 0.79 ± 0.21g, mean ± SEM, p < 0.0001); without any change on day 4 of infusion (aCSF: 4.43 ± 0.32g; pair-fed: 3.97± 0.33g; leptin: 3.36 ± 0.42g, mean ± SEM).
On day 4 of leptin infusion, POMC and NT mRNA levels in the MBH were significantly increased when compared with aCSF control (p < 0.01 for POMC, p < 0.05 for NT) and pair-fed (p < 0.01 for POMC, p < 0.05 for NT) groups (Fig 2). In pair-fed rats, there was no change in either POMC or NT mRNA levels as compared to that of aCSF control. In contrast, on day 16 of infusion, both POMC and NT mRNA levels were comparable among aCSF, pair-fed and leptin groups (Fig 2).
Fig. 2.
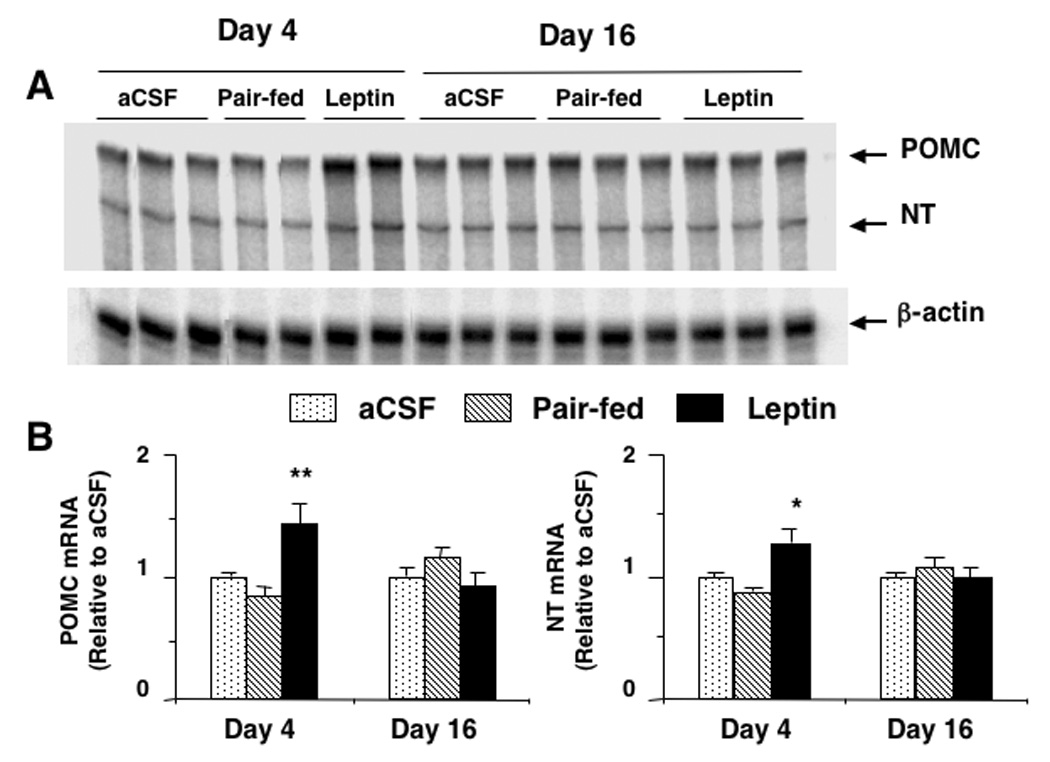
Proopiomelanocortin (POMC) and neurotensin (NT) gene expression as determined by ribonuclease protection assay in the hypothalamus after 4 or 16 days of leptin infusion. (A): representative phosphorimages showing the level of POMC mRNA, NT mRNA and β-actin mRNA in the hypothalamus. (B): results obtained by phosphor imaging showing the changes in POMC and NT mRNA levels. The values were first normalized to β-actin mRNA levels and then expressed as relative to aCSF control. Values represent the mean ± SEM. Day 4: n = 7, 8 and 7 in aCSF, pair-fed and leptin groups, respectively. Day 16: n = 9, 7 and 10 in aCSF, pair-fed and leptin groups, respectively. * p < 0.05 and ** p < 0.01 vs all other groups on day 4.
Discussion
The present study shows that POMC- and NT-producing neurons in the hypothalamus become resistant to chronic leptin infusion in that on day 4 of leptin infusion POMC and NT mRNA levels were increased, but on day 16 of leptin infusion, when food intake was recovered, POMC and NT mRNA levels did not differ from that of aCSF control. Because α-melanocyte stimulating hormone, a product of POMC gene, and NT are known to inhibit food intake [7,9,31,35,40], increased POMC and NT neuronal activity seen on day 4 of leptin infusion is most likely involved in decreased food intake during early period of leptin infusion. Similarly, leptin resistance in POMC and NT neurons may contribute in food intake recovery seen during later period of leptin infusion. Notably, although food intake in the leptin infused rats was mostly recovered by day 16 but it still remained significantly lower than control animals. Whereas the underlying mechanisms are currently unknown but if hyper-responsiveness of the melanocortin system as reported in case of DIO mice [10] did occur in our animals, then normal levels of POMC observed at this time could inhibit feeding. This requires further investigation. Nevertheless, these findings along with our previous demonstration of leptin resistance in the NPY neurons [34] suggest the development of leptin resistance in distinct orexigenic and anorectic peptide producing neurons of the hypothalamus, which may, at least in part, underlie the development of resistance to the satiety action of leptin following chronic leptin infusion.
The mechanism(s) behind the development of leptin resistance in the hypothalamic neurons, particularly in the POMC and NT neurons, following chronic increase in hypothalamic leptin tone is not clearly understood. In this regard, JAK2-STAT3 pathway is established to be the major pathway of leptin signaling in the hypothalamus [11,15,42]. Bates et al [3] have shown that disruption of long-form of leptin receptor (Ob-Rb)-STAT3 signaling by mutation of Tyr1138 in Ob-Rb results in reduction of POMC gene expression. Munzberg et al. [25] have suggested that gene expression of leptin-responsive POMC neurons in the hypothalamus requires STAT3 activation. However, the possibility of an impaired STAT3 signaling as a cause of leptin resistance, at least, in the POMC neurons is not supported by our previous demonstration of intact STAT3 activation in the hypothalamus during 16 days of central leptin infusion [30]. Amongst other signaling pathways, leptin signaling through the phosphatidylinositol-3 kinase (PI3K)-phosphodiesterase 3B (PDE3B)-cAMP pathway has gained significant importance [27,35,48]. We have recently demonstrated that this pathway is impaired following chronic leptin infusion [36] in association with an increased expression of suppressor of cytokine signaling 3 (SOCS3) [30], a negative regulator of cytokine signaling including that of leptin [4,19]. Notably, SOCS3 has been implicated in the development of leptin resistance in the hypothalamus [26], and brain specific SOCS3 deficiency [24] or haploinsufficiency in SOCS3 [14] resists the development of leptin resistance in DIO. Also, SOCS3 overexpression in hypothalamic neuronal cell line expressing POMC reverses the stimulating effect of leptin on PI3K activity [37]. It remains to be seen if impaired PI3K-PDE3B-cAMP pathway and increased SOCS3 contribute to the development of leptin resistance in the POMC and NT neurons (present study) and NPY neurons [34]. However, activation of JAK2-STAT3 pathway [30] may be responsible for regulation of energy expenditure and body weight, since the body weight remained decreased despite a recovery in food intake.
In addition to changes in the hypothalamus, the animals showed hyperleptinimia, hypoinsulinemia and reduced WAT weight, and maintained reduced body weight following chronic leptin infusion as described previously [34]. Whereas the reason behind the maintenance of reduce body weight despite normalization in food intake is not known, the direct inhibitory effect of leptin in lipogenesis and stimulation of lipolysis and fatty acid oxidation in adipocytes [1,45,46] may contribute to the reduced body weight following chronic leptin infusion. Previous studies have shown that chronic leptin administration (or overexpression) leads to a selective loss of adipose tissue while lean body mass is preserved [12,28]. Thus, decrease in WAT weight suggests that this type of response may be occurring in our rat model of chronic central leptin infusion, and which may explain the development of “leptin resistance” to the satiety action. Alternatively, melanocortin/SNS-mediated leptin-induced lipolysis could occur and remains to be examined. Notably, during the later period of leptin infusion, pair-fed animals gained weight as compared to leptin group, which may be due to decreased energy expenditure in pair-fed animals as opposed to increased energy expenditure in leptin group.
In summary, we have demonstrated that POMC and NT neurons develop leptin resistance in association with the development of resistance to the satiety action of leptin following chronic central leptin infusion. This study along with previous evidence of leptin resistance in NPY neurons suggest the development of leptin resistance, at least in some distinct neuronal system, in the hypothalamus in this rat model of chronic leptin infusion.
Acknowledgements
This work was supported by NIH RO1 Grant DK61499. Thanks to A. F. Parlow and the NIDDK National Hormone & Pituitary Program, Torrance, CA, for supplying the recombinant murine leptin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bai Y, Zhang S, Kim KS, Lee JK, Kim KH. Obese gene expression alters the ability of 30A5 preadipocytes to respond to lipogenic hormones. J Biol Chem. 1996;271:13939–13942. doi: 10.1074/jbc.271.24.13939. [DOI] [PubMed] [Google Scholar]
- 2.Banks WA. Leptin transport across the blood-brain barrier: implications for the cause and treatment of obesity. Curr Pharm Des. 2001;7:125–133. doi: 10.2174/1381612013398310. [DOI] [PubMed] [Google Scholar]
- 3.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 4.Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 5.Bluher S, Ziotopoulou M, Bullen JW, Jr, Moschos SJ, Ungsunan L, Kokkotou E, Maratos-Flier E, Mantzoros CS. Responsiveness to peripherally administered melanocortins in lean and obese mice. Diabetes. 2004;53:82–90. doi: 10.2337/diabetes.53.1.82. [DOI] [PubMed] [Google Scholar]
- 6.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 7.Cone RD. The Central Melanocortin System and Energy Homeostasis. Trends Endocrinol. Metab. 1999;10:211–216. doi: 10.1016/s1043-2760(99)00153-8. [DOI] [PubMed] [Google Scholar]
- 8.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 9.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 10.Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 12.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. Jama. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 14.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 15.Hubschle T, Thom E, Watson A, Roth J, Klaus S, Meyerhof W. Leptin-induced nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei involved in body weight regulation. J Neurosci. 2001;21:2413–2424. doi: 10.1523/JNEUROSCI.21-07-02413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakubowski M, Roberts JL. Multiplex solution hybridization-ribonuclease protection assay for quantification of different ribonucleic acid transcripts from snap-frozen neuroendocrine tissues of individual animals. J. Neuroendocrinol. 1992;4:79–89. doi: 10.1111/j.1365-2826.1992.tb00349.x. [DOI] [PubMed] [Google Scholar]
- 17.Kahn D, Abrams GM, Zimmerman EA, Carraway R, Leeman SE. Neurotensin neurons in the rat hypothalamus: an immunocytochemical study. Endocrinology. 1980;107:47–54. doi: 10.1210/endo-107-1-47. [DOI] [PubMed] [Google Scholar]
- 18.Kislauskis E, Bullock B, McNeil S, Dobner PR. The rat gene encoding neurotensin and neuromedin N. Structure, tissue-specific expression, and evolution of exon sequences. J Biol Chem. 1988;263:4963–4968. [PubMed] [Google Scholar]
- 19.Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113(Pt 16):2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 20.Levin BE, Dunn-Meynell AA. Reduced central leptin sensitivity in rats with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2002;283:R941–R948. doi: 10.1152/ajpregu.00245.2002. [DOI] [PubMed] [Google Scholar]
- 21.Levine AS, Kneip J, Grace M, Morley JE. Effect of centrally administered neurotensin on multiple feeding paradigms. Pharmacol Biochem Behav. 1983;18:19–23. doi: 10.1016/0091-3057(83)90244-7. [DOI] [PubMed] [Google Scholar]
- 22.Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord. 2000;24:639–646. doi: 10.1038/sj.ijo.0801209. [DOI] [PubMed] [Google Scholar]
- 23.Luttinger D, King RA, Sheppard D, Strupp J, Nemeroff CB, Prange AJ., Jr The effect of neurotensin on food consumption in the rat. Eur J Pharmacol. 1982;81:499–503. doi: 10.1016/0014-2999(82)90116-9. [DOI] [PubMed] [Google Scholar]
- 24.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 25.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 26.Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- 27.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 28.Ogawa Y, Masuzaki H, Hosoda K, Aizawa-Abe M, Suga J, Suda M, Ebihara K, Iwai H, Matsuoka N, Satoh N, Odaka H, Kasuga H, Fujisawa Y, Inoue G, Nishimura H, Yoshimasa Y, Nakao K. Increased glucose metabolism and insulin sensitivity in transgenic skinny mice overexpressing leptin. Diabetes. 1999;48:1822–1829. doi: 10.2337/diabetes.48.9.1822. [DOI] [PubMed] [Google Scholar]
- 29.Ohinata K, Shimano T, Yamauchi R, Sakurada S, Yanai K, Yoshikawa M. The anorectic effect of neurotensin is mediated via a histamine H1 receptor in mice. Peptides. 2004;25:2135–2138. doi: 10.1016/j.peptides.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Pal R, Sahu A. Leptin signaling in the hypothalamus during chronic central leptin infusion. Endocrinology. 2003;144:3789–3798. doi: 10.1210/en.2002-0148. [DOI] [PubMed] [Google Scholar]
- 31.Sahu A. Evidence suggesting that galanin (GAL), melanin-concentrating hormone (MCH), neurotensin (NT), proopiomelanocortin (POMC) and neuropeptide Y (NPY) are targets of leptin signaling in the hypothalamus. Endocrinology. 1998;139:795–798. doi: 10.1210/endo.139.2.5909. [DOI] [PubMed] [Google Scholar]
- 32.Sahu A. Quantification of NPY mRNA by ribonuclease protection assay. Methods Mol. Biol. 2000;153:219–230. doi: 10.1385/1-59259-042-X:219. [DOI] [PubMed] [Google Scholar]
- 33.Sahu A, Carraway RE, Wang YP. Evidence that neurotensin mediates the central effect of leptin on food intake in rat. Brain Res. 2001;888:343–347. doi: 10.1016/s0006-8993(00)03107-3. [DOI] [PubMed] [Google Scholar]
- 34.Sahu A. Resistance to the satiety action of leptin following chronic central leptin infusion is associated with the development of leptin resistance in neuropeptide Y neurones. J Neuroendocrinol. 2002;14:796–804. doi: 10.1046/j.1365-2826.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 35.Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol. 2003;24:225–253. doi: 10.1016/j.yfrne.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Sahu A, Metlakunta AS. Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion. J Neuroendocrinol. 2005;17:720–726. doi: 10.1111/j.1365-2826.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- 37.Sahu A, Metlakunta AR, Sahu M. Program No.60.10. 2006 Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience; 2006. Suppressor of cytokine signaling-3 interacts with the phosphatidylinositol 3-kinase pathway of leptin signaling in the hypothalamus. Online. [Google Scholar]
- 38.Scarpace PJ, Matheny M, Zolotukhin S, Tumer N, Zhang Y. Leptin-induced leptin resistant rats exhibit enhanced responses to the melanocortin agonist MT II. Neuropharmacology. 2003;45:211–219. doi: 10.1016/s0028-3908(03)00158-8. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D., Jr Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2:589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 41.Stanley BG, Hoebel BG, Leibowitz SF. Neurotensin: effects of hypothalamic and intravenous injections on eating and drinking in rats. Peptides. 1983;4:493–500. doi: 10.1016/0196-9781(83)90054-2. [DOI] [PubMed] [Google Scholar]
- 42.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 43.Trinder P. Determination of blood glucose using an oxidase-peroxidase system with a non-carcinogenic chromogen. J Clin Pathol. 1969;22:158–161. doi: 10.1136/jcp.22.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, Sybertz EJ, Strader CD, Davis HR., Jr Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang MY, Lee Y, Unger RH. Novel form of lipolysis induced by leptin. J Biol Chem. 1999;274:17541–17544. doi: 10.1074/jbc.274.25.17541. [DOI] [PubMed] [Google Scholar]
- 46.Wang ZW, Zhou YT, Lee Y, Higa M, Kalra SP, Unger RH. Hyperleptinemia depletes fat from denervated fat tissue. Biochem Biophys Res Commun. 1999;260:653–657. doi: 10.1006/bbrc.1999.0918. [DOI] [PubMed] [Google Scholar]
- 47.Widdowson PS, Upton R, Buckingham R, Arch J, Williams G. Inhibition of food response to intracerebroventricular injection of leptin is attenuated in rats with diet-induced obesity. Diabetes. 1997;46:1782–1785. doi: 10.2337/diab.46.11.1782. [DOI] [PubMed] [Google Scholar]
- 48.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 49.Ziotopoulou M, Mantzoros CS, Hileman SM, Flier JS. Differential expression of hypothalamic neuropeptides in the early phase of diet-induced obesity in mice. Am J Physiol Endocrinol Metab. 2000;279:E838–E845. doi: 10.1152/ajpendo.2000.279.4.E838. [DOI] [PubMed] [Google Scholar]