

Abstract
Enoyl-ACP reductase (ENR), the product of the FabI gene, from Bacillus anthracis (BaENR) is responsible for catalyzing the final step of bacterial fatty acid biosynthesis. A number of novel 2-pyridone derivatives were synthesized and shown to be potent inhibitors of BaENR.
Fatty acids are an essential source of energy for organisms from all taxa. Fatty acid synthesis in mammals is substantially different from that in bacteria. In mammals, the fatty acid synthesis involves a single multifunctional enzyme–acyl carrier protein (ACP) complex. In bacteria, the synthesis utilizes several small monofunctional enzymes that operate in conjunction with ACP–associated substrates.1 Thus, it is possible to selectively target key enzymes in the bacterial fatty acid biosynthesis.2
The final and rate–determining step of chain elongation in the bacterial fatty acid biosynthesis is the reduction of enoyl–ACP to an acyl–ACP, which is catalyzed by the enzyme–enoyl–acyl carrier protein reductase. Considerable research over the past few years has shown that ENR is the target of a number of known antibacterial agents, including isoniazid,3 diazaboranes,4 and triclosan.5 Targeting ENR is an attractive approach for the development of novel antibacterials, which has been validated by the recent discovery of several other small molecule inhibitors,6 including those that exhibit potent in vitro activity against clinical isolates of methicillin-resistant S. epidermidis and S. aureus.7
As part of an on-going program to develop novel therapeutics for anthrax, we recently reported that the diphenyl ether–triclosan and a number of its aryl ether derivatives inhibit BaENR.8 Although triclosan is a potent inhibitor of BaENR (IC50 = 0.5 µM, MIC = 3.1 µg/mL),9 a major caveat in developing triclosan or its derivatives as drugs is the metabolic liability of the phenolic hydroxyl group.10 In addition, triclosan-like diphenyl ethers are fairly lipophilic molecules and pose serious solubility problems. In order to overcome these structural drawbacks, it is important to design compounds in which the phenolic group of ring A of triclosan is replaced by a more metabolically stable functionality. We report herein our efforts toward addressing these issues by developing novel scaffolds that replace the phenolic ring of triclosan with other heterocyclic rings that retain the essential structural features of triclosan required for interaction with ENR, such as π-stacking of the aromatic ring with NAD+, hydrogen bonding of the phenolic OH group with the ribose, as well as optimal flexibility of the two rings in order to allow complementary interactions with the active site.9
Our structure optimization studies were based on predicted binding interactions of 2-pyridones with the enzyme active site. Triclosan and synthesized pyridones were docked into the BaENR crystal structure9 using the GOLD docking program.11 It was observed that the 2-pyridones dock to BaENR in the same binding pocket as that of triclosan, and maintain similar H–bonding interactions with the residues in the active site. Figure 1 shows the similarities in the binding geometry of triclosan and a representative 2-pyridone, 2. Figure 2 shows the crystal structure of triclosan bound to BaENR and the GOLD docking conformations of compounds 2 and 35 in the active site. The X-ray structure of triclosan bound to BaENR shows that the phenolic hydroxyl group on ring A is involved in two hydrogen bonds, one with Tyr157 (OH) and the other to the 2′-hydroxyl group of nicotinamide ribose (shown in Fig. 2A).9 These interactions appear to be preserved in the GOLD docking conformation of the pyridones as well, and show that the oxygen on the carbonyl group of ring A is involved in a hydrogen bonding interaction with Tyr157 (OH) and the NAD+ (shown in Fig. 2B and 2C). As seen in Figure 1, the ring A pocket of the pyridones is surrounded by Val 154, Val 201, Ile 207, and Phe 204, and thus appears to be dominated by a high amount of hydrophobicity.
Figure 1.

Schematic representation of residues in the binding pockets of rings A and B of A) triclosan B) compound 2.
Figure 2.

(A) Crystal structure of triclosan bound to BaENR. ENR atoms are colored by atom type, NAD+ is green, and triclosan is pink. (B) GOLD docking conformation of 2 against the crystal structure of BaENR. ENR atoms are colored by atom type, NAD+ is green, and 2 is cyan. Distances between atoms that are close enough to be within hydrogen bonding range are shown in green. (C) GOLD docking conformation of 35 against the crystal structure of BaENR. ENR atoms are colored by atom type, NAD+ is green, and 35 is light pink.
The synthesis of compounds 1–3, 5, and 9 involved selective N-alkylation of the commercially available 4-benzyloxy-2(1H)-pyridone with the corresponding benzyl halides according to the procedure described by Conreaux et al. (Scheme 1).12 Reduction of 3 gave the amino compound 4, which was further converted to the acetamide 8. While alkaline hydrolysis of the benzonitrile 5 resulted in the carboxylic acid 6, hydrolysis in the presence of hydrogen peroxide gave the carboxamide 7. Isoxazole 10 was prepared by treating 9 with ethyl chlorooximidoacetate in the presence of a base.13 Rapid and selective O-debenzylation of 4-benzyloxy-2(1H)-pyridone derivatives occurred when treated with Pd/C under atmospheric pressure of hydrogen to give compounds 11 and 12. Compound 13 was obtained by treatment of 12 with BBr3.
Scheme 1.

Reagents and conditions: (a) KOtBu, TBAI, THF, ArCH2X, 0–25 °C, 16 h; (b) NaBH4, Cu(OAc)2, THF, 0 °C, 40 min; (c) 25% NaOH, EtOH, reflux, 20 h; (d) 35% H2O2, 3N NaOH, EtOH, 30 °C, 20 h; (e) Ac2O, DMAP, Et3N, CH2Cl2, 0–25 °C, 3 h; (f) KOtBu, TBAI, THF, propargyl bromide, 0–25 °C, 16 h; (g) ethyl chlorooximidoacetate, Et3N, rt, 5 h; (h) Pd/C, H2, MeOH, rt, 10–15 min; (i) BBr3, CH2Cl2, −78 °C to rt, 12 h.
The above 4-hydroxy-pyridones were elaborated into a number of derivatives via a series of alkylation reactions (Scheme 2). Compound 14 was prepared by benzylation of 4-hydroxy-pyridone with 4-cyanobenzyl bromide. Naphthyl derivatives 15 and 16 were obtained by using a similar procedure. The common synthetic intermediates 17 were prepared by alkylating the 4-hydroxy-pyridones with 1,3-dibromopropane. Compounds 18–20 were prepared in turn by N-alkylation of carbazole with these intermediates. Compounds 21–23 were synthesized using a similar protocol.
Scheme 2.

Reagents and conditions: (a) NaH, DMF/THF, 4-CN-PhCH2Br, 0–25 °C, 12 h; (b) NaH, DMF, 1,3-dibromopropane, 0–25 °C, 6 h; (c) NaH, DMF, carbazole, 0–25 °C, 12 h; (d) For 21: NaH, DMF, indole, rt, 16 h; For 22: benzotriazole, DMF, K2CO3, rt, 12 h; For 23: imidazole, KOH, THF, 85 °C, 16 h; (e) NaH, DMF/THF, 1- or 2-bromomethyl naphthalene, 0–25 °C, 12 h.
The synthesis of the 3-substituted 2-pyridones and N-oxide derivatives is shown in scheme 3. Pyridones 33–35 were synthesized by acetic anhydride mediated rearrangement of the corresponding N-oxides, followed by acidic hydrolysis.14 The N-oxides were synthesized from the corresponding pyridines by m-CPBA oxidation. While the ethers 27 and 28 were prepared by standard coupling reactions, intermediate 29 was obtained by treating 2-benzoyloxy-5-bromopyridine with benzyltrimethyltin under Stille reaction conditions.
Scheme 3.

Reagents and conditions: (a) benzyl alcohol, NaH, THF, rt, 12 h; (b) For 27: KOH-CuCO3, 2-chlorophenol, 180 °C, 15 h; For 28: 2,4-dichlorophenol, KOtBu, DMF, rt, 3 h, then at 45 °C under vacuum, Cu(OTf)2·PhCH3, DMF, reflux, 16 h; (c) trimethylbenzylstannane, Pd(PPh3)4, DMF, 80 °C, 12 h; (d) m-chloroperbenzoic acid, CHCl3, rt, 12 h; (e) Ac2O, reflux, 5 h, 1N HCl, 100 °C, 12 h; (f) KOH-CuCO3, 2,4-dichlorophenol, 180 °C, 15 h.
The compounds synthesized were evaluated for their BaENR inhibitory and anitibacterial activities.15 Inhibitor 2 appears to be the best compound in this series and was considered as a lead. As expected from the interactions of hydrophobic residues located around the 3-position of the pyridone ring in the active site, replacing the benzyloxy group at this position with a more polar hydroxy group led to a substantial decrease in the ENR inhibitory activity (Table 1. compounds 11–13). A four–fold improvement in the enzyme inhibitory activity was observed when a chlorine atom is present at the 2′-position on the aromatic ring B (compare 1 vs 2). A similar improvement in the binding affinity of triclosan derivatives to the BaENR active site was recently observed by us.8 Attachment of an electron withdrawing cyano group to the aromatic ring of the benzyloxy moiety resulted in 14, which showed an ENR inhibitory activity lower than the lead compound. Thus, we explored the activities of compounds with other hydrophobic substitutents at the 3-position of the pyridone ring. Introduction of 1- and 2-naphthyl groups resulted in compounds 15 and 16 whose enzymatic activity was comparable to the lead compound. Introduction of a bulkier carbazole unit, tethered with a three-carbon chain on the other hand, decreased the BaENR inhibitory activity (Table 1, compounds 18–20). Again, the importance of having a chlorine atom at the 2′-position on ring B to improve the inhibitory activity of these compounds is evident by comparing the activities of 18 and 20. Replacing the carbazole unit with a smaller indole moiety resulted in two–fold improvement in the ENR inhibitory activity (compound 21), while a benzotriazolyl substitution resulted in the compound (22) with an ENR inhibitory activity comparable to that of the lead compound. Introduction of an imidazolyl unit did not improve the activity.
Table 1.
BaENR inhibitory activities of compounds.
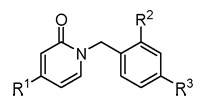 | ||||
|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | IC50 (µM) |
| 11 | OH | H | H | > 100a |
| 12 | OH | H | OMe | > 100a |
| 13 | OH | H | OH | > 100a |
| 1 | BnO | H | H | 6.8 ± 0.8 |
| 2b | BnO | Cl | H | 1.5 ± 0.1 |
| 14 | 4-CN-PhCH2O | Cl | H | 2.7 ± 0.4 |
| 15 | 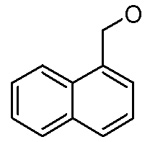 |
Cl | H | 1.5 ± 0.8 |
| 16 | 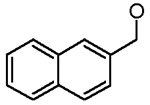 |
Cl | H | 1.1 ± 0.1 |
| 18 |  |
Cl | H | > 6c |
| 19 | 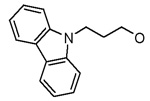 |
H | OMe | > 3d |
| 20 | 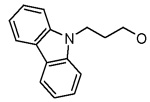 |
H | H | > 100a |
| 21 |  |
Cl | H | 0.8 ± 0.2 |
| 22 |  |
Cl | H | 1.6 ± 0.3 |
| 23 | 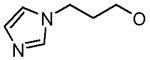 |
Cl | H | 28.0 ± 6.2 |
| 5 | BnO | Cl | CN | 7.0 ± 1.1 |
| 7 | BnO | Cl | CONH2 | 3.6 ± 0.3 |
| 6b | BnO | Cl | COOH | 23.8 ± 2.5 |
| 3 | BnO | Cl | NO2 | 3.7 ± 1.1 |
| 4 | BnO | Cl | NH2 | 0.8 ± 0.1 |
| 8 | BnO | Cl | NHAc | 1.8 ± 0.5 |
BaENR inhibition was less than 30% at 100 µM.
MIC values against ΔANR B. anthracis14 for compound 2: 16 µg/mL, for compound 6: 74 µg/mL. MICs for all other compounds in the table are > 80 µg/mL.
The inhibitor precipitated at concentrations > 6 µM.
The inhibitor precipitated at concentrations > 3 µM.
We explored the SAR of the 2-pyridones by functionalizing ring B. From the docking conformations of the lead compound into the BaENR X-ray crystal structure (Figure 2B), we anticipated that hydrogen bond donors/acceptors at the 4′-position would be ideally positioned to interact with either Ala 97 or Arg 99. Hence, we synthesized compounds 3–8 with various functional groups at the 4′-position of the ring B. Compound 4, bearing an amino group at the 4′-position turned out to be the best compound with an IC50 of 0.8 µM. Conversion of the amino functionality into an acetamide (compound 8) reduced the BaENR inhibitory activity by half. Thus, it appears that the presence of an electron-donating group at the 4′-position is able to enhance the interaction of ligands with the enzyme active site. Attempts to replace the aromatic ring B of these 2-pyridones with an acetylene (compound 9) or an isoxazole (compound 10) were not successful in improving the activity (Table 2).
Table 2.
BaENR inhibitory activities of compounds.
| Compd. a | Structure | IC50 (µM) |
|---|---|---|
| 9 | 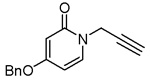 |
> 100b |
| 10 |  |
> 100b |
| 35 | 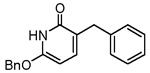 |
0.7 ± 0.4 |
| 33 | 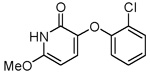 |
18.8 ± 4.2 |
| 34 | 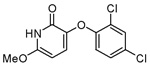 |
23.7 ± 11.7 |
| 30 |  |
> 100b |
| 37 |  |
21.4 ± 7.7 |
MIC values against ΔANR B. anthracis were >100 µg/mL.
BaENR inhibition was less than 30% at 100 µM.
We briefly explored the activities of C-substituted 2-pyridones that are structurally similar to the N-substituted 2-pyridones discussed above (compounds 33–35). These C-substituted pyridones are capable of existing in their enol form as hydroxypyridines, and thus closely mimic triclosan in structure. The activities of these compounds are shown in Table 2. It is gratifying to note that the novel C-substituted 2-pyridone, 35 showed a 10–fold improvement in ENR inhibitory activity over its N-substituted analog 1. The GOLD docking conformation of 35 in Figure 2C suggests a nearly identical orientation of the C-substituted 2-pyridones compared to the N-substituted pyridones. Although the origin of improved activity of compound 35 is not completely clear at this stage, the pyridone NH and the nicotinamide ring are about 3.6 Å apart and thus ligand binding stabilization from an N–H⋯π interaction cannot be ruled out.16 Moderate ENR inhibition was observed by the 3-phenoxy-2-pyridones 33 and 34. Among the pyridine N-oxides, compound 37 exhibited modest ENR inhibition, while compound 30 was inactive.
In conclusion, we have identified certain 2-pyridone derivatives as novel, small-molecule inhibitors of bacterial enoyl-ACP reductase (ENR) from Bacillus anthracis. Compound 2 showed good ENR-inhibitory activity as well as reasonable antibacterial activity, thus providing a lead compound for further development. Compounds 4 and 21 show nearly a two–fold improvement in ENR inhibitory activity over compound 2. Compound 35, a ‘reversed’ pyridone, is also an encouraging lead for the development of a new class of ENR inhibitors. Current efforts focus on further improvement of BaENR inhibition and improving antibacterial activities of these compounds.
Supplementary Material
Acknowledgment
This work was funded by a grant from NIH/NIAID (U19 AI056575).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Rock CO, Jackowski S. Biochem. Biophys. Res. Commun. 2002;292:1155. doi: 10.1006/bbrc.2001.2022. [DOI] [PubMed] [Google Scholar]; (b) Wakil SJ. Biochemistry. 1989;28:4523. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 2.(a) Campbell JW, Cronan JE., Jr Annu. Rev. Microbol. 2001;55:305. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]; (b) Heath RJ, White SW, Rock CO. Prog. Lipid Res. 2001;40:467. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 3.Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr, Sacchettini JC. Science (New York, N.Y.) 1998;279:98. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 4.Levy CW, Baldock C, Wallace AJ, Sedelnikova S, Viner RC, Clough JM, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. J. Mol. Biol. 2001;309:171. doi: 10.1006/jmbi.2001.4643. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 5.(a) McLeod R, Muench SP, Rafferty JB, Kyle DE, Mui EJ, Kirisits MJ, Mack DG, Roberts CW, Samuel BU, Lyons RE, Dorris M, Milhous WK, Rice DW. Int. J. Parasitol. 2001;31:109. doi: 10.1016/s0020-7519(01)00111-4. [DOI] [PubMed] [Google Scholar]; (b) ref 8 and references cited therein. [Google Scholar]
- 6.(a) Kitagawa H, Kumura K, Takahata S, Iida M, Atsumi K. Bioorg. Med. Chem. Lett. 2007;15:1106. doi: 10.1016/j.bmc.2006.10.012. [DOI] [PubMed] [Google Scholar]; (b) Kitagawa H, Ozawa T, Takahata S, Iida M, Saito J, Yamada M. J. Med. Chem. 2007;50:4710. doi: 10.1021/jm0705354. and references cited therein. [DOI] [PubMed] [Google Scholar]; (c) Moir DT. Curr. Drug Targets Infect. Disord. 2005;5:297. doi: 10.2174/1568005054880154. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 7.Karlowsky JA, Laing NM, Baudry T, Kaplan N, Vaughan D, Hoban DJ, Zhanel GG. Antimicrob. Agents Chemother. 2007;51:1580. doi: 10.1128/AAC.01254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tipparaju SK, Mulhearn DC, Klien GM, Chen Y, Tapadar S, Bishop MH, Yang S, Chen J, Ghassemi M, Santarsiero BD, Cook JL, Johlfs M, Mesecar AD, Johnson ME, Kozikowski AP. ChemMedChem. 2008 April; In press. [Google Scholar]
- 9.Klein GM, Bernard DS, Tipparaju SK, Pegan S, Bishop MH, Kozikowski AP, Mesecar AD. Biochemistry. 2007 Accepted with minor revisions. [Google Scholar]
- 10.Wang LQ, Falany CN, James MO. Drug Metab. Dispos. 2004;32:1162. doi: 10.1124/dmd.104.000273. [DOI] [PubMed] [Google Scholar]
- 11.The synthesized pyridones were docked into the B.anthracis ENR active site from the crystal structure with PDB code 2QIT. GOLD docking was performed using the default parameters and the ChemScore scoring function. GOLD reference:Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609. doi: 10.1002/prot.10465.
- 12.Conreaux D, Bossharth E, Monteiro N, Desbordesb P, Balmea GV. Tetrahedron Lett. 2005;46:7917. [Google Scholar]
- 13.Kozikowski AP, Adamczyk M. J. Org. Chem. 1983;48:366. [Google Scholar]
- 14.Shone RL, Coker VM, Moormann AE. J. Heterocycl. Chem. 1975;12:389. [Google Scholar]
- 15.Detailed description of methods for biological assay is provided in supplementary information.
- 16.Meyer EA, Castellano RK, Diederich F. Angew. Chem. Int. Ed. 2003;42:1210. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.