

Abstract
We have investigated the influence of polymer structure on the erosion profiles of multilayered polyelectrolyte assemblies fabricated from sodium poly(styrene sulfonate) (SPS) and three different hydrolytically degradable polyamines. We synthesized three structurally related poly(β-amino ester)s (polymers 1−3) having systematic variations in both charge density and hydrophobicity. These changes in structure did not influence film thickness significantly, but polymer structure was found to play an important role in defining the rates at which multilayered assemblies fabricated from these materials eroded in physiologically relevant media. Films 60 nm thick fabricated from polymer 1 and SPS eroded completely in 50 hours when incubated in PBS buffer at 37 °C, as determined by ellipsometry. Analogous films fabricated from polymers 2 and 3 eroded and released SPS into solution over significantly longer time periods ranging from approximately 150 hours (ca. 6 days) to 370 hours (ca. 15 days), respectively. These differences are consistent with a systematic increase in the hydrophobicity of polymers 1−3 as well as the relative rates at which these polymers degrade hydrolytically. This work demonstrates that it is possible to tailor the rates at which thin, multilayered polyelectrolyte assemblies release incorporated anionic polyelectrolytes over a large range of time periods simply by changing the structure of the degradable polyamine used to fabricate a film. The principles reported here may therefore contribute to the design of multilayered assemblies that permit a broad range of spatial and temporal control over the release of therapeutic agents from coated surfaces.
Introduction
The layer-by-layer deposition of oppositely charged polyelectrolytes on surfaces is a convenient and versatile method for the fabrication of multilayered polyelectrolyte assemblies.1-4 The polyvalent nature of the electrostatic interactions that contribute to the assembly of these films often yields robust, ionically crosslinked thin films that are stable under the conditions in which they are used. This general stability, combined with the range of other properties imparted through the variety of materials that can be incorporated,1-4 has prompted the evaluation of these materials in numerous biomedical and biotechnological contexts.3,5-21 For example, several groups have reported the fabrication of films designed to be stable at physiological pH and ionic strength that promote, enhance, or resist the adhesion of cells on coated surfaces.9,13-20 We22-26 and others27-39 have sought to develop principles that contribute to the controlled instability of these materials in physiological media by designing into the structures of polyelectrolytes and multilayered films functionality that can be disrupted or degraded. This work has resulted in the fabrication of assemblies that erode and release polyelectrolytes incorporated into the structure of a film in response to changes in environmental pH and ionic strength,27-33 the action of enzymes,34-36 or the presence of small molecules that disrupt supramolecular interactions in a film.37-39
We have demonstrated in several past studies that synthetic polyamine 1 can be used to fabricate multilayered assemblies that erode gradually and release incorporated anionic polyelectrolytes under physiologically relevant conditions (e.g., PBS buffer, pH 7.4, 37 °C).22-26 For example, multilayered assemblies 100 nm thick fabricated using polymer 1 have been used to sustain the release of synthetic polyanions,22,24 transcriptionally active plasmid DNA constructs,23-25 and therapeutically relevant anionic polysaccharides26 from a variety of coated surfaces. Polymer 1 can be rendered cationic by protonation and it is hydrolytically degradable in aqueous media by virtue of the esters located in the polymer backbone. This polymer is a member of a larger class of synthetic materials known as poly(β-amino ester)s40 that can be synthesized by the conjugate addition of amine-containing compounds to diacrylates. Several past studies have used this synthetic procedure to synthesize small collections41-43 or large libraries44-46 of structurally diverse linear41,43-46 and hyperbranched42 degradable polyamines. An important outcome of this prior work has been the demonstration that the chemical structure of these polymers influences significantly several physical and functional properties, including 1) the ability (or inability) to self-assemble with polyanions in solution,41,43-47 2) the ability to promote the delivery of DNA to cells,41,43-46 and, of particular importance to the work reported here, 3) the range of rates and conditions under which different polymers degrade by ester hydrolysis in aqueous media.41,42

Multilayered films fabricated from polymer 1 typically erode over periods of time ranging from 24 to 48 hours at physiological pH, temperature, and ionic strength.22-26 In the context of controlled release, these assemblies are therefore limited to the sustained release of incorporated material over relatively short time periods. The development of principles that can be used to reduce erosion rates or otherwise delay release for longer periods of time would contribute to the design of these assemblies for a broader range of therapeutic and controlled release applications. We sought to explore the influence of polymer structure on the fabrication and erosion profiles of multilayered films by varying systematically the hydrophobicity and charge densities of the polycations incorporated into these assemblies. Here, we report the fabrication, physical characterization, and erosion profiles of 60−100 nm thick multilayered films constructed from sodium poly(styrene sulfonate) (SPS) and three different structurally related poly(β-amino ester)s, polymers 1−3. We find large differences in the erosion and release profiles of these assemblies in physiological media. Films fabricated from polymers 2 and 3 erode more slowly than polymer 1 and release SPS from the surface of coated substrates for periods ranging up to 150 hours (ca. 6 days) and 370 hours (ca. 15 days), respectively. These differences in erosion profiles correlate directly with relative differences in the hydrophobicities of polymers 1−3 as well as the rates at which these polymers degrade. Thus, varying the structure of these degradable polyamines expands significantly the range of erosion and release profiles that are accessible using these conformal thin films. A more thorough understanding of the range of different structure/property relationships that govern the behavior of these erodible materials should contribute, in the longer term, to the design of more sophisticated assemblies that permit tunable control over the release of anionic polyelectrolytes from surfaces.
Materials and Methods
General Considerations
1H and 13C NMR spectra were recorded on a Bruker AC+ 300 spectrometer. Chemical shift values are reported in ppm and are referenced to residual protons from solvent. Gel permeation chromatography (GPC) was performed using a Waters 515 HPLC pump (Waters Corporation, Milford, MA), a Rheodyne Model 7725 injector with a 20-μL injection loop, and two Waters Styragel HT 6E columns in series. THF containing 0.1 M triethylamine was used as the eluent at a flow rate of 1.0 mL/min. Data were collected using a Waters 2410 Refractive Index Detector and processed using the Waters Empower software package. Molecular weights are reported relative to monodisperse polystyrene standards. Silicon substrates (e.g., 0.5 × 2.0 cm) used for the fabrication of multilayered films were cleaned with methylene chloride, ethanol, methanol, and deionized water, and dried under a stream of filtered compressed air. Surfaces were then activated by etching with an oxygen plasma for 5 minutes (Plasma Etch, Carson City, NV) prior to film deposition. The optical thicknesses of films deposited on silicon substrates were determined using a Gaertner LSE ellipsometer (632.8 nm, incident angle = 70°). Data were processed using the Gaertner Ellipsometer Measurement Program. Relative thicknesses were calculated assuming an average refractive index of 1.577 for the multilayered films. Thicknesses were determined in at least 5 different standardized locations on each substrate and are presented as an average (with standard deviation) for each film. All films were dried under a stream of nitrogen prior to measurement. UV-visible absorbance values for PBS solutions used to determine film release kinetics were recorded on a Beckman Coulter DU520 UV/vis Spectrophotometer (Fullerton, CA). Absorbance values were recorded at a wavelength of 226 nm. The pH of buffers used for erosion and hydrolysis experiments was recorded using a pH meter and, for the preparation of deuterated buffers, is reported as pH.
Materials
4,4’-Trimethylenedipiperidine, poly(sodium 4-styrenesulfonate) (SPS, MW = 70,000), and sodium acetate buffer were purchased from Aldrich Chemical Company (Milwaukee, WI). 1,4-Butanediol diacylate and 1,6-hexanediol diacrylate were purchased from Alfa Aesar Organics (Ward Hill, MA). 1,8-Octanediol diacrylate (CAS# 10526−04−2) was synthesized from 1,8-octanediol and acryloyl chloride and purified by column chromatography using a procedure similar to that reported for the synthesis of structurally related diacrylates.48 Test grade n-type silicon wafers were purchased from Si-Tech, Inc. (Topsfield, MA). Quartz microscope slides were purchased from Chemglass (Vineland, NJ). Commercially available samples of linear poly(ethylene imine) (LPEI, MW = 25,000) were obtained from Polysciences, Inc. (Warrignton, PA). Phosphate buffered saline was prepared by dilution of commercially available concentrate (EM science, Gibbstown, NJ). All materials were used as received without further purification unless noted otherwise. Deionized water (18 MΩ) was used for washing steps and to prepare all buffer and polymer solutions. All buffers and polymer solutions were filtered through a 0.2 μm membrane syringe filter prior to use unless noted otherwise. Compressed air used to dry films and coated substrates was filtered through a 0.4 μm membrane syringe filter.
General Polymerization Procedure
Polymers 2 and 3 were synthesized in analogy to methods previously reported for the synthesis of polymer 1.41 Briefly, 4,4’-trimethylenedipiperidine (3 mmol) and the appropriate diacrylate (3 mmol) were weighed into separate vials and dissolved in anhydrous THF (5 mL). The solution containing the diamine was added to the solution containing the diacrylate via pipette. The reaction mixture was heated to 50 °C and stirred for 48 hrs. The resulting reaction products were concentrated by rotary evaporation, precipitated into hexanes, and dried under vaccum to yield each polymer as a white powder. 1H NMR data for polymer 2: (CDCl3, 300.135 MHz) δ (ppm) = 4.07 (br t, 4H); 2.87 (br, m, 4H); 2.67 (br m, 4H); 2.50 (br m, 4H); 1.98 (br m, 4H); 1.64 (br m, 8H); 1.1−1.4 (br m, 16H). 1H NMR data for polymer 3: (CDCl3, 300.135 MHz) δ (ppm) = 4.07 (br t, 4H); 2.88 (br, m, 4H); 2.68 (br m, 4H); 2.52 (br m, 4H); 1.96 (br m, 4H); 1.63 (br m, 8H); 1.1−1.4 (br m, 20H). 13C NMR data for polymer 2: 13C NMR data for polymer 2: (CDCl3, 75.41 MHz) δ (ppm) = 172.94, 64.48, 54.16, 53.98, 36.95, 35.84, 32.60 (br), 28.70, 25.76, 24.08. 13C NMR data for polymer 3: (CDCl3, 75.41 MHz) δ (ppm) = 172.97, 64.65, 54.18, 53.98, 36.96, 35.84, 32.61 (br), 29.31, 28.78, 26.02, 24.09.
Preparation of Polyelectrolyte Solutions
Solutions of polymers 1, 2, and 3 used for dipping (5 mM with respect to the molecular weight of the polymer repeat unit) were prepared in sodium acetate buffer (100 mM, pH = 5.1) and filtered through a 0.2 μm membrane syringe filter prior to use. Solutions of LPEI and SPS used for the fabrication of LPEI/SPS precursor layers (20 mM with respect to the molecular weight of the polymer repeat unit) were prepared using a 50 mM NaCl solution in 18 MΩ water. LPEI solutions contained 5mM HCl to aid polymer solubility. SPS solutions used for the deposition of polymer/SPS layers (20 mM with respect to the molecular weight of the polymer repeat unit) were prepared in water containing 0.067 mM HCl.
Fabrication of Multilayered Films
Films were deposited on planar silicon substrates pre-coated with a multilayered film composed of 10 bilayers of LPEI and SPS (terminated with a topmost layer of SPS) to ensure a suitably charged surface for the adsorption of polymer as previously described.23,24 These precursor layers were fabricated using an automated dipping robot (Riegler & Kirstein GmbH, Potsdam, Germany). Multilayered films fabricated from polymers 1−3 and SPS were fabricated on these precursor layers manually using an alternating dipping procedure according to the following general protocol: 1) Substrates were submerged in a solution of polycation for 5 minutes, 2) substrates were removed and immersed in an initial water bath for 1 minute followed by a second water bath for 1 minute, 3) substrates were submerged in a solution of polyanion for 5 minutes, and 4) substrates were rinsed in the manner described above. This cycle was repeated until the desired number of polycation/SPS bilayers (typically eight) had been deposited. For experiments aimed at characterizing film growth profiles by ellipsometry, films were dried after every two cycles of the above procedure using filtered compressed air. Films to be used in erosion experiments were either used immediately or were dried under a stream of filtered compressed air and stored in a vacuum dessicator until use. All films were fabricated at ambient room temperature.
Erosion of Multilayered Films and Evaluation of Release Kinetics
Experiments designed to investigate the erosion profiles of multilayered polycation/SPS films were performed in the following general manner: Film-coated substrates were placed in a plastic UV-transparent cuvette and 1.0 mL of phosphate buffered saline (PBS, pH = 7.4, 137 mM NaCl) was added to cover completely the film-coated portion of the substrate. The samples were incubated at 37 °C and removed at predetermined intervals for analysis by ellipsometry. For experiments designed to monitor a decrease in film thickness, optical thickness values were determined in at least 5 different predetermined locations on the substrate and the sample was returned immediately to the buffer solution or a cuvette containing fresh buffer.
Kinetics of Ester Hydrolysis
1H NMR experiments designed to determine kinetics for the hydrolysis of the esters in polymers 1−3 in aqueous solution were performed in the following manner. Polymer (10 mg) was dissolved in deuterated sodium acetate buffer (1.0 mL, 0.5 M, pH=5.1), 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (2 mg) was added as an internal standard, and this solution was transferred to a glass NMR tube. The NMR tube was heated to 50 °C in an oil bath and removed periodically for analysis by 1H NMR spectroscopy. The decrease of the resonance corresponding to the methylene protons adjacent to the ester functionality (at 4.16 ppm) was monitored and the extent of hydrolysis was determined by integrating this signal versus the trimethylsilyl protons of the internal standard.
Characterization of Surface Topography by Atomic Force Microscopy (AFM)
Film topography and surface roughness were obtained from height data imaged in tapping mode on a Nanoscope Multimode atomic force microscope (Digital Instruments, Santa Barbara, CA), using scan rates of 20 μm/s to obtain 256 × 256 pixel images. Silicon cantilevers with a spring constant of 40 N/m and a radius of curvature of less than 10 nm were used (model NSC15/NoAl, MikroMasch USA, Inc., Portland, OR). For each sample, at least two different 10 μm × 10 μm scans were obtained at randomly chosen points near the center of the film at each time point. Height data were flattened using a 3rd-order fit. Root-mean squared surface roughness (Rrms) was calculated over the scan area using the Nanoscope® IIIa software package (Digital Instruments, Santa Barbara, CA) and is reported as an average with standard deviation from scans in two or three different locations.
Results and Discussion
Initial Studies: Influence of Film Thickness and Polymer Molecular Weight on Erosion
We demonstrated previously that multilayered polyelectrolyte assemblies fabricated using polymer 1 erode gradually when incubated in physiologically relevant media, and that these thin films can be used to sustain the release of synthetic and therapeutically relevant anionic polyelectrolytes from surfaces.22-26 The specific erosion and release profiles reported in these past studies were highly dependent on the nature and structure of the incorporated anionic polyelectrolyte. For example, for films incorporating sodium poly(styrene sulfonate) (SPS), AFM and ellipsometry data are consistent with a well-defined erosion process that occurs in a gradual, uniform manner.24 However, for films fabricated using plasmid DNA, the mechanism of film erosion is more complex and involves the formation of nanoparticles on the surfaces of coated substrates.24 We selected SPS as a model polyanion on the basis of this previous work to facilitate the extraction of basic structure/property relationships in this current study.
Irrespective of the structure of the anionic polyelectrolytes incorporated into a film, one common characteristic of previously reported assemblies fabricated from polymer 1 is the relatively short time periods over which they erode.22-26 For example, the erosion of assemblies fabricated from eight alternating layers of polymer 1 and SPS (referred to hereafter as ‘bilayers’) generally occurs over a period of 40−50 hours when these assemblies are incubated in PBS buffer at 37 °C.22,24 On the basis of the well-defined physical erosion profiles discussed above for this polycation/polyanion pair, we hypothesized that thicker films (i.e., those composed of more than eight bilayers) would erode more slowly and release SPS into solution over longer time periods. To determine the relationship between film thickness and erosion profiles, we fabricated three different films consisting of 10-, 20-, and 30-bilayers of polymer 1/SPS on planar silicon substrates. All films were fabricated using an alternate dipping procedure optimized previously for this polyelectrolyte system24 and substrates were pre-coated with a thin (ca. 20 nm) multilayered film consisting of 10 bilayers of linear poly(ethylenimine)/SPS to create a surface suitable for the adsorption of polymer 1.23,24 Figure 1 shows a plot of the optical thickness, determined by ellipsometry, as a function of time for these three films incubated in PBS at 37 °C.
Figure 1.

Plot of ellipsometric thickness v. time for multilayered films fabricated from 10 (◆), 20 (■), and 30 (▲) bilayers of polymer 1 and SPS incubated at 37 °C in phosphate buffer (pH = 7.2). Silicon substrates were pre-coated with 10 bilayers (ca. 20 nm) of an LPEI/SPS film prior to experiment (see text).
As shown in Figure 1, the thicknesses of these 10-, 20-, and 30-bilayer films decreased from initial values of 100 nm, 220 nm, and 370 nm, respectively, to terminal thicknesses of ca. 30 nm after approximately 50 hours. Thus, although the addition of more bilayers substantially increases the thickness of a film, all films erode completely over the same period of time regardless of their initial thickness. The reasons for this behavior are not entirely clear, and additional experiments will be required to characterize and evaluate the complex erosion behavior of these thicker films more completely. We do conclude in the context of the current study, however, that increasing film thickness through the incorporation of additional polyelectrolyte layers is not a practical means through which to prolong film erosion or to extend the release of SPS for time periods longer than 50 hours.
We also considered that increasing the molecular weight of the polycations incorporated into a film might result in changes in erosion profiles. To examine the potential influence of polycation molecular weight on the erosion of polymer 1/SPS assemblies, we fabricated films composed of 10 bilayers of SPS and two samples of polymer 1 having different number average molecular weights (Mn = 10,000 or 20,000). We observed no significant differences in the erosion profiles of these films over this range of molecular weight (data not shown). Unfortunately, however, it is difficult to draw substantive conclusions from these experiments due to the substantial overlap in the molecular weight distributions of these two step growth polymers (PDI ∼ 2.5), as well as uncertainties associated with identifying the weight fractions of these distributions (i.e., high molecular weight or low molecular weight chains) that could potentially be incorporated differentially into a film during the fabrication process. As such, all subsequent studies aimed at investigating the influence of polymer structure on the erosion profiles of polymer 1/SPS assemblies were focused on varying systematically the structure of the repeat unit of polymer 1, as described below.
Polymer Synthesis and Characterization
Poly(β-amino ester)s can be synthesized readily by the conjugate addition of either primary amines or bis(secondary amine)s to diacrylate compounds.41,44 For example, polymer 1 is synthesized by the conjugate addition of the bis(secondary amine) 4,4’-trimethylenedipiperidine to 1,4-butanediol diacrylate (Eq 1, m = 1).41 In this study, we chose to hold constant the diamine segment of the repeat unit and vary the structure of polymer 1 by changing systematically the length of the linear hydrocarbon chain linking the ester units. Polymers 2 and 3 were synthesized by the conjugate addition of 4,4’-trimethylenedipiperidine to 1,6-hexanediol diacrylate (Eq 1, m = 2) and 1,8-octanediol diacrylate (Eq 1, m = 3), respectively, to yield a series of structurally related polymers differing only with respect to the 4-, 6-, and 8-carbon linkers between the esters in the repeat units of the polymers. Thus, these polymers vary systematically in both charge density (upon protonation) and hydrophobicity. The molecular weights (Mn) of the polymers used in the investigations below were determined to be 7400 (polymer 1), 7600 (polymer 2), and 16 700 (polymer 3) g/mol using gel permeation chromatography calibrated against monodisperse polystyrene standards.

Past studies have demonstrated the influence of solution pH and backbone hydrophobicity on the rates of hydrolysis for a range of different poly(β-amino ester)s in aqueous media.41,42 Zhong et al. used 1H NMR spectroscopy to demonstrate that, for a series of hyperbranched poly(β-amino ester)s, more hydrophobic polymers degrade by hydrolysis more slowly than hydrophilic polymers in aqueous solution.42 These investigators also demonstrated that degradation occurs more rapidly at pH 7.4 than at pH 5.0,42 consistent with a previous report describing the degradation of linear polymer 1.41 The insolubility of polymers 1−3 in aqueous solution at near-neutral pH values prevented the analysis of hydrolysis kinetics at pH 7.4 by NMR spectroscopy. To evaluate the kinetics of ester hydrolysis by NMR, we incubated samples of each polymer in deuterated sodium acetate buffer (0.5 M, pH 5.1) at 50 °C and monitored the disappearance of the ester resonance at 4.16 ppm by 1H NMR spectroscopy. As shown in Figure 2, the half-lives for the disappearance of ester functionality in polymers 1, 2, and 3 were approximately 145 hours, 200 hours, and 230 hours, respectively, under these conditions. These data represent relatively small changes in the rates of hydroysis but correlate with incremental increases in the hydrophobicities of these polymers.
Figure 2.

Kinetics of ester hydrolysis for polymers 1 (◆), 2 (■), and 3 (▲) in deuterated sodium acetate buffer (pH = 5.1) at 50 °C, determined by 1H NMR spectroscopy. Standard error associated with NMR peak integration is 5%.
The correlations between pH and hydrolysis kinetics discussed above for other poly(β-amino ester)s suggest that the hydrolysis of polymers 1−3 should occur more rapidly at pH 7.441,42 (i.e., the pH at which the erosion of multilayered materials fabricated from these materials is typically conducted). In an attempt to evaluate the kinetics of hydrolysis at pH 7.4, we incubated thin films of each polymer (ca. 80 nm thick, spin-coated onto gold-coated silicon wafers) in PBS buffer at 37 °C and monitored the disappearance of ester functionality using polarization modulation infrared reflection adsorption spectroscopy (PM-IRRAS). The results of these experiments correlated qualitatively with the relative rates of hydroysis indicated by 1H NMR spectroscopy, but rates were considerably faster and we observed larger differences in the relative rates than those shown in Figure 2. Unfortunately, we were unable to draw substantive quantitative conclusions based on these IR data (not shown) due to uncertainties associated with the diffusion and loss of polymers or degradation products out of these films during incubation and the potential influence of gradual changes in film thickness on degradation kinetics.
Fabrication, Characterization, and Erosion of Multilayered Films
Multilayered films were fabricated from SPS and polymers 1, 2, or 3 on silicon substrates using the alternate dipping procedure described above. As shown in Figure 3, the growth of each film was linear with respect to the number of polycation/SPS bilayers incorporated, and uniform optical thicknesses of approximately 60 nm were achieved after the deposition of eight bilayers. These nearly identical thickness and growth profiles suggest that the variations in the charge densities and hydrophobicities of polymers 1−3 do not influence substantially the nature of the layer-by-layer assembly process. Characterization of the microscale and nanoscale topographies of assemblies fabricated from polymers 2 and 3 by atomic force microscopy (AFM) revealed that the surfaces of these assemblies were smooth (RMS roughness = 1.0 to 5.8 nm) and generally devoid of any significant cracks, pits, or surface defects at these length scales (data not shown). These data are consistent with results we reported previously for the characterization of polymer 1/SPS films.24
Figure 3.
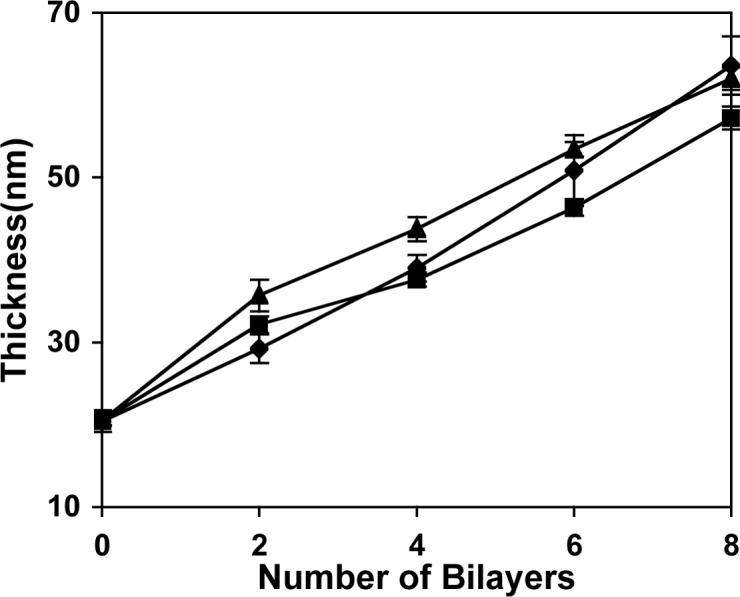
Plot of ellipsometric thickness versus the number of polycation/SPS bilayers incorporated for multilayered polyelectrolyte assemblies fabricated from polymers 1 (◆), 2 (■), and 3 (▲) on planar silicon substrates. Substrates were pre-coated with 10 bilayers (ca. 20 nm) of an LPEI/SPS film as an adhesive layer prior to experiment.
We incubated multilayered assemblies fabricated from eight bilayers of SPS and polymers 1, 2, or 3 in PBS buffer at 37 °C to investigate the influence of polycation structure on the erosion of these films in physiologically relevant media. We observed striking differences in the erosion profiles of these three different materials (Figure 4). Films fabricated from polymer 1 eroded completely in approximately 48 hours, consistent with our previous results23,24 and those shown above in Figure 1. However, films fabricated from polymer 2 eroded more slowly over a period of approximately 150 hours (ca. 6 days), and, in further contrast, films fabricated from polymer 3 required up to 370 hours (ca. 15 days) to erode completely. The erosion profiles shown in Figure 4 are not completely linear over the entire course of each experiment. However, it is apparent from inspection of these data that the substitution of polymers 2 and 3 for polymer 1 in these assemblies leads to approximately three-fold and seven-fold increases in the total times required for these films to erode completely. Closer inspection of the initial linear portions of each curve yields calculated decreases in the optical thicknesses of these films of 1.65, 0.69, and 0.31 nm/hr for films fabricated from polymers 1−3, respectively (Figure 4). The rates of erosion for each film change over time as film thicknesses approach the approximate thickness of the LPEI/SPS films applied as pre-coatings on the silicon substrates used in these experiments. These results are consistent with results reported previously for the erosion of polymer 1/SPS films and may result from the interpenetration of polymer 1/SPS layers and non-degradable LPEI/SPS layers that necessarily occurs at this interface.24
Figure 4.
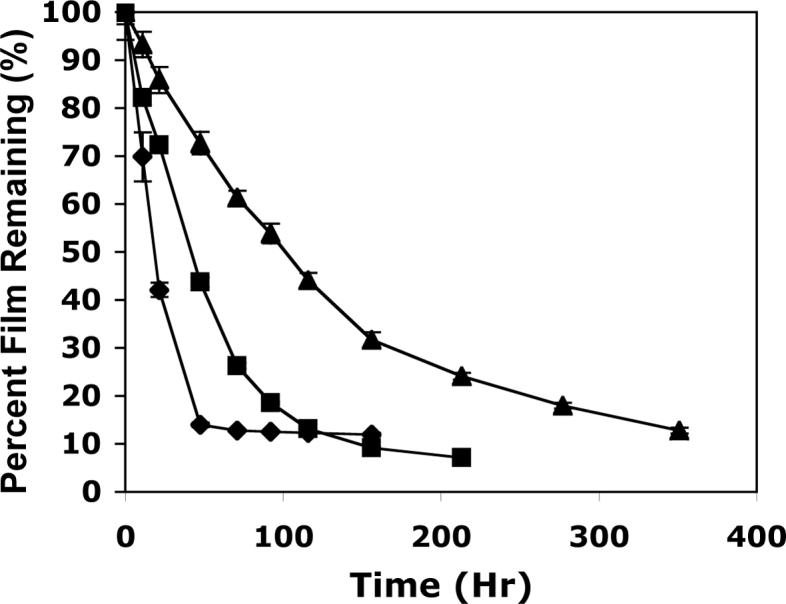
Plot of film erosion versus time for polycation/SPS bilayers fabricated from polymers 1 (◆), 2 (■), and 3 (▲) incubated in PBS buffer at 37 °C. Optical film thicknesses were determined using ellipsometry and are expressed as a percentage of the original thickness at each time point. The films used in this experiment were constructed from eight bilayers of polyamine and SPS and were ca. 60 nm thick. The planar silicon substrates used were pre-coated with 10 bilayers (ca. 20 nm) of an LPEI/SPS film as an adhesive layer prior to experiment.
Taken together, the data shown in Figures 3 and 4 demonstrate that changes in the charge densities and repeat unit structures of polymers 1−3 do not influence film growth or optical thickness significantly, but that these relatively minor changes in polymer structure do play an important role in governing the erosion profiles of these materials. The lengthening of erosion times observed for these materials is consistent with the systematic increase in the hydrophobicities of polymers 1, 2, and 3 as well as the relative rates at which these polymers degrade hydrolytically, as discussed above (Figure 2). We note here that the samples of polymers 1 and 2 used to fabricate these films have similar molecular weights (Mn ≈ 7500) but that the molecular weight of polymer 3 used in this study is approximately two times higher (Mn = 16 700). We have thus far been unable to synthesize polymer 3 having an identical molecular weight and cannot, therefore, completely discount the potential influence of molecular weight on the extended erosion times observed using polymer 3 on the basis of the current experiments. We note, however, that there is substantial overlap between the molecular weight distributions of these three materials (PDI ∼ 2.5−3.0) and the experiments described above using two different samples of polymer 1 with similarly overlapping molecular weight distributions suggest that molecular weight may not play a significant role in determining erosion rates under these conditions. We note further that the large difference in erosion profiles observed for polymers 1 and 2 (which do have equivalent number average molecular weights) provides strong evidence in support of the direct influence of polymer structure on the large changes observed in these erosion profiles.
The erosion data presented above are consistent with a mechanism of erosion for these films that involves at least partial hydrolysis of polymers 1−3. However, further inspection of the data in Figures 2 and 4 reveals that the relative differences in the rates of erosion for films fabricated from polymers 1, 2, and 3 (Figure 4) are larger than the differences in the relative rates of polymer hydrolysis for the polymers themselves in solution (Figure 2). These two data sets represent data for two different materials (polymers in solution versus multilayered polyelectrolyte films) conducted at two different solution conditions (PBS buffer at pH 7.4 versus sodium acetate buffer at pH 5.1), and it is therefore difficult to draw direct comparisons and conclusions. However, we speculate that the large differences in the erosion rates observed for the films in Figure 4 could arise from differences in the hydrophobicity of polymers 1−3 and, thus, the solubility and diffusivity of any oligomers that would be formed upon polymer hydrolysis. It is important to note that the erosion of these films is likely governed by a complex combination of physical and chemical factors and that several additional parameters will also need to be investigated before a complete description of the erosion behavior of these materials can be formulated.
The erosion of films fabricated from polymers 1−3 also results in the controlled and sustained release of SPS into aqueous solution. We characterized the kinetics of the release of SPS from these films directly by monitoring periodically the absorbance (at 226 nm, the absorbance maximum of SPS) of PBS solutions used to incubate and erode these films. Figure 5 shows a representative plot of absorbance v. time for the erosion of three films fabricated from eight bilayers of polymers 1, 2, or 3 and SPS. All three films used in this experiment were ca. 60 nm thick; differences in the final solution absorbance values measured for each film likely reflect small differences in the dimensions (and, thus, the surfaces areas) of the silicon substrates on which the original films were deposited. The differences in these release curves are consistent with the erosion profiles presented in Figure 4 and demonstrate directly that it is possible to tailor the rates at which these nanoscale polyelectrolyte assemblies release incorporated anionic polyelectrolytes into solution over time periods ranging from 48 hours to greater than 15 days simply by changing the structure of the degradable polyamine used to fabricate the films.
Figure 5.

Plot of absorbance at 226 nm vs. time showing the release of SPS from multilayered polyelectrolyte films fabricated from SPS and polymers 1 (◆), 2 (■), and 3 (▲). Films were deposited on planar silicon substrates pre-coated with 10 bilayers of an LPEI/SPS film and incubated in PBS buffer at 37 °C. Markers represent absorbance values recorded for the incubation buffer and correspond to the concentration of SPS released into solution; error bars are in most cases smaller than the symbols used. The films used in this experiment were constructed from eight bilayers of polyamine and SPS and were ca. 60 nm thick.
Summary and Conclusions
We have investigated the influence of polymer structure on the fabrication and erosion profiles of multilayered polyelectrolyte assemblies fabricated from sodium poly(styrene sulfonate) (SPS) and three different hydrolytically degradable polyamines. We synthesized a series of structurally related poly(β-amino ester)s having systematic variations in charge density and hydrophobicity by virtue of the linear 4-, 6-, and 8-carbon linkers situated between the esters in the repeat units of these materials. These relatively minor changes in polymer structure were found to play an important role in determining the rates at which multilayered assemblies fabricated from these materials erode when incubated in physiologically relevant media. While films 60 nm thick fabricated from polymer 1 eroded completely in approximately 48 hours in PBS buffer at 37 °C, analogous films fabricated from polymers 2 and 3 eroded over significantly longer time periods [approximately 150 hours (ca. 6 days) and 370 hours (ca. 15 days), respectively]. These substantial increases in erosion times are consistent with the systematic increase in the hydrphobicity of polymers 1−3 as well as the relative rates at which these polymers degrade hydrolytically in aqueous solution.
This work demonstrates that it is possible to tailor the rates at which polyelectrolyte assemblies release incorporated anionic polyelectrolytes over a large range of time periods simply by changing the structure of the degradable polyamine used to fabricate the film. In this study, SPS serves as a model anionic polyelectrolyte. In the broader context of controlled release, polymer 1 has been used previously to fabricate multilayered assemblies that sustain the release of transcriptionally active DNA and therapeutically relevant anionic polysaccharides under physiologically relevant conditions. The structure/property relationships resulting from this investigation demonstrate the versatility of this class of degradable materials and suggest that they may prove useful in therapeutic contexts that require control over the rate and duration of release of materials from coated surfaces.
Acknowledgment
Financial support was provided by the National Institutes of Health (R21 EB02746), the Arnold and Mabel Beckman Foundation, the National Science Foundation (through the UW Materials Research Science and Engineering Center), and the University of Wisconsin. We are grateful to the NSF (CHE-9208463) and the NIH (NIH 1 S10 RR0 8389-01) for support of the UW NMR spectroscopy facilities.
References
- 1.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 2.Bertrand P, Jonas A, Laschewsky A, Legras R. Macromol Rapid Comm. 2000;21:319–348. [Google Scholar]
- 3.Peyratout CS, Dahne L. Angew Chem Int Ed Engl. 2004;43:3762–83. doi: 10.1002/anie.200300568. [DOI] [PubMed] [Google Scholar]
- 4.Hammond PT. Adv. Mater. 2004;16:1271–1293. [Google Scholar]
- 5.Jin W, Shi XY, Caruso F. J Am Chem Soc. 2001;123:8121–8122. doi: 10.1021/ja015807l. [DOI] [PubMed] [Google Scholar]
- 6.Tiourina OP, Antipov AA, Sukhorukov GB, Larionova NL, Lvov Y, Mohwald H. Macromol Biosci. 2001;1:209–214. [Google Scholar]
- 7.Lvov Y, Antipov AA, Mamedov A, Mohwald H, Sukhorukov GB. Nano Letters. 2001;1:125–128. [Google Scholar]
- 8.Thierry B, Kujawa P, Tkaczyk C, Winnik FM, Bilodeau L, Tabrizian M. J. Am. Chem. Soc. 2005;127:1626–1627. doi: 10.1021/ja045077s. [DOI] [PubMed] [Google Scholar]
- 9.Thierry B, Winnik FM, Merhi Y, Silver J, Tabrizian M. Biomacromolecules. 2003;4:1564–1571. doi: 10.1021/bm0341834. [DOI] [PubMed] [Google Scholar]
- 10.Thierry B, Winnik FM, Merhi Y, Tabrizian M. J Am Chem Soc. 2003;125:7494–5. doi: 10.1021/ja034321x. [DOI] [PubMed] [Google Scholar]
- 11.Groth T, Lendlein A. Angew Chem Int Ed Engl. 2004;43:926–928. doi: 10.1002/anie.200301708. [DOI] [PubMed] [Google Scholar]
- 12.Dai ZF, Heilig A, Zastrow H, Donath E, Mohwald H. Chem-Eur J. 2004;10:6369–6374. doi: 10.1002/chem.200400579. [DOI] [PubMed] [Google Scholar]
- 13.Tryoen-Toth P, Vautier D, Haikel Y, Voegel JC, Schaaf P, Chluba J, Ogier J. J Biomed Mater Res. 2002;60:657–667. doi: 10.1002/jbm.10110. [DOI] [PubMed] [Google Scholar]
- 14.Berg MC, Yang SY, Mendelsohn JD, Hammond PT, Rubner MF. Abstr Pap Am Chem S. 2003;225:U663–U664. [Google Scholar]
- 15.Richert L, Lavalle P, Vautier D, Senger B, Stoltz JF, Schaaf P, Voegel JC, Picart C. Biomacromolecules. 2002;3:1170–1178. doi: 10.1021/bm0255490. [DOI] [PubMed] [Google Scholar]
- 16.Mendelsohn JD, Yang SY, Hiller J, Hochbaum AI, Rubner MF. Biomacromolecules. 2003;4:96–106. doi: 10.1021/bm0256101. [DOI] [PubMed] [Google Scholar]
- 17.Yang SY, Mendelsohn JD, Rubner MF. Abstr Pap Am Chem S. 2002;224:U429–U429. [Google Scholar]
- 18.Yang SY, Mendelsohn JD, Rubner MF. Biomacromolecules. 2003;4:987–994. doi: 10.1021/bm034035d. [DOI] [PubMed] [Google Scholar]
- 19.Richert L, Boulmedais F, Lavalle P, Mutterer J, Ferreux E, Decher G, Schaaf P, Voegel JC, Picart C. Biomacromolecules. 2004;5:284–294. doi: 10.1021/bm0342281. [DOI] [PubMed] [Google Scholar]
- 20.Salloum DS, Olenych SG, Keller TCS, Schlenoff JB. Biomacromolecules. 2005;6:161–167. doi: 10.1021/bm0497015. [DOI] [PubMed] [Google Scholar]
- 21.Chung AJ, Rubner MF. Langmuir. 2002;18:1176–1183. [Google Scholar]
- 22.Vazquez E, Dewitt DM, Hammond PT, Lynn DM. J Am Chem Soc. 2002;124:13992–3. doi: 10.1021/ja026405w. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 24.Fredin NJ, Zhang J, Lynn DM. Langmuir. 2005;21:5803–5811. doi: 10.1021/la050596+. [DOI] [PubMed] [Google Scholar]
- 25.Jewell CM, Zhang J, Fredin NJ, Lynn DM. J. Control. Release. 2005;106:214–223. doi: 10.1016/j.jconrel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Wood KC, Boedicker JQ, Lynn DM, Hammond PT. Langmuir. 2005;21:1603–9. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 27.Sukhishvili SA, Granick S. J Am Chem Soc. 2000;122:9550–9551. [Google Scholar]
- 28.Dubas ST, Farhat TR, Schlenoff JB. J Am Chem Soc. 2001;123:5368–5369. doi: 10.1021/ja015774+. [DOI] [PubMed] [Google Scholar]
- 29.Dubas ST, Schlenoff JB. Macromolecules. 2001;34:3736–3740. [Google Scholar]
- 30.Schuler C, Caruso F. Biomacromolecules. 2001;2:921–926. doi: 10.1021/bm010052w. [DOI] [PubMed] [Google Scholar]
- 31.Sukhishvili SA, Granick S. Macromolecules. 2002;35:301–310. [Google Scholar]
- 32.Caruso F, Rodda E, Furlong DF, Niikura K, Okahata Y. Anal Chem. 1997;69:2043–2049. doi: 10.1021/ac961220r. [DOI] [PubMed] [Google Scholar]
- 33.Shutava T, Prouty M, Kommireddy D, Lvov Y. Macromolecules. 2005;38:2850–2858. [Google Scholar]
- 34.Serizawa T, Yamaguchi M, Akashi M. Macromolecules. 2002;35:8656–8658. [Google Scholar]
- 35.Serizawa T, Yamaguchi M, Akashi M. Angew Chem Int Edit. 2003;42:1115–1118. doi: 10.1002/anie.200390293. [DOI] [PubMed] [Google Scholar]
- 36.Etienne O, Schneider A, Taddei C, Richert L, Schaaf P, Voegel JC, Egles C, Picart C. Biomacromolecules. 2005;6:726–33. doi: 10.1021/bm049425u. [DOI] [PubMed] [Google Scholar]
- 37.Sato K, Imoto Y, Sugama J, Seki S, Inoue H, Odagiri T, Hoshi T, Anzai J. Langmuir. 2005;21:797–9. doi: 10.1021/la048059x. [DOI] [PubMed] [Google Scholar]
- 38.Inoue H, Anzai J. Langmuir. 2005 doi: 10.1021/la0508341., published on the world wide web: doi: 10.1021.
- 39.Inoue H, Sato K, Anzai J. Biomacromolecules. 2005;6:27–9. doi: 10.1021/bm0495856. [DOI] [PubMed] [Google Scholar]
- 40.Lynn DM, Anderson DG, Akinc AB, Langer R. Degradable Poly(beta-amino ester)s for Gene Delivery. In: Amiji M, editor. Polymeric Gene Delivery: Principles and Applications. CRC Press; New York: 2004. pp. 227–241. [Google Scholar]
- 41.Lynn DM, Langer R. J Am Chem Soc. 2000;122:10761–10768. [Google Scholar]
- 42.Zhong Z, Song Y, Engbersen JFJ, Lok MC, Hennink WE, Feijen J. J. Control. Release. 2005 doi: 10.1016/j.jconrel.2005.06.022., published on the world wide web: doi:10.1016/j.jconrel.2005.06.022.
- 43.Akinc A, Anderson DG, Lynn DM, Langer R. Bioconjugate Chem. 2003;14:979–988. doi: 10.1021/bc034067y. [DOI] [PubMed] [Google Scholar]
- 44.Lynn DM, Anderson DG, Putnam D, Langer R. J Am Chem Soc. 2001;123:8155–6. doi: 10.1021/ja016288p. [DOI] [PubMed] [Google Scholar]
- 45.Akinc A, Lynn DM, Anderson DG, Langer R. J Am Chem Soc. 2003;125:5316–5323. doi: 10.1021/ja034429c. [DOI] [PubMed] [Google Scholar]
- 46.Anderson DG, Lynn DM, Langer R. Angew Chem Int Ed Engl. 2003;42:3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 47.Berry D, Lynn DM, Sasisekharan R, Langer R. Chem Biol. 2004;11:487–498. doi: 10.1016/j.chembiol.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 48.Skinner WA, Hayford J, Shellenberger TE, Colwell WT. J. Med. Chem. 1966;9:605–607. [Google Scholar]