

Abstract
Two amino acid derived (leucinol and N-methyl pyrrolidinol) chiral ionic liquids are synthesized and characterized both in monomeric and polymeric forms. Leucinol based chiral cationic surfactant is room a temperature ionic liquid (RTIL), and pyrrolidinol based chiral cationic surfactant melts at 30-35 °C to form ionic liquid (IL). The monomeric and polymeric ILs are thoroughly characterized to determine critical micelle concentration, aggregation number, polarity, optical rotation and partial specific volume. Here in, we present the first enantioseparation using chiral IL as pseudostationary phase in capillary electrophoresis. Chiral separation of two acidic analytes, (±)-alpha-bromo-phenylacetic acid (±)-(α-BP-AA) and (±)-2-(2-chlorophenoxy)propanoic acid(±)-(2-PPA) can be achieved with both monomers and polymers of undecenoxy carbonyl-L-pryrrolidinol bromide (L-UCPB) and undecenoxy carbonyl-L-leucinol bromide (L-UCLB) at 25 mM surfactant concentration using phosphate buffer at pH 7.50. The chiral recognition seems to be facilitated by the extent of interaction of the acidic analytes with the cationic head group of chiral selectors. Polysodium N-undecenoxy carbonyl-L-leucine sulfate (poly-L-SUCLS) and polysodium N-undecenoxy carbonyl-L-leucinate (poly-L-SUCL) were compared at high and low pH for the enantioseparation of (±)-(2-PPA). At pH 7.5, poly-L-SUCLS, poly-L-SUCL and ±-2-PPA are negatively charged resulting in no enantioseparation. However, chiral separation was observed for (±)-(2-PPA) using poly-L-SUCLS at low pH (pH 2.00) at which analyte is neutral. The comparison of chiral separation of anionic and cationic surfactants demonstrates that the electrostatic interaction between the acidic analyte and cationic micelle plays a profound role in enantioseparation.
The separation of chiral compounds is currently the center of great interest.1 This interest can be attributed largely to a heightened awareness that enantiomers of a racemic drug usually display markedly different pharmacological activities.2,3 The human body metabolizes individual enantiomers by separate pathways to produce different pharmacological effects. Presently, a majority of commercially available drugs are synthetic and chiral. Most of these chiral drugs are obtained as a mixture of two enantiomers during synthesis.4 In order to avoid the possible undesirable effects of enantiomeric impurity in chiral drug, it is inevitable that only therapeutically active form be marketed. Hence there is a continuous need to develop technologies that have the ability to separate enantiomers.
Very recently, ionic liquids (ILs) have found great applications in efficient and environmentally benign chemical processing and chemical analysis.5,6 By definition the ionic liquids (ILs) are organic salts with melting points (MP) below 100 °C or more often even lower than room temperature.7-11 These compounds posses dual capability of dissolving both polar and nonpolar species and the most useful feature is that they do not evaporate even at high temperatures.12-15 Most commonly, ILs are based on nitrogen-rich alkyl substituted heterocyclic cations, with a variety of anions (e.g., 1-ethyl-3-methylimidazolium tetrafluoroborate). Although, the reasons for low melting point of ILs are not clear, it is stated that, ILs consist of bulky inorganic anions with delocalized charged organic cations, which prevents the formation of a stable crystal lattice or random molecular packing resulting in lower melting points.16 Due to these remarkable characteristics, ionic liquids have been used as, medium for liquid-liquid extractions,17-19 mobile phase additives in high performance liquid chromatography (HPLC),20,21 electrolytes in capillary electrophoresis (CE),22-26 matrixes for matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS),27,28 stationary phases for gas chromatography (GC)29-32 and as modifiers in micellar electrokinetic chromatography (MEKC).33,34 However, there is no report in the literature about the use of ILs as chiral selector in CE.
Cationic surfactants are referred to as compounds containing at least one long hydrophobic chain attached to a positively charged nitrogen. These quaternary ammonium group containing surfactants are well known for displaying emulsifying properties, antimicrobial activity, components in cosmetic formulations, anti corrosive effects, phase transfer catalyst and as chiral induction medium (if chiral cationic surfactant) in organic reactions.35-41 As with the case of chiral anionic surfactants, amino acid based (both monomeric and polymeric) and ephedrine based (monomeric) chiral cationic surfactants have been used as chiral selectors in MEKC.42,43 However, unlike chiral anionic polymeric surfactants, chiral cationic polymeric surfactants have not found great application so far, and only one report of chiral cationic polymeric surfactants as pseudostationary phase (PSP) in MEKC is reported.42
In this study, we report the synthesis, characterization and application of novel IL type surfactants and their polymers for chiral separation of acidic analytes in MEKC. Acidic analytes due to inherent negative charge poorly interact with most commonly employed chiral anionic surfactants at basic pH. As a result, still a large number of acidic analytes could not be resolved by MEKC. The cationic surfactant, undecenoxy carbonyl-L-leucinol bromide (L-UCLB) is an ionic liquid at room temperature, while undecenoxy carbonyl-L-pyrrolidinol bromide (L-UCPB) is a greasy solid that melts to form ionic liquid at 30-35 °C. In our case, quarternized nitrogen (chiral head group) is surrounded by hydrophobic tail and leucinol or pyrrolidinol side chain, which presumably prevent the proper packing of the cations and anions in regular three-dimensional patterns to form ionic liquids.
Current report is the first demonstration of MEKC chiral separation of several anionic compounds such as phenoxypropionic acid herbicide (±)-(2-PPA) and a very useful synthetic intermediate ±-alpha-bromophenylacetic acid (±)-(α-BP-AA)44,45 using two synthetic chiral ionic liquids L-UCLB and L-UCPB as well their polymers. Chiral separation of acidic analyte is compared using polymeric anionic surfactants containing similar head group under both acidic and basic pH conditions.
MATERIALS AND METHODS
Standards and Chemicals
The analytes (±)-alpha-bromophenylacetic acid (±)-(α-BP-AA) and (±)-2-(2-chlorophenoxy)propanoic acid (±)-(2-PPA) were obtained as racemic mixture from Sigma Chemical Co (St. Louis, MO) and Aldrich (Milwaukee, WI), respectively. Chemicals used for the synthesis of surfactants included ω-undecylenyl alcohol, triphosgene, pyridine, dichloromethane, 2-bromoethylamine hydrobromide, L-leucinol, N-methylpryrrolidinol, 96% formic acid, 37% formaldehyde, and isopropanol (HPLC grade) were also obtained from Aldrich (Milwaukee, WI) and were used as received.
Synthesis and Characterization of Monomeric Surfactants and Micelle Polymers
Choloroformate has been synthesized as reported earlier46 by reacting triphosgene with unsaturated alcohol (step 1, Fig 1). The carbamate functionalized alkenyl bromide (step 2, Fig 1) was synthesized by dropwise addition of (10 mmoles) choloroformate over equimolar aqueous solution of 2-bromoethylamine hydrobromide and Na2CO3 and were stirred for 2 hrs. The resulting solution was extracted twice with dichloromethane, which then was washed three times with H2O, dried over Na2SO4 and concentrated by evaporating solvent to yield product 1(89-93%). The N,N-dimethyl leucinol (product 2, step B, Fig 1) was synthesized by reductive alkylation of primary amine of leucinol using the well-known Eschweiler-Clark reaction (Yield 55-70%).47-49 The chiral ionic liquids were synthesized by refluxing the carbamate functionalized alkenyl bromide (product 1) with N,N-dimethyl leucinol or N-methylpryrrolidinol for 48 hrs in isopropanol (IPA). After 48 hrs, the reaction mixture was concentrated by evaporating IPA, and the resulting fluid was dissolved in water and extracted with ethyl acetate. The aqueous solution of ionic liquids (products 3 and 4, Figure 2) were lyophilized (Yield 40-55%) at −50 °C collector temperature and 0.05 mBar pressure for 14 days (to ensure complete removal of water from both products). 1H-NMR spectra of L-UCPB, L-UCLB and their polymers were recorded on a Varian Unity+ 300 MHz spectrometer using D2O as the solvent. The surfactants were characterized and checked for purity by MALDI-TOF-MS (Fig 3A-B), 1H-NMR and elemental analysis. L-UCPB, 1H-NMR: δ 0.759-0.893 (b, 6H), 1.170 (m, 12H), 1.471 (m, 2H), 1.767 (m, 2H), 1.883 (b, 1H), 2.085 (m, 2H), 3.06 (b, 2H), 3.239-3.613 (b, 8H), 3.777-3.844 (m, 1H), 4.052 (d, J= 14.7, 2H), 4.379 (b, 2H), 4.789 (m, 2H), 5.626 (m, 1H). Anal. Calcd. for C20H39N2O3Br + 2H2O: C, 50.95; H, 9.19; N, 5.94; Found: C, 51.56; H, 10.07; N, 5.88. L-UCLB, 1H-NMR: δ 1.170 (b, 12H), 1.468 (b, 2H), 1.766-1.992 (b, 4H), 1.992-2.164 (b, 2H), 3.032 (b, 2H), 3.147 (b, 2H), 3.472 (b, 3H), 3.506-3.619 (b, 3H), 3.830 (b, 2H), 4.376 (b, 2H), 4.804 (m, 2H), 5.658 (m, 1H). Anal. Calcd. for C22H45N2O3Br + 2H2O: C, 52.68; H, 9.85; N, 5.59; Found: C, 52.14; H, 9.00; N, 6.87.
Figure 1.

Synthesis of the carbamate functionalized (A) alkenyl bromide and (B) N, N-dimethyl-L-leucinol.
Figure 2.

Synthesis and polymerization of leucinol and pryrrolidinol derived ionic liquid and their polymers.
Figure 3.
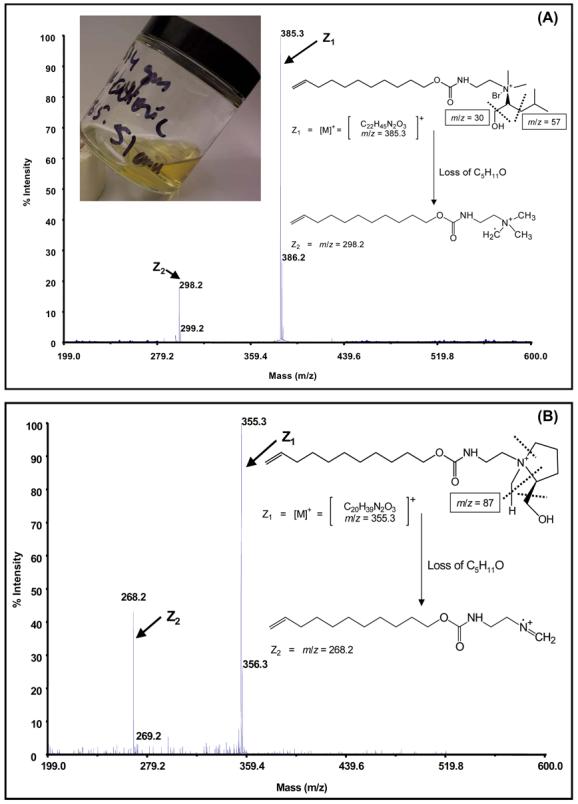
MALDI-TOF mass spectra (positive mode) with proposed cleavages and the corresponding fragment masses for IL type surfactants (A) undecenoxy carbonyl-L-leucinol bromide (L-UCLB) and (B) undecenoxy carbonyl-L-pryrrolidinol bromide (L-UCPB) after freeze drying on a lyophilizer at −50 °C collector temperature and 0.05 mbar pressure for 14 days. The mass spectra were obtained using α-cyano-4-hydroxycinnamic acid as MALDI-matrix.
The critical micelle concentration (CMC) was determined using a sigma 703 Digital Tensiometer (KVS Instruments USA, Monroe, Connecticut), by the Du NoÜy ring method at room temperature. Polymerization of the synthesized ionic liquids was achieved by 60Co γ-irradiation (8 Mrad/h) of 100 mM aqueous solution for 30 hrs. To ensure complete polymerization, 30 hrs of continuous γ-irradiation was applied. The 1H-NMR indicated the disappearance of double bond protons signal in the region of 4.8-5.0 and 5.7-5.9 ppm. After irradiation, the polymeric surfactant solutions were filtered and dialyzed against triply deionized water using regenerated cellulose (RC) dialysis membrane (Spectrum Laboratories, Inc, Rancho Dominguez, CA, USA) with a 1000 Da molecular mass cutoff for 24 hrs. Finally, the dialyzed solutions were lyophilized to obtain the dried polymeric surfactants. Further characterization, such as aggregation number and polarity of the amphiphilic ionic liquids (monomers and polymers) were determined by using pyrene emission vibronic fine structure method.46 The partial specific volume of both monomer and polymer was determined using previously reported procedure.46 The optical rotation of monomeric and the polymeric surfactants was obtained by an AUTOPOL III automatic polarimeter (Rudolph Research Analytical, Flanders, New Jersey) by measuring the optical rotation at 589 nm of a 10 mg/mL solution of each monomer and polymer in triply deionized water at 25 °C.
MEKC Instrumentation
All experiments were performed using an Agilent CE system (Agilent Technologies, Palo Alto, California) equipped with 0-30 kV high-voltage power supply, a diode array detector for UV detection and Chemstation software (V 9.0) for system control and data acquisition. The fused-silica capillary was obtained from Polymicro Technologies (Phoenix, AZ). The total length of the capillary used with an Agilent CE system was 64.5 cm (56.0 cm from inlet to detector, 50 μm ID, 350 μm OD), prepared by burning about 3 mm polyimide coating to create a detection window.
Capillary Electrophoresis Procedures and Calculations
The capillaries for all MEKC experiments were prepared by flushing with 1M NH4OH for 60 min at 50 °C followed by 30 min rinse with triply deionized water at 20 °C. Between each injection, the capillary was flushed with 0.1 M NH4OH and H2O for 3 min each. Separations began after a 2 min rinse with the running buffer, followed by a 5 min flush with the running buffer containing ionic liquids. All separations were performed at − 20 kV and at 20 °C. All surfactants (both monomers and polymers) were run with a new capillary (cut to the same length from the same capillary bundle) and was preconditioned using the identical flushing procedure as mentioned above. Chiral resolution (Rs) of acidic analytes (±)-(2-PPA) and (±)-α–BP-AA were calculated by Chemstation software using the peak width at half height method:
W50 (1) and W50 (2) are the widths at 50% height for peak 1 and 2, respectively. The selectivity (α) is defined as t2/t1, where t1 and t2 are the migration times of the first and second eluting enantiomers. Methanol was used as the t0 marker and was measured from the time of injection to the first deviation from the baseline. Dodecanophenone was used as tracer for tmc at 100 mM surfactants concentration of each monomer and polymer. The effective electrophoretic mobility of the monomers and polymers of ionic liquids was calculated by following equation:
Where, μep, μeof and μapp are effective electrophoretic mobility, electroosmotic mobility and apparent electrophoretic mobility respectively. The negative sign of μapp and μeof is due to the fact that monomeric and polymeric ionic liquids coat the capillary wall and result in anodic electroosmotic flow, therefore negative voltage (−20 kV) has to be applied for separation.
Preparation of MEKC Buffers and Analyte Solutions
For all MEKC experiments, the final background electrolyte (BGE) consisted of a 25 mM each of Na2HPO4/NaH2PO4 buffered at pH 7.5. The desired pH value was obtained by using 1 M NaOH. The pH of BGE was adjusted before the addition of ILs (monomers and polymers). This BGE solution is finally filtered through a 0.45 μm Nalgene syringe filter (Rochester, NY). The running MEKC buffer solution was prepared by addition of 25 mM IL type surfactants to the BGE, followed by ultrasonication for about 25-30 min. The analytes prepared in 50/50 (v/v) of MeOH:H2O at various concentrations were injected at a pressure of 50 mbar for 1-5s. The dodecanophenone was prepared in 100% MeOH at 3 mg/mL (stock solution), diluted to 1.8 mg/mL in 60:40 MeOH:H2O and injected at a pressure of 50 mbar for 10s
RESULTS AND DISCUSSION
Physicochemical Properties
Table 1 represents the physicochemical properties of the synthesized enantiomerically pure chiral surfactants L-UCLB [room temperature ionic liquid (RTIL)] and L-UCPB [ionic liquid (IL), MP 30-35 °C] as well as their polymers, poly-L-UCLB and poly-L-UCPB. The L-UCLB exhibited higher polarity, lower CMC and partial specific volume (V̄), significantly higher optical rotation but similar aggregation number (A) compared to L-UCPB. Similar trend was also observed for the poly-L-UCLB and poly-L-UCPB, except that the A value was higher for the former polymer. Comparing physicochemical properties of monomeric and polymeric cationic surfactants, it can be noticed that A is always lower for the polymers than monomers, while polarity and V̄ is always higher for polymeric surfactants.
Table 1.
Physicochemical properties of the monomers and polymers of chiral amino acid derived cationic surfactants undecenoxy carbonyl-L-leucinol bromide (L-UCLB) and undecenoxy carbonyl-L-pryrrolidinol bromide (L-UCPB).
|
Characteristic of the ionic liquid type monomeric surfactants |
L-UCPB | L-UCLB |
|---|---|---|
| Critical micelle concentration (CMC) a) [mM] |
1.15 ± (0.01)* | 0.84 ± (0.05)* |
| Aggregation numberb) | 95 ± (0.09)* | 97 ± 0.04 |
| Polarity (I1/I3) ratio c) | 1.095 ± (0.001)* | 1.180 ± 0.040 |
| Optical rotationd) | −2.35 ± (0.02)* | +21.67 ± 0.03 |
| Partial specific volumee) | 0.8281 ± (0.0036)* | 0.7185 ± 0.00 |
| Electroosmotic mobility μeof (cm2V−1S−1)f) |
−2.83 × 10−4 (± 1.56 × 10−5)* |
−2.42 × 10−4 (± 5.31 × 10−6)* |
| Effective electrophoretic mobility μep (cm2V−1S−1)f) |
2.08 × 10−4 (± 1.54 × 10−5)* |
1.94 × 10−4 (± 7.49 × 10−7)* |
| Migration-time window (tmc/to)f) |
3.79 (± 0.20)* |
5.09 (± 0.38)* |
|
Characteristic of the polymeric surfactants |
poly-L-UCPB | poly-L-UCLB |
| Aggregation numberb) | 34 ± (0.780)* | 25 ± (0.034)* |
| Polarity (I1/I3) ratioc) | 1.219 ± (0.001)* | 1.22 ± (0.007)* |
| Optical rotationd) | −7.84 ± (0.04)* | +17.45 ± (0.64)* |
| Partial specific volumee) | 0.8408 ± (0.0075)* | 0.7634 ± (0.0008)* |
| Electroosmotic mobility μeof (cm2V−1S−1)f) |
−2.54 × 10−4 (± 3.67 × 10−6)* |
−2.34 × 10−4 (± 3.12 × 10−6)* |
| Effective electrophoretic mobility μep (cm2V−1S−1)f) |
2.02 × 10−4 (± 2.96 × 10−6)* |
1.91 × 10−4 (± 3.52 × 10−6)* |
| Migration-time window (tmc/to)f) |
4.87 (± 0.16)* |
5.38 (± 0.53)* |
Critical micelle concentration is determined by the surface tension measurements.
Aggregation number is determined by the florescence quenching experiment using pyrene as a probe and cetyl pyridinium chloride as a quencher.
Polarities of the surfactants are determined using ratio of the fluorescence intensity (I1/I3) of pyrene.
Optical rotation of 10 mg/mL of monomer and micelle polymers were determined in triply deionized water were obtained at 589nm [sodium D line].
Partial specific volumes were determined by the density measurements at different surfactant concentrations.
The μep values for all monomer and polymeric ionic liquids were determined using methanol as to marker and dodecanophenone as tmc tracer. Experimental conditions: 64.5 cm (56 cm effective length) × 50 μm ID capillary with an applied voltage of −20 kV at 25 °C using a running buffer of 25 mM each of NaH2PO4 and Na2HPO4, 100 mM monomer and polymeric ionic liquids; dodecanophenone introduction, 50 mbar for 10 s (1.5 mg /mL in 50:50 MeOH/H2O).
Standard deviations are given in parentheses.
Fig 3. shows the MALDI-TOF MS of both L-UCLB (A) and L-UCPB (B) in positive mode. Both L-UCLB and L-UCPB surfactants showed the molecular ion peak (base peak) at mass-to-charge ratios (m/z) of 385.3 and 355.3, respectively along with a fragment generated by the loss C5H11O. For L-UCLB the masses at 386.2 and 299.2 m/z, for L-UCPB the masses at 356.3 and 268.2 m/z are generated due to the 13C isotope related to the molecular ion and the generated fragment ion, respectively. The generation of the cationic fragments (Z2) for both ionic liquids as shown in Fig 3 is in accord with the previous observations that most of the fragments generated from cationic surfactants bear preformed cations (contain quaternary nitrogen of the cationic surfactants).50,51
The electrophoretic parameters of monomeric and polymeric ionic liquids were also examined (Table 1) at 100 mM surfactant concentrations (at lower surfactant concentration tmc marker was not observed even after 3 hrs). The reversed electroosmotic flow (−μeof) and effective electrophoretic (μep) mobilities of both poly-L-UCPB and poly-L-UCLB were slightly lower, while migration time window (tmc/t0) was greater compared to their respective monomers. In addition, the monomer and polymer of L-UCLB compared to the monomer and polymer of L-UCPB have lower μep and provided larger tmc/t0.
Enantioseparation of Acidic Analytes
The optimization of chiral resolution of (±)-(α-BP-AA) and (±)-(2-PPA) was performed by studying pH of the background electrolyte (BGE), type and concentration of BGE, organic modifiers and surfactant concentration. After optimizing the chiral MEKC conditions, chiral separation of (±)-(α-BP-AA) and (±)-(2-PPA) were compared using L-UCPB, L-UCLB and their respective polymers (poly-L-UCPB and poly-L-UCLB) to get insight on the factors affecting analyte-micelle interactions and ultimately chiral separation.
Enantioseparation of ±-alpha-bromophenylacetic acid
Figure 4(A) and 4(C) show the chiral separation of (±)-(α-BP-AA) at optimum separation conditions with L-UCPB and L-UCLB, respectively. Since (±)-(α-BP-AA) has dissociable carboxylic acid group with pKa = 2.40 (±0.10) therefore, the effect of pH on enantioseparation was evaluated from pH 2.00-8.50 (data not shown). Although chiral resolution (Rs) at lower pH range (4.00-6.00) do not differ drastically, maximum Rs was obtained at pH 7.5 but no Rs at pH 2.00 and at pH > 7.5, Rs deteriorates (data not shown). One plausible explanation of Rs deterioration at pH > 7.5 could be the excess hydroxide ions (originated from the use of NaOH to adjust the BGE pH), which competes with the anionic chiral analyte for the positively charged ionic liquid at basic pH. The absence of any Rs at pH 2.00 and lower Rs at intermediate pH suggests that electrostatic interaction indeed contributes significantly to chiral recognition. It has been reported in the literature that in the presence of certain organized media (e.g., micelles), the pKa of the organic acid is altered up to more than 4 pH units.43,52 Therefore, it is reasonable to believe that the amphiphilic ionic liquids might have increased the pKa of (±)-(α-BP-AA) such that maximum ionization occurs around pH 7.50. Hence, greater electrostatic interaction with the positively charged ionic liquids provided maximum chiral Rsat pH 7.50. On the other hand, L-UCPB and L-UCLB concentrations, as well as the use of organic modifiers (e.g., methanol, acetonitrile) did not show any significant variations in Rs.
Figure 4.
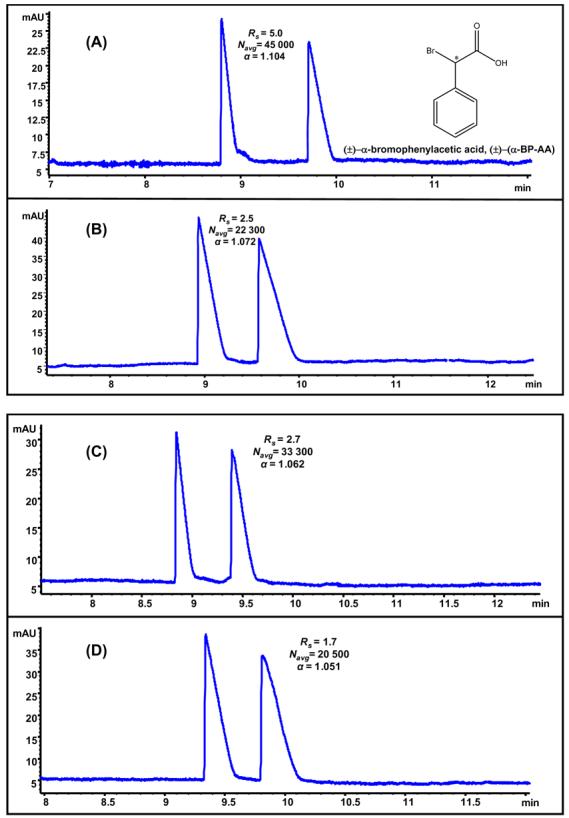
Comparison of 25 mM L-UCPB (A), poly-L-UCPB (B), 25 mM L-UCLB (C) and poly-L-UCLB (D) for enantioseparation of (±)-alpha-bromophenylacetic acid [(±)-(α-BP-AA), 2.5 mg/mL in MeOH/H2O]. MEKC conditions: 50 mM NaH2PO4/ Na2HPO4, pH 7.5, pressure injection 50 mbar 5s, −20 kV, 20 °C, UV detection at 214 nm.
As depicted, L-UCPB provided almost twice as high chiral Rs for (±)-(α-BP-AA) compared to L-UCLB (Figure 4A vs. 4C). One possible explanation for enhanced chiral resolution provided by L-UCPB over L-UCLB could be due to the rigid ring system of L-UCPB, which apparently allows maximum interaction via three-point interaction with ±-α-Br-Ph-AA.53 The Rs trend is consistent to the findings of Thiobodeaux et al.,54 who observed that surfactants derived from L-proline (a rigid amino acid) provided better chiral separation for rigid chiral molecules (e.g., BNP). The analyte (±)-(α-BP-AA) has a chiral center, which is adjacent to a bromo group and a carboxylate group. Thus, it appears that the chiral recognition was greatly facilitated not only by electrostatic interactions between carboxylate group of the analyte and the positively charge nitrogen,but the presence of bromo group adjacent to the chiral center might also hydrogen bond with the –OH group of the ionic liquids (Figure 2).
Comparing the electropherograms in 4(A) vs. 4(C) and 4(B) vs. 4(D), it is obvious that monomers of both L-UCPB and L-UCLB provided better chiral resolution, selectivity and efficiency compared to the corresponding polymers. The probable reason behind this observation could be the polydispersity of the polymers,55 which usually is the case when surfactants are polymerized at concentration higher than the CMC.55,56
Enantioseparation of ±-2-(2-Chlorophenoxy)propanoic acid
As discussed above, in the case of (±)-(α-BP-AA) maximum chiral Rs was obtained at pH 7.50 and no Rs at pH 2.00. O'Keeffe et al.57 and Haynes et al.58 have reported the separation of (±)-(2-PPA) at pH 5.00-6.00 with a cationic substituted β-cyclodextrin. Similar to the case of (±)-(α-BP-AA) separation, the variation in surfactant concentration and addition of organic modifier showed no significant effects on chiral Rs of (±)-(2-PPA).
Figure 5(A) and 5(C) show the chiral separation of (±)-(2-PPA) at optimum MEKC parameters using L-UCPB and L-UCLB, respectively. The non-rigid leucine based (L-UCLB) chiral selector (Fig 5C) provided significantly higher chiral Rs of (±)-(2-PPA) than L-UCPB. This resolution trend is opposite to the separation of (±)-(α-BP-AA) (Fig 4A, 4C). As stated, the proximity of bromo and carboxylate group to the chiral center of (±)-(α-BP-AA) as well as the rigidity of the chiral selector was thought to be the key factors ensuring maximum enantioselectivity. However, in case of (±)-(2-PPA), the chloro group on the benzene ring is farther away from the chiral center. Furthermore, the non-rigidity of L-UCLB might have resulted in favorable hydrogen bonding interactions between the chloro group on the benzene ring and the primary alcohol of the L-leucinol. Comparing 5(A) vs. 5(B) and 5(C) vs. 5(D), it is clear that monomers and polymers of L-UCPB and L-UCLB show very similar stereoselective interactions with (±)-(2-PPA) as evident from the Rs and α values.
Figure 5.
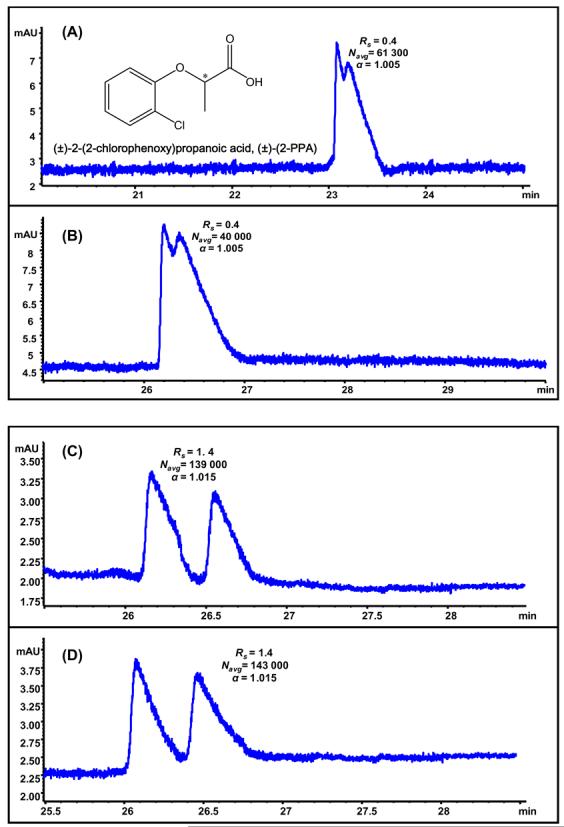
Comparison of 25 mM L-UCPB (A), poly-L-UCPB (B), 25 mM L-UCLB (C) and poly-L-UCLB (D) for enantioseparation of 2-(2-chlorophenoxy)propanoic acid[(±)-(2-PPA), 0.5 mg/mL in MeOH/H2O]. MEKC conditions are same as in Fig. 4.
It is interesting to compare the enantioseparation capability between two polymeric chiral anionic surfactants [polysodium N-undecenoxy carbonyl-L-leucine sulfate (poly-L-SUCLS) and polysodium N-undecenoxy carbonyl-L-leucinate (poly-L-SUCL)] with the chiral cationic surfactants discussed earlier for racemic anionic analyte. The chiral separation of (±)-(2-PPA) with both sulfated and carboxylated head group polymeric surfactants were investigated at basic pH (Fig 6A-B). As we have mentioned earlier that anionic compounds are usually difficult to separate with anionic surfactant due to the electrostatic repulsion between similar charges. Hence, as expected no chiral resolution was obtained for (±)-(2-PPA) at pH 8.00. Since poly-L-SUCLS has sulfated head group, it can be used at any pH without any solubility problem. Therefore, we performed MEKC at pH 2.00 (Fig 6C) in order to minimize dissociation of carboxylic acid group of (±)-(2-PPA) (pKa 3.11 ± 0.10). As can be seen in Fig 6C, partial chiral separation of (±)-(2-PPA) was achieved at pH 2.00. However, we could not improve this chiral Rs any further even after fine-tuning of the MEKC parameters (data not shown). Hence, comparing chiral separation of (±)-(2-PPA) with poly-L-UCLB (Fig 5D), poly-L-SUCLS (Fig 6A,C) and poly-L-SUCL (Fig 6B), it is clear that indeed electrostatic attraction interaction plays a dominant role in chiral recognition. In addition, structural features (e.g., rigidity and charges) of both analyte and chiral selector also seem to affect the chiral recognition.
Figure 6.
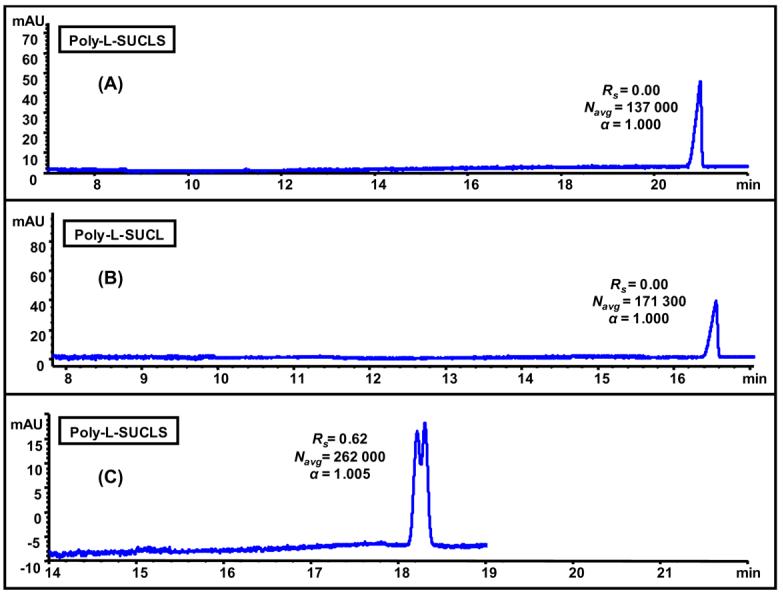
Comparison of 25 mM poly-L-SUCLS (A), 25 mM poly-L-SUCL (B), and 50 mM poly-L-SUCLS (C) for enantioseparation of chiral phenoxypropionic acid [(±)-(2-PPA), 0.5 mg/mL in MeOH/H2O]. MEKC conditions (A, B): pH 8.00, 25 mM NH4OAc/ 25 mM TEA, 15 °C, pressure injection 50 mbar s, +20 kV applied for separations, UV detection at 200 nm. (C)MEKC conditions same as 5(A) except pH 2.00, 25 mM NaH2PO4/ 25 mM CH3COONa and −20 kV applied for separations.
CONCLUSIONS
This paper is the first demonstration of successful chiral separation of acidic analytes with synthetic chiral ILs in CE. Both L-UCLB and L-UCPB ionic liquid type surfactants were thoroughly characterized before and after the polymerization. It was found that chiral separation of the acidic analytes with the chiral ILs and their polymers is strongly dependent on the presence of opposite charge as well as the structural compatibility between chiral selector and the analyte. Even though we did not demonstrate the enantioseparation of large number of acidic analytes, we still believe that our findings will guide the future research in MEKC separation of acidic analytes with intelligently designed synthetic chiral ionic liquids.
ACKNOWLEDGEMENT
This work was supported by grant from the National Institutes of Health (Grant No. GM 62314).
References
- 1.Bladon C. Pharmaceutical Chemistry: Therapeutic Aspects of Biomacromolecules. 5th ed. John Wiley & Sons; New York: 2002. [Google Scholar]
- 2.Coleman M. Human Drug Metabolism: An Introduction. John Wiley & Sons; England: 2005. [Google Scholar]
- 3.Williams DA, Lamke TL, Foye WO. Foye's Principles of Medicinal Chemistry. Lippincott Williams & Wilkins; Philadelphia: 2002. [Google Scholar]
- 3.Silverman RB. The Organic Chemistry of Drug Design and Drug Action. Academic Press; San Diego: 2004. [Google Scholar]
- 5.Blanchard LA, Hancu D, Beckman EJ, Brennecke JF. Nature. 1999;399:28. [Google Scholar]
- 6.Stalcup AM, Cabovska B. J. Liq. Chromatogr Relat. Tech. 2004;27:7–9. [Google Scholar]
- 7.Wasserscheid P, Keim W. Angew. Chem. Int. Ed. 2000;39:3772–3789. doi: 10.1002/1521-3773(20001103)39:21<3772::aid-anie3772>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Sheldon R. Chem. Commun. 2001:2399–2407. doi: 10.1039/b107270f. [DOI] [PubMed] [Google Scholar]
- 9.Dupont J, de Souza RF, Saurez PAZ. Chem. Rev. 2002;102:3667–3692. doi: 10.1021/cr010338r. [DOI] [PubMed] [Google Scholar]
- 10.Wasserscheid P, Welton T. Ionic Liquids in Synthesis. Wiley-VCH: Weinheim; Germany: 2003. [Google Scholar]
- 11.Anderson JL, Armstrong DW, Wei GT. Anal. Chem. 2006;78:2893–2902. doi: 10.1021/ac069394o. [DOI] [PubMed] [Google Scholar]
- 12.Brennecke JF, Maginn EJ. AIChE J. 2001;47(11):2384–2389. [Google Scholar]
- 13.Chauvin Y, Olivier-Bourbigou H. Chemtech. 1995;25:26–30. [Google Scholar]
- 14.Earle NJ, Seddon KR. Pure Appl. Chem. 2000;72:1391–1398. [Google Scholar]
- 15.Welton T. Chem. Rev. 2000;99:2071–2083. doi: 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- 16.Del Po'polo MG, Voth GA. J. Phys. Chem. B. 2004;108:1744–1752. [Google Scholar]
- 17.Carda-Broch S, Berthod A, Armstrong DW. Anal. Bioanal. Chem. 2003;375:191–199. doi: 10.1007/s00216-002-1684-1. [DOI] [PubMed] [Google Scholar]
- 18.Holbrey JH, Seddon KR. J. Chem. Soc. Dalton Trans. 1999:2133–2139. [Google Scholar]
- 19.Carmichael AJ, Earle MJ, Holbrey JD, McCormac PB, Seddon KR. Org. Lett. 1999;1:997–1000. [Google Scholar]
- 20.Poole CF, Kersten BR, Ho SSJ, Coddens ME, Furton KJ. J. Chromatogr. 1986;352:407–425. [Google Scholar]
- 21.Poole SK, Shetty PH, Poole CF. Anal. Chim. Acta. 1989;218:241–264. [Google Scholar]
- 22.Huang X, Luckey JA, Gordon MJ, Zare RN. Anal. Chem. 1989;61:766–770. [Google Scholar]
- 23.Harrold MP, Wojtusik MJ, Riviello J, Henson PJ. Chromatrogr. 1993;640:463–471. [Google Scholar]
- 24.Quang C, Khaledi MG. Anal. Chem. 1993;65:3354–3358. doi: 10.1021/ac00071a003. [DOI] [PubMed] [Google Scholar]
- 25.Yanes EG, Gratz SR, Stalcup AM. Analyst. 2000;125:1919–1923. doi: 10.1039/b004530f. [DOI] [PubMed] [Google Scholar]
- 26.Yanes EG, Gratz SR, Baldwin MJ, Robinson SE, Stalcup AM. Anal. Chem. 2001;73:3838–3844. doi: 10.1021/ac010263r. [DOI] [PubMed] [Google Scholar]
- 27.Armstrong DW, Zhang L, He L, Gross ML. Anal. Chem. 2001;73:3679–3686. doi: 10.1021/ac010259f. [DOI] [PubMed] [Google Scholar]
- 28.Carda-Broch S, Berthod A, Armstrong DW. Rapid Comm. Mass Spec. 2003;17:553–560. doi: 10.1002/rcm.931. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong DW, He L, Liu YS. Anal. Chem. 1999;71:3873–3876. doi: 10.1021/ac990443p. [DOI] [PubMed] [Google Scholar]
- 30.Berthod A, He L, Armstrong DW. Chromatographia. 2001;53:63–68. [Google Scholar]
- 31.Anderson JL, Armstrong DW. Anal. Chem. 2003;75:4851–4858. doi: 10.1021/ac0345749. [DOI] [PubMed] [Google Scholar]
- 32.Ding J, Welton T, Armstrong DW. Anal. Chem. 2004;76:6819–6822. doi: 10.1021/ac049144c. [DOI] [PubMed] [Google Scholar]
- 33.Mwonngela SM, Numan A, Gill N, Agbaria RA, Warner IM. Anal. Chem. 2003;75:6089–6096. doi: 10.1021/ac034386i. [DOI] [PubMed] [Google Scholar]
- 34.Laamanen PL, Lahtinen SBM, Matilainen R. J. Chromatogr. A. 2005;1095:164–171. doi: 10.1016/j.chroma.2005.07.111. [DOI] [PubMed] [Google Scholar]
- 35.Cross J, Singer EJ, editors. Cationic Surfactants. Marcel Dekker, INC; New York: 1994. [Google Scholar]
- 36.Dwars T, Paetzold E, Oehme G. Angew. Chem. Int. Ed. 2005;44:7174–7199. doi: 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg SI, Baba N, Green RL. J. Am. Chem. Soc. 1978;100:6768–6769. [Google Scholar]
- 38.Borocci S, Ceccacci F, Galantini L, Mancini G, Monti D, Scipioni A, Venanzi M. Chirality. 2003;15:441–447. doi: 10.1002/chir.10230. [DOI] [PubMed] [Google Scholar]
- 39.Davidson TA, Mondal K, Yang X. J. Colloid Interface Sci. 2004;276:468–502. doi: 10.1016/j.jcis.2004.03.053. [DOI] [PubMed] [Google Scholar]
- 40.Diego-Castro MJ, Hailes HC, Lawrence MJ. J. Colloid Interface Sci. 2001;234:122–126. doi: 10.1006/jcis.2000.7285. [DOI] [PubMed] [Google Scholar]
- 41.Diego-Castro MJ, Hailes HC. J. Chem. Soc. Chem. Commun. 1998;15:1549–1550. [Google Scholar]
- 42.Dobashi A, Hamada M, Yamaguchi J. Electrophoresis. 2001;22:88–96. doi: 10.1002/1522-2683(200101)22:1<88::AID-ELPS88>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 43.Dey J, Mohanty A, Roy S, Khatua D. J. Chromatogr. A. 2004;1048:127–132. doi: 10.1016/j.chroma.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 44.Hamdouchi C, Martinez CS, Gruber J, Prado MD, Lopez J, Rubio A, Heinz BA. J. Med. Chem. 2003;46(20):4333–4341. doi: 10.1021/jm020583i. [DOI] [PubMed] [Google Scholar]
- 45.Procopio A, Alcaro S, Cundari S, De Nino A, Ortuso F, Sacchetta P, Pennelli A, Sindona G. J. Med. Chem. 2005;48(19):6084–6089. doi: 10.1021/jm0504609. [DOI] [PubMed] [Google Scholar]
- 46.Rizvi SAA, Shamsi SA. Electrophoresis. 2003;24:2514–2526. doi: 10.1002/elps.200305516. [DOI] [PubMed] [Google Scholar]
- 47.Eschweiler W. Ber. 1905;38:880. [Google Scholar]
- 48.Clarke HT, Gillespie HB, Weisshaus SZ. J. Am. Chem. Soc. 1933;55:4571–4587. [Google Scholar]
- 49.Farkas E, Sunman CJ. J. Org. Chem. 1985;50:1110–1112. [Google Scholar]
- 50.Morrow AP, Kassim OO, Ayorinde FO. Rapid Commun. Mass Spectrom. 2001;15:767–770. doi: 10.1002/rcm.293. [DOI] [PubMed] [Google Scholar]
- 51.Tuiman AA, Cook KD, Magid LJ. J. Am. Soc. Mass Spectrom. 1990;1:85–91. doi: 10.1016/1044-0305(90)80009-C. [DOI] [PubMed] [Google Scholar]
- 52.Bertschinger AT, Perry CS, Galland A, Prannkerd RJ, Charman WN. J. Pharm. Sci. 2003;92:2217–2228. doi: 10.1002/jps.10479. [DOI] [PubMed] [Google Scholar]
- 53.Davankov VA. Pure Appl. Chem. 1997;69:1469–1474. [Google Scholar]
- 54.Thibodeaux SJ, Billiot E, Warner IM. J. Chromatogr. A. 2002;966:179–186. doi: 10.1016/s0021-9673(02)00747-1. [DOI] [PubMed] [Google Scholar]
- 55.Tarus J, Agbaria RA, Morris K, Mwongela S, Numan A, Simuli L, Fletcher KA, Warner IM. Langmuir. 2004;20:6887–6895. doi: 10.1021/la036349s. [DOI] [PubMed] [Google Scholar]
- 56.Mileva E. J. Colloid Interface Sci. 2000;232:211–218. doi: 10.1006/jcis.2000.7205. [DOI] [PubMed] [Google Scholar]
- 57.O′Keeffe F, Shamsi SA, Darcy R, Schwinte P, Warner IM. Anal. Chem. 1997;69:4773–4782. doi: 10.1021/ac970370e. [DOI] [PubMed] [Google Scholar]
- 58.Hanes JL, III, Shamsi SA, O'Keeffe F, Darcy R, Warner IM. J. Chromatogr. A. 1998;803:261–271. doi: 10.1016/s0021-9673(97)01212-0. [DOI] [PubMed] [Google Scholar]