

Abstract
The X-ray crystal structures of human purine nucleoside phosphorylase (PNP) with bound inosine or transition state analogues show His257 within hydrogen-bonding distance to the 5′-hydroxyl. The mutants His257Phe, His257Gly, and His257Asp exhibited greatly decreased affinity for Immucillin-H (ImmH), binding this mimic of an early transition state as much as 370-fold (Km/Ki) less tightly than native PNP. In contrast, these mutants bound DADMe-ImmH, a mimic of a late transition state, nearly as well as the native enzyme. These results indicate that His257 serves an important role in the early stages of transition state formation. Whereas mutation of His257 resulted in little variation in the PNP·DADMe-ImmH·SO4 structures, His257Phe·ImmH·PO4 showed distortion at the 5′-hydroxyl, indicating the importance of H-bonding in positioning this group during progression to the transition state. Binding isotope effect (BIE) and kinetic isotope effect (KIE) studies on the remote 5′-3H for the arsenolysis of inosine with native PNP revealed a BIE of 1.5% and an unexpectedly large intrinsic KIE of 4.6%. This result is interpreted as a moderate electronic distortion toward the transition state in the Michaelis complex with continued development of a similar distortion at the transition state. The mutants His257Phe, His257Gly, and His257Asp altered the 5′-3H intrinsic KIE to −3%, −14%, and 7%, respectively, while the BIEs contributed 2%, 2%, and −2%, respectively. These surprising results establish that forces in the Michaelis complex, reported by the BIEs, can be reversed or enhanced at the transition state.
Keywords: purine nucleoside phosphorylase, mutagenesis, binding isotope effect, kinetic isotope effect, transition state, binding distortion, transition state geometry, dynamics
Determination of the transition state structure of enzymatic reactions allows for the design of tight binding transition state analogue inhibitors. These transition state mimics can subvert the energetics that govern formation of the enzymatic transition state to achieve binding orders of magnitude stronger than that of substrate. The transition state structures of enzymatic reactions have frequently been established by measuring kinetic isotope effects (KIEs1) from the competitive reaction of isotopically labeled substrates. These isotope effects on kcat/Km, often referred to as V/K KIEs (1), are a powerful tool in establishing bond vibrational differences between reactants free in solution and at the transition state. Unfortunately, they do not distinguish isotope effects arising from chemistry at the transition state from those generated in the Michaelis complex (Figure 1a). Equilibrium binding isotope effects (BIEs), however, can be measured in separate experiments to determine the extent to which binding contributes to the V/K KIE (2, 3). Measurement of both BIEs and KIEs permits resolution of binding distortion and bond distortions due to chemistry towards the transition state.
Figure 1.

a) The relationship among BIE, V/K KIE, and intrinsic KIE using purine nucleoside phosphorylase (PNP) as a model. Ino = inosine, Hx = hypoxanthine, R1As = ribose 1-arsenate. b) Arsenolysis reaction catalyzed by purine nucleoside phosphorylase including the SN1-like transition state. Unlike the analogous phosphorolysis reaction, arsenolysis is irreversible due to the instability of the ribose 1-arsenate product, which rapidly hydrolyzes. N7 of the leaving group has been depicted as being protonated at the transition state, as this has been demonstrated to be a common mechanistic feature in PNP and other nucleoside phosphorylases and hydrolases (37).
The determination of BIEs to help interpret KIE data has recently been applied to thymidine phosphorylase (TP), in which a large secondary 3H KIE of 6.1%, remote from the reaction center, had been found (4). The corresponding 3H BIE was subsequently measured to be 6.0%, accounting for the entire V/K KIE (2). Human purine nucleoside phosphorylase (HsPNP) catalyzes the mechanistically similar reversible phosphorolysis of purines (e.g., inosine) to yield ribose 1-phosphate and free nucleobase (e.g., hypoxanthine). By determining the KIEs for the HsPNP-catalyzed arsenolysis reaction (Figure 1b), the transition state structure was solved, indicating that the reaction proceeds via an SN1-like mechanism (5). As in TP, a large remote secondary 5′-3H KIE of 6.2% was determined for HsPNP. The same question is therefore raised: is this KIE due primarily to binding interactions or to changes exclusive to the transition state?
Insight into the origin of the remote KIE in the PNP reaction may be obtained by examination of structural features in proximity to C-5′. Human and bovine PNP structures obtained from X-ray crystallography show that the 5′-hydroxyl of the substrate and transition state analogues is within hydrogen-bonding distance of His257 (6, 7). It has been hypothesized that this residue is responsible for positioning O-5′ in line with O-4′ and the nucleophilic oxygen of phosphate (8). This “oxygen stack” is proposed to contribute to catalysis dynamically, with vibrational compression of the three oxygen atoms providing electron density that increases the leaving group ability of the purine base through stabilization of the oxacarbenium-like transition state (Figure 2). We have also investigated the role of His257 through mutagenesis to evaluate the corresponding kinetic and structural impacts. X-ray crystal structures with bound ImmH and DADMe-ImmH, transition state analogues for HsPNP, reveal distortion of the 5′-OH when H-bonding to this group is removed. The 5′-3H V/K KIEs and BIEs on the native and mutant enzymes were determined to establish the relative contributions to catalysis provided by formation of the Michaelis complex and subsequent changes at the transition state.
Figure 2.
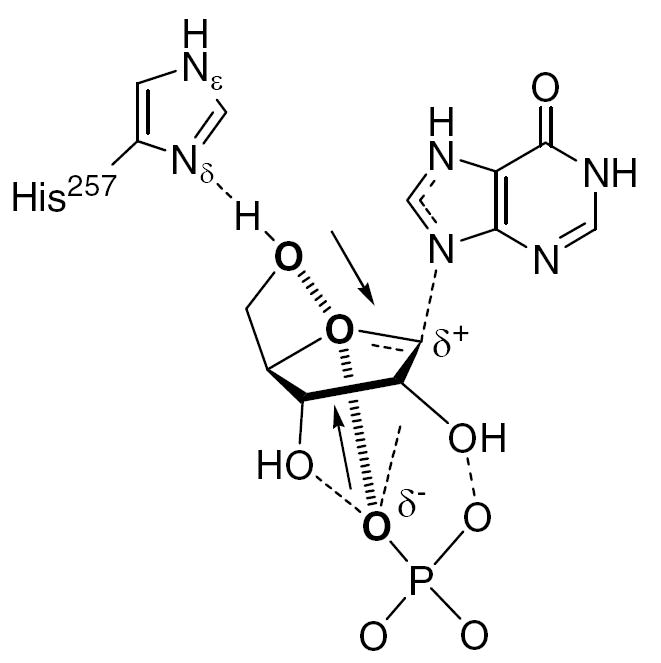
Proposed role of His257 in formation of the transition state, featuring dynamic compression of the O5′–O4′–OP “oxygen stack”. The “oxygen stack” is represented by hashed bonds connecting bolded atoms, and arrows indicate promoting vibrational modes. Dashed bonds represent hydrogen bonds or partial bonds. Dynamic motion in the enzyme active site pushes O-5′ and the phosphate oxygen towards the ring oxygen, leading to increased electron density in the center of reactivity. This contributes electron density to weaken the N9–C1′ bond, enhances hypoxanthine’s leaving group ability, and forms the developing ribooxacarbenium ion.
Materials and Methods
Site-Directed Mutagenesis
The PCR product for HsPNP was cloned into the pCR-T7/CT-TOPO vector (Invitrogen), using samples and methods described earlier (5, 9). The resulting plasmid was transformed into TOP10F′ chemically competent E. coli cells (Invitrogen) and grown overnight on LB-agar plates containing 100 μg/mL ampicillin. Plasmids isolated from positive transformants were characterized by restriction analysis using HindIII and XbaI (New England Biolabs). The sequence of the HsPNP gene, including a stop codon prior to the C-terminal histidine tag of the TOPO vector, was confirmed by automated DNA sequencing (Albert Einstein College of Medicine).
Mutants were prepared according to the protocol of the QuikChange® Site-Directed Mutagenesis Kit (Stratagene). Oligonucleotide pairs that were used to introduce mutations in the forward and reverse directions are listed below, with the mutated nucleotides underlined. Primers used for the His257Gly mutant were 5′-CTGGAGAAGGCCAACGGTGAAGAAGTCTTAGCA-3′ (forward) and 5′-TGCTAAGACTTCTTCCACCGTTGGCCTTCTCCAG-3′ (reverse). Primers used for the His257Phe mutant were 5′-GCCTGGAGAAGGCCAACTTTGAAGAAGTCTTAGCAGCTG-3′ (forward) and 5′-CAGCTGCTAAGACTTCTTCAAAGTTGGCCTTCTCCAGGC-3′ (reverse). Primers used for the His257Asp mutant were 5′-CTGGAGAAGGCCAACGATGAAGAAGTCTTAGCAGC-3′ (forward) and 5′-GCTGCTAAGACTTCTTCATCGTTGGCCTTCTCCCAG-3′ (reverse). All mutations were confirmed by automated DNA sequencing.
Overexpression and Purification of PNP Mutants
The H257G and H257F recombinant plasmids were transformed into BL21Star(DE3) chemically competent E. coli (Invitrogen), and the His257Asp recombinant plasmid was transformed into BL21AI(DE3) competent cells (Invitrogen). Positive transformants from overnight incubation on LB-agar plates containing 100 μg/mL ampicillin were used to inoculate 10 mL cultures grown overnight in LB containing 100 μg/mL ampicillin at 37 °C with shaking at 200 rpm. The overnight cultures were poured into 1 L LB medium containing 100 μg/mL ampicillin and grown at 37 °C with shaking at 225 rpm until an OD600 of 0.5–0.7 was reached. Cells were induced for overexpression for 3–4 h at 37 °C by addition of either 0.1% lactose (His257Gly and His257Phe) or 0.2% l-arabinose (His257Asp). Cells were harvested at 4000 rpm for 30 min and stored at 4 °C overnight. Cell pellets were resuspended in 5–6 mL of lysis buffer (50 mM Tris-HCl, pH 8.0, containing one Complete protease inhibitor cocktail tablet (Roche Applied Science) per 50 mL and ~ 1 mg of DNase I) per gram of pellet weight, and lysed by sonication (5 min at 20% duty cycle, power level 6) on ice. The cell lysate was clarified by centrifugation at 18,000 rpm for 30 min.
The clarified cell lysate was loaded at 2 mL/min onto a 140-mL column of Q-Sepharose Fast Flow resin (Amersham), pre-equilibrated with start buffer (50 mM Tris-HCl, pH 8.0). After washing with 2 column volumes of start buffer at 4.6 mL/min, the desired protein was eluted by a linear salt gradient (start buffer containing 0–0.25 M NaCl, applied over 6 column volumes). Fractions containing the mutant enzyme were identified by SDS-PAGE, pooled, and concentrated by ultrafiltration with a 400-mL Amicon stirred cell (10 kDa molecular weight cut-off (MWCO), Millipore), followed by a 15-mL centrifugal filter (10 kDa MWCO, Millipore). The concentrated enzyme solution (approximately 20 mg/mL by A280, assuming ε280 = 28,830 M-1 cm-1 (10)) was then rapidly-frozen in liquid nitrogen and stored at -80 °C.
Purified proteins were found to be >95% pure by SDS-PAGE, and the presence of the correct amino acid substitutions was confirmed by electrospray ionization mass spectrometry (ESI-MS): His257Gly – 32,061 Da (calc’d 32,067 Da); His257Phe – 32,154 Da (calc’d 32,157 Da); His257Asp – 32,123 Da (calc’d 32,125 Da).
Overexpression and Purification of Histidine-Tagged Wild-Type HsPNP
Native human PNP was expressed and purified as the N-terminal histidine-tagged fusion as previously described (5).
Crystallization
The HsPNP·DADMe-ImmH·SO4 crystal complexes were prepared by hanging-drop vapor diffusion at 18 °C by mixing the protein solution (2 μL of 20 mg/mL) containing DADMe-ImmH (1:1.5 stoichiometry) with an equal volume of the reservoir solution containing 100 mM sodium citrate, pH 5.0, and 1.4 M (NH4)2SO4 (Hampton), and equilibrating against 1.0 mL of the reservoir solution. The HsPNP·ImmH·PO4 crystal complexes were prepared by sitting-drop vapor diffusion by mixing the protein solution (1 μL of 20 mg/mL) containing ImmH and PO4 (each 1.5 stoichiometry with respect to protein) with an equal volume of the reservoir solution containing 100 mM NaOAc, pH 4.6, and 4.0 M NH4OAc, and equilibrating against 80 μL of the reservoir solution. Crystals appeared in 2 days and grew to the maximum size of 0.3 × 0.3 × 0.3 mm3. Diffraction from the HsPNP mutant crystals is consistent with the space group R32 (a = b = 142 Å, c = 166 Å), with a monomer in the asymmetric unit (Vm = 4.7 Å3/Da; 76 % solvent content).
Data Collection and Structure Refinement
HsPNP·DADMe-ImmH·SO4 mutant crystals were transferred to cryoprotectant composed of crystallization solution containing 20% glycerol and flash cooled at -178 °C. HsPNP·ImmH·PO4 mutant crystals were flash cooled without cryoprotection. X-ray diffraction data were collected at 1.1 Å wavelength on a Quantum 315 CCD detector using synchrotron radiation at beamline X29A at the Brookhaven National Synchrotron Light Source. Data were reduced using the HKL package (11). HsPNP mutants were crystallized in the same space group as native apoprotein. The structures of native HsPNP apo form co-crystallized with DADMe-ImmH (PDB ID: 1RSZ3) or ImmH (PDB ID: 1RR6) were used as starting models for the refinement using CNS (12) or REFMAC5 (13), respectively. Mutated side chains were built into the density map using program O (14) and COOT (15). The final models with DADMe-ImmH include residues 3–284, DADMe-ImmH, and 2 sulfate ions for each monomer, while the final models with ImmH contained residues 1-286, one ImmH, and one phosphate for each monomer. The model displays good stereochemistry as determined by PROCHECK (16). The structures have been submitted to the Protein Data Bank, PDB accession codes 2A0W, 2A0X, and 2A0Y for His257Gly, His257Phe, and His257Asp, respectively, with DADMe-ImmH and 2OC9, 2ON6, and 2OC4, respectively, with ImmH. Data collection and structure refinement statistics are shown in Table 1.
Table 1.
Data Collection and Structural Refinement Statistics
| PNP Mutant | His257Gly | His257Phe | His257Asp | |||
|---|---|---|---|---|---|---|
| Inhibitor | DADMe | ImmH | DADMe | ImmH | DADMe | ImmH |
| Ion | SO4 | PO4 | SO4 | PO4 | SO4 | PO4 |
| PDB ID | 2A0W | 2OC9 | 2A0X | 2ON6 | 2A0Y | 2OC4 |
| Space group | R32 | R32 | R32 | R32 | R32 | R32 |
| Unit cell (Å) | ||||||
| a | 141.881 | 142.441 | 141.764 | 143.011 | 142.467 | 143.386 |
| b | 141.881 | 142.441 | 141.764 | 143.011 | 142.467 | 143.386 |
| c | 166.399 | 168.765 | 166.172 | 168.324 | 166.029 | 168.457 |
| Data Collection | ||||||
| Resolution (Å) | 50-2.28 | 30-2.60 | 50-2.28 | 30-2.50 | 50-2.28 | 30-2.60 |
| No. of reflections total | 267,740 | 128,876 | 265,893 | 151,918 | 262,079 | 142,541 |
| unique | 29,413 | 19,978 | 29,401 | 21,634 | 29,445 | 20,554 |
| Completeness (%) | 99.9(99.9) | 97.2 (75.8) | 99.9(100) | 94.5(83.3) | 99.9 (99.6) | 99.3(94.6) |
| Rmerge (%) | 4.6 (19.6) | 6.9(39.9) | 6.2 (36.4) | 6.6(37.8) | 5.1 (56.4) | 7.1(55.1) |
| Structural Refinement | ||||||
| Rcryst (%) | 23.3 | 19.4 | 22.7 | 20.5 | 24.3 | 20.0 |
| Rfree(%) | 25.4 | 24.0 | 25.1 | 24.6 | 26.7 | 24.7 |
| No. of amino acids | 282 | 284 | 282 | 286 | 282 | 286 |
| No. of ligands | 1 DADMe | 1 ImmH | 1 DADMe | 1 ImmH | 1 DADMe | 1 ImmH |
| 2 SO4 | 1 PO4 | 2 SO4 | 1 PO4 | 3 SO4 | 1 PO4 | |
| No. of water | 79 | 48 | 60 | 50 | 46 | 53 |
| RMS Deviations | ||||||
| bond (Å) | 0.007 | 0.024 | 0.007 | 0.022 | 0.007 | 0.029 |
| angle (deg) | 1.33 | 2.2 | 1.33 | 2.14 | 1.31 | 2.62 |
| Ramachandran Plot | ||||||
| most favor regions | 89.5 | 87.6 | 90.8 | 88.7 | 87.9 | 84 |
| add. allowed | 10.1 | 10.7 | 8.8 | 10.5 | 11.7 | 13.9 |
| gen. allowed | 0.0 | 1.3 | 0.0 | 0.4 | 0.0 | 1.7 |
| disallowed | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 |
Steady-State Kinetic Assays and Inhibition Studies
Catalytic activity was measured in 50 mM potassium phosphate, pH 7.4, at 25 °C, using a xanthine oxidase coupled assay, as previously described (17), but with the inclusion of 5.0 mM dithiothreitol. The molar extinction coefficient for uric acid formation at 293 nm was taken to be 12,900 M-1 cm-1 (18). Histidine-tagged native and untagged mutant protein concentrations were determined by absorbance at 280 nm using extinction coefficients of 31,650 M-1 cm-1 and 30,160 M-1 cm-1, respectively, which were calculated from their protein sequences (19). Inosine and inhibitor concentrations were determined spectrophotometrically using ε260 = 7,100 M-1 cm-1 (pH 6) (20) and ε261 = 9,540 M-1 cm-1 (pH 7) (21), respectively. Inhibitor dissociation constants for the phosphorolysis of inosine were based on initial and equilibrium reaction rate measurements with varied inhibitor concentrations (typically 2.0 nM to 2.0 μM). Reactions were performed in the presence of 5.0 mM dithiothreitol at fixed inosine concentrations of 1.0 mM, 5.0 mM, 5.0 mM, and 15.0 mM for native HsPNP, His257Phe, His257Gly, and His257Asp, respectively. Reactions were initiated by the addition of PNP to final concentrations of 0.5 nM to 1.5 nM. In most cases the inhibitor concentration was at least ten-fold greater than the enzyme concentration, as required for simple analysis of slow-onset tight-binding inhibition (22). When this condition could not be satisfied, corrections were made to compensate for the concentration of bound inhibitor (23).
Synthesis of [5′-3H]Inosine and [5′-14C]Inosine
To a solution of 500 μCi [5′- 3H]uridine (33 nmol, 15 Ci/mmol, Moravek Biochemicals) in 50 μL of 10 mM Tris-HCl, pH 8.0, were added 1.0 μL of E. coli thymidine phosphorylase (1 unit, Sigma), 5.0 μL HsPNP (~11 units of 32 mg/mL), and 1.0 μL of 1.0 mM hypoxanthine (Sigma). Note that E. coli TP accepts uridine as an alternative substrate. After incubation for 10 min at room temperature, [5′-3H]inosine was obtained in >98% purity by reversed-phase HPLC (5% methanol in 50 mM ammonium formate, pH 4.0, at 1 mL/min) using an analytical C18 column (Deltapak, Waters). The specific radioactivity of the starting material suggests it is a mixture of 5′-R and 5′-S monolabeled epimers. [5′-14C]Inosine was prepared from [6-14C]glucose as previously described (5).
Kinetic Isotope Effect Measurements
KIEs were determined as described by Lewandowicz and Schramm (5). [5′-3H]Inosine and [5′-14C]inosine were mixed at a ratio of 3:1 and subjected to arsenolysis using 50 mM Tris-HCl (pH 7.5), 50 mM Na2HAsO4 (Sigma), 250 μM carrier inosine, and HsPNP. Reactions, monitored by reversed-phase HPLC equipped with a photodiode array detector, were allowed to proceed until approximately 30% conversion. A one-third volume aliquot was removed and allowed to react to 100% completion. The remainder was divided into two equal volumes and quenched by application to charcoal spin-columns (100 mg of activated charcoal in a Qiagen DNA purification spin-column, previously washed with 1 mL of 10 mM d-ribose in 10% ethanol). The labeled ribose was eluted with 5 × 0.5 mL of 10 mM d-ribose in 10% ethanol, mixed with 7 mL scintillation fluid (National Diagnostics Liquiscint), and counted for at least 5 cycles of 10 min each.
The count rate was averaged over all cycles, and the 3H and 14C emissions were separated according to the spectra of standard 14C samples in identical matrixes according to Equations 1 and 2,
| (1) |
| (2) |
where (14C channel ratio) is the ratio of 14C counts in channels A and B for a control compound, [1-14C]glucose. The spectral windows are set such that no counts from 3H appear in channel B. The ratios of 3H to 14C were determined for both the 30% reactions and the 100% reaction, and the KIEs, adjusted to 0% reaction, were calculated according to Equation 3,
| (3) |
where f is the fraction of reaction progress as determined by HPLC and Rf and Ro are the ratios of heavy to light isotope after partial and complete reaction, respectively. The forward commitment factor for HsPNP of 0.147 was previously determined (5).
Equilibrium Binding Isotope Effect Measurements
BIEs were determined as described by Lewis and Schramm using the ultrafiltration method (24, 25). To a solution of 50 mM Tris-HCl (pH 7.4), 500 mM ammonium sulfate, and 16–18 μM inosine (> 5:1 ratio of [5′-3H]inosine:[5′-14C]inosine by dpm), 18–20 μM HsPNP was added to a final volume of 325 μL. Three 100 μL aliquots were removed and added to the upper wells of the ultrafiltration apparatus and 22 psi N2 was applied for 60–90 min or until approximately half of the solution had passed through the dialysis membrane (10 kDa MW retention limit) into the lower well. From both upper and lower wells, 25 μL of solution was sampled using a Hamilton syringe and added to 1 mL of water in a scintillation vial. Ten mL of scintillation fluid (National Diagnostics Liquiscint) was added and the samples were counted for at least 5 cycles of 5 min each.
Spectral deconvolution of 3H and 14C was performed as described previously using a 14C standard in a matrix identical to the BIE samples. The BIE was then calculated as the quotient of the ratio of 14C to 3H bound to the enzyme (Rb) and free in solution (Rf). Rb was determined according to Equation 4:
| (4) |
where f is the fraction of substrate unbound in the upper well, which is taken as the ratio of 3H below the membrane to that above, Rm is the 14C to 3H ratio above the membrane, Rf is the ratio below the membrane, and Rb is the isotopic ratio of bound substrate.
Commitment to Catalysis
The isotope-trapping method, similar to previously reported methods, was used to determine commitment to catalysis (4, 5). A pulse solution containing 50 mM Tris-HCl (pH 7.4), 260 μM [5’-14C]inosine (ca. 105 cpm), and 30 μM His257Phe, 200 μM His257Gly, or 200 μM His257Asp was prepared and allowed to equilibrate at room temperature for 2 min. Ten microliters of this solution was then added to 200 μL of a chase solution containing 50 mM Tris-HCl (pH 7.4), 10 mM unlabeled inosine, and varying concentrations of sodium arsenate (1 μM to 100 mM). After approximately 6 s, the reaction was quenched by the addition of 50 μL of 1 M HCl. [5’-14C]Ribose generated in the reaction was isolated by passing through charcoal columns (~150 mg 1:6 charcoal/cellulose), preequilibrated with 20 mM ribose in 10% EtOH, and eluted with 2.75 mL of this solution. Seven milliliters of scintillation fluid was added, and the samples were counted for 1 min each. Controls measured the amount of ribose formed under the same conditions but either without enzyme or without arsenate. The forward commitment factor was determined by plotting [14C]ribose/[14C]inosinereleased against [AsO4], where [14C]inosinereleased is the inosine in the pulse solution not converted to ribose, given by Equation 5:
| (5) |
where [Ino]p is the total inosine in the pulse solution, and Km and [E ] are the respective Michaelis constant and enzyme concentration for the mutant protein.
Results and Discussion
To examine the contribution of His257 to the unusually large 5′-3H kinetic isotope effect observed in the PNP reaction and thereby assess its importance in binding and catalysis, this residue was mutated individually to glycine, phenylalanine, and aspartate. The His257Gly mutant was expected to eliminate H-bonding to the 5′-hydroxyl group of the substrate and to reduce the steric effects in its immediate proximity, possibly allowing it to rotate with less restriction. The His257Phe mutant would also remove H-bonding to the 5′-hydroxyl group but would retain steric bulk and hydrophobicity in this region of the active site. Finally, the His257Asp mutant would retain the possibility of H-bonding to the 5′-hydroxyl group, while introducing only a minor decrease in size. These mutants were tested for their catalytic and structural properties, as well as their isotopic discrimination.
Steady-State Kinetics of His257 Mutants
The effects of His257 mutation on the steady-state kinetics of inosine phosphorolysis were determined (Table 2), and all three mutants exhibited compromised catalysis in Km (inosine), kcat, and kcat/Km. In comparison to the native enzyme, His257Phe gave an eight-fold increase in Km and a nine-fold decrease in kcat. As this mutant lacks H-bonding ability yet retains significant catalytic activity, it is clear that the H-bond between His257 and the 5′-hydroxyl of inosine is not essential. This notion is further supported by the observation that 5′-deoxyinosine is a substrate for PNP (26). The kinetic values of His257Phe are similar to those obtained by Ealick and coworkers for His257Ala (Table 2) (27). Therefore, it appears that PNP has some tolerance for hydrophobic side chain substitutions at this position, likely due to the existence of hydrophobic neighboring residues within the ribose-binding cleft, including Val260, Leu261, and Phe200, as well as Phe159 from the adjacent monomer.
Table 2.
Steady-State Kinetic Parameters of HsPNP and His257 Mutants
| Enzyme | Km (μM) | kcat (S-1) | kcat/Km (M-1 S-1) | |||
|---|---|---|---|---|---|---|
| Value | Fold Change | Value | Fold change | Value | Fold change | |
| Native PNPa | 40 | 1 | 56 | 1 | 1.4 × 106 | 1 |
| His257Alaa | 210 | 5 | 16 | 4 | 7 × 104 | 20 |
| His257Phe | 320 ± 12 | 8 | 6.55 ± 0.07 | 9 | (2.05 ± 0.10) × 104 | 68 |
| His257Gly | 750 ± 60 | 19 | 1.75 ± 0.04 | 32 | (2.3 ± 0.2) × 103 | 610 |
| His257Asp | 1350 ± 130 | 34 | 1.30 ± 0.05 | 41 | (9.6 ± 1.3) × 102 | 1,460 |
Values obtained from reference (27).
Removal of the side chain on residue 257, as in the His257Gly mutant, results in a more pronounced catalytic impairment, with 19-fold and 32-fold changes in Km and kcat, respectively. The resulting 610-fold decrease in catalytic efficiency (kcat/Km) compared to native HsPNP is likely due to the elimination of stabilizing interactions either with substrate via H-bonding or with neighboring residues via hydrophobic packing.
Intriguingly, His257Asp, the mutant that conserved the 5′-OH H-bond with the native enzyme, suffered the greatest impact on catalysis. The increase in Km from 40 μM to 1.35 mM, combined with the 41-fold decrease in kcat, resulted in a drop in catalytic efficiency by a factor of 1,400. The substitution of a carboxyl group for the imidazole side chain of histidine introduces a functional group with a lower pKa. As a result, the electrostatic environment surrounding the 5′-OH is made more negative, and the H-bond strength between this hydroxyl group and residue 257 may have been altered. That the negative charge in the active site is not well tolerated is supported by the observation that activity in this mutant increases at more acidic pH values (maximal at pH 6.3), where the neutral form of the side chain is more favorable.
Inhibition of His257 Mutants by Transition State Analogues
Immucillin-H (ImmH; Figure 3) and DADMe-Immucillin-H (DADMe-ImmH) are tight-binding transition state analogue inhibitors of HsPNP, exhibiting slow-onset inhibition with dissociation constants (Ki*) of 58 and 11 pM, respectively. The greater affinity for DADMe-ImmH is attributed to placement of the cationic charge at the anomeric position and to increased separation between the sugar ring and leaving group (2.6 Å vs. 1.6 Å for ImmH), characteristics which allow DADMe-ImmH to more closely resemble HsPNP’s dissociative oxacarbenium-ion transition state (Figure 1b) (28). When DADMe-ImmH was tested with each of the His257 mutants, it was found to give Ki* values between 270 and 950 pM (Table 3). After accounting for differences in Km among the variants, this yields Km/Ki values of 337,000 to 2,800,000, all within a factor of 11 to the native value of 3,700,000. Thus, DADMe-ImmH binds nearly as tightly to the mutants as to native HsPNP. In contrast, differences of as much as 370-fold in Km/Ki were observed with ImmH (Table 3). Simple competitive inhibition occurred with these mutants with ImmH, in contrast to the slow-onset inhibition kinetics exhibited by the native enzyme (Figure 4). This finding reveals the importance of His257 in the early progress towards the transition state, which is mimicked by the molecular electrostatic structure of ImmH. As mentioned earlier, the H-bond to His257 has been proposed to steer the 5′-OH into alignment with O-4′ and the oxygen of the approaching nucleophile. In the later stages of the reaction coordinate, which is better mimicked by DADMe-ImmH, this residue apparently plays a diminished role, and interactions at the reaction center become more significant. Specifically, the ion pair between sulfate (a mimic of the phosphate anion) and the DADMe-ImmH cation is more favorable than that with ImmH (N–O distances of 3.1 Å and 3.7 Å, respectively).
Figure 3.
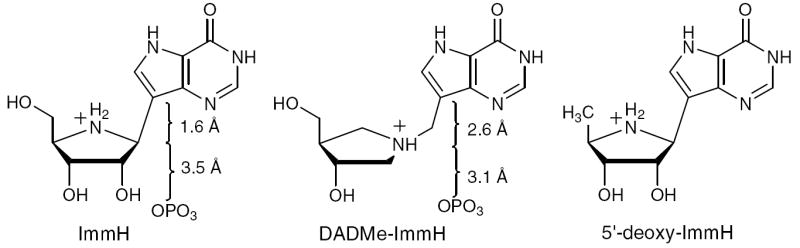
Transition state analogue inhibitors of HsPNP. ImmH and DADMe-ImmH are shown with their respective C-1′–C-9 (N-1′–C-9) and C-1′–OP distances. Inhibition by 5′-deoxy-ImmH was also tested in this study for comparison with the His257 mutants.
Table 3.
Dissociation Constants of Transition State Analogues with HsPNP and His257 Mutants
| DADMe-ImmH
|
ImmH
|
5′-deoxy-ImmH
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ki*a |
Km/Ki |
Ki | Ki* |
Km/Ki |
Kib |
Km/Ki |
||||
| Enzyme | Value | Fold | Value | Fold | Value | Fold | ||||
| Native | 10.7 ± 1.1 pM | 3,700,000 | 1 | a | 57.9 ± 1.5 pM | 690,000 | 1 | 20.0 ± 0.9 nM | 2,000 | 1 |
| His257Phe | 950 ± 60 pM | 337,000 | 11 | 172 ± 16 nM | b | 1,860 | 370 | 19.8 ± 1.0 nM | 16,200 | 8 |
| His257Gly | 270 ± 20 pM | 2,800,000 | 1 | 11.0 ± 0.9 nM | b | 68,100 | 10 | 7.7 ± 0.6 nM | 97,000 | 93 |
| His257Asp | 900 ± 100 pM | 1,500,000 | 2 | 86 ± 7 nM | b | 15,700 | 45 | 54 ± 3 nM | 25,000 | 12 |
The weak inhibition phase (Ki) was observed but too short to quantitate, so only the slow-onset, tight-binding phase (Ki*) is reported.
No slow-onset phase was observed.
Figure 4.
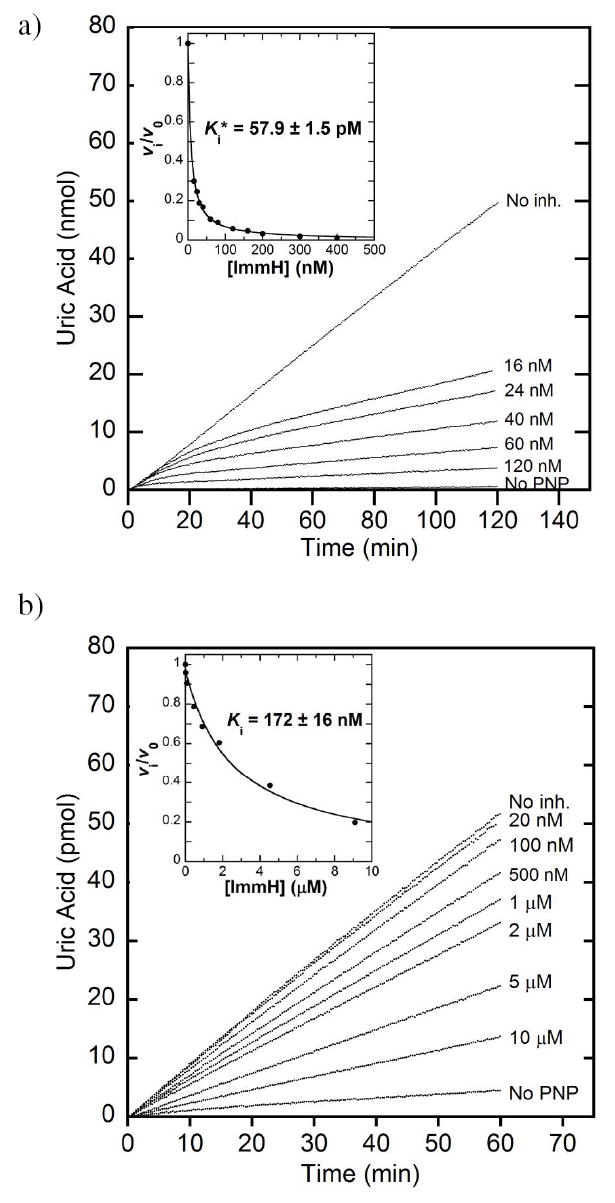
Inhibition of native HsPNP and His257Phe by ImmH. a) Slow-onset inhibition measured for native HsPNP in the presence of ImmH at the concentrations indicated. Hypoxanthine formation is monitored by conversion to uric acid by a xanthine oxidase coupled assay. Inset: The rate during 100–120 min of incubation with inhibitor (vi) relative to that of an uninhibited sample (v0) is plotted against the inhibitor concentration to calculate the dissociation constant, Ki*. Panel b shows the corresponding plots for His257Phe with ImmH.
To verify the importance of the interaction between His257 and the 5′-hydroxyl for tight-binding inhibition with ImmH, 5′-deoxy-ImmH (Figure 3) was tested with HsPNP and the mutant proteins. This inhibitor bound to native PNP with 345-fold less affinity (Ki = 20 nM) than ImmH and did not exhibit slow-onset inhibition (Table 3). However, the native Km/Ki of 2,000 for 5′-deoxy-ImmH is the same as that for His257Phe with ImmH. Intriguingly, the mutant proteins bound 5′-deoxy-ImmH up to nine-fold better than ImmH, perhaps reflecting favorable hydrophobic interactions to the 5′-methyl group.
Structures of His257 Mutants Complexed with DADMe-ImmH and ImmH
Each His257 mutant was crystallized in the presence of DADMe-ImmH and sulfate, a mimic of the phosphate nucleophile. Structures of these mutants, obtained at 2.3-Å resolution, were solved by molecular replacement using the previously obtained structure of the native protein.2 All three are highly similar to native HsPNP, with RMS-deviations of 0.14–0.17 Å in the backbone α-carbons, and with only small differences in the active site (Figure 5a). In mutant and native PNP structures with DADMe-ImmH, the orientation of the 5′-OH of the bound transition state analogue is unchanged. As has been previously observed with the homologous PNP from Mycobacterium tuberculosis (29), the 60° O5′-C5′-C4′-C4″ dihedral angle3 of DADMe-ImmH positions the 5′-OH in a syn orientation with respect to the ring, thereby placing O-5′ within 3.1 Å of C-4″ (Figure 5b). This angle is conserved even in the His257Gly and His257Phe mutants, where no H-bond to O-5′ is apparent. Similarly, this 60° dihedral angle is found in the structure of native HsPNP complexed with inosine (30), indicating that the orientation of the 5′-OH is the same for bound inosine and DADMe-ImmH. The invariant dihedral angle in these bound ligands suggests that the orientation of the hydroxyl may be a preferred geometry for these molecules and may not be altered by the surrounding protein environment. This notion is supported by structural studies on inosine and other nucleosides, in which a syn conformation has been found to be statistically predominant for the molecule free in solution (31-35); however, the energetic barrier to full rotation at this position of inosine is estimated to be less than 9 kcal/mol in water, which is of similar magnitude to the barrier governing cyclohexane chair-flipping (36).
Figure 5.

Crystal structures of HsPNP and His257 mutants complexed with DADMe-ImmH and SO4. a) The superposition of the monomeric structures of the four HsPNP His257 variants reveals very few differences in the position of DADMe-ImmH (cyan) at the active sites. Secondary structural elements are colored red for α-helices, yellow for β-sheets, and green for random coils. b) The dihedral angle between O5′–C5′ and C4′–C4″ in PNP-bound DADMe-ImmH. The model is presented viewing down the C5′–C4′ bond.
The His257 mutants were also crystallized with ImmH and phosphate, and the resulting structures were solved by molecular replacement with the native structure.2 In contrast to the case with DADMe-ImmH, the mutant complexes bind ImmH in differing orientations (Figure 6a). The ImmH-bound His257Gly complex did not differ significantly from native (Figure 6b), though the O5′-C5′-C4′-N4′ dihedral angle closed slightly from 59° to 57° (Figure 7). The His257Phe mutant was found to bind ImmH in a poorly ordered manner (Figure 6d). Electron density was absent for the 5′-hydroxyl group, and the inhibitor could be adequately modeled with the iminoribitol moiety rotated, such that the 5′-hydroxyl points toward the phosphate molecule. Additionally, whereas the phenyl ring of Phe257 was shifted away from DADMe-ImmH (pink in Figure 6d), it was found to have moved closer to ImmH (blue in Figure 6d). The His257Asp mutant (Figure 6c), though maintaining the overall protein structure, greatly altered the orientation of the 5′-hydroxyl, decreasing the dihedral angle to 14° (Figure 7). This places O-5′ directly above N-4′ in a nearly eclipsed geometry reminiscent of the hypothetical “oxygen stack”. The aspartate residue in this mutant may have polarized the 5′-hydroxyl, to increase its electronegativity and promote its interaction with the positively charged N-4′. When the structural changes imposed on ImmH due to mutation of His257 are taken in light of the poor binding of this transition state analogue observed in the inhibition studies, it is clear that His257 is important for the enzyme as the reaction approaches the transition state.
Figure 6.

Crystal structures of HsPNP and His257 mutants complexed with ImmH and PO4. a) The superposition of the monomeric structures of the four HsPNP His257 variants reveals differences in the orientation of ImmH (cyan) at the active sites. Secondary structural elements are colored red for α-helices, yellow for β-sheets, and green for random coils. The overlays of the active site regions of native HsPNP and b) His257Gly, c) His257Asp, and d) His257Phe show small changes in the region surrounding the 5′-hydroxyl of the bound inhibitor. Side chains of selected active site residues within 3.2 Å of ImmH have been included. Carbon atoms of these side chains and of the inhibitor are depicted in green for native HsPNP and in cyan for the mutant proteins. Phosphate molecules bound to the native and mutant variants are colored yellow and black, respectively. Water molecules have been excluded for clarity. e) The superposition of His257Phe·DADMe-ImmH·SO4 (magenta), and His257Phe·ImmH·PO4 (cyan), showing the differences in the positions of Phe257 and the ligand. For the Phe mutant in d) and e), only one of the two most probable orientations of ImmH has been illustrated; in this case, the phosphate ligand cannot be conclusively located.
Figure 7.
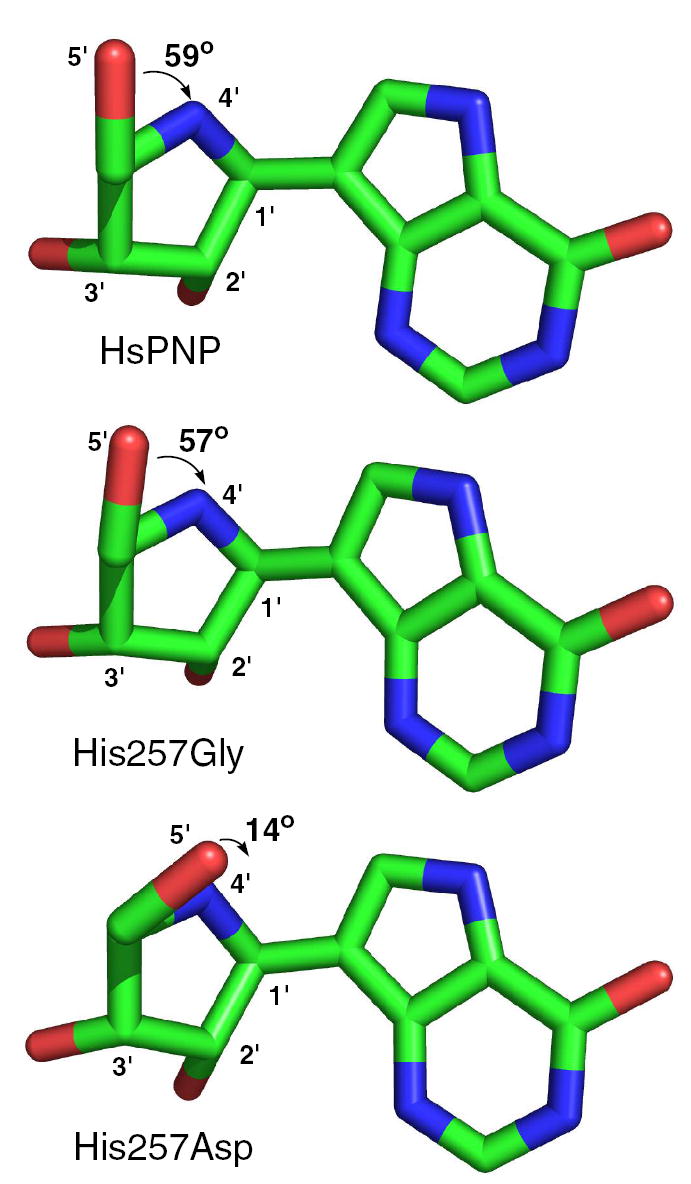
Dihedral angle between O5′–C5′ and C4′–N4′ of ImmH bound to native HsPNP, His257Gly, and His257Asp. The models are presented viewing down the C5′–C4′ bond.
Intrinsic Kinetic Isotope Effects from V/K Kinetic Isotope Effects and Binding Isotope Effects
Kinetic isotope effects have been useful in the elucidation of transition state structures for N-ribosyl transferase reactions (37, 38). Although V/K KIEs are used in determining these structures, they do not differentiate isotopically sensitive binding events from chemistry at the transition state. Binding isotope effects are usually small and are often ignored, especially when considering large isotope effects. However, when secondary deuterium or tritium isotope effects are used for establishing a transition state structure, BIEs are valuable to distinguish bond distortion and polarization upon binding from that occurring in the transition state (39).
The importance of distinguishing BIEs from V/K KIEs is particularly applicable for remote isotope effects. Although KIEs are not expected for atoms that are several bonds removed from the reactive center, remote 5′-3H KIEs have been measured in several N-ribosyltransferases and hydrolases, including TP (4), pertussis toxin, cholera toxin, diphtheria toxin (40-43), nucleoside hydrolase (44), and mammalian PNPs (5, 45). The V/K 5′-3H KIE of 5.4% previously determined for HsPNP was calculated as an “intrinsic” KIE of 6.2% with the assumption that there are no isotopically sensitive steps aside from the chemical event. This large KIE has been attributed to distortions in the vibrational modes of the C5′–H bonds that occur during progression from unbound substrate to the transition state (5). The BIE analysis on TP by Birck and Schramm (2, 4), however, highlights the importance of determining the relative contributions to the V/K KIE that are attributable to isotopic discrimination during formation of the Michaelis complex.
The intrinsic KIE from the chemical nature of the transition state is a function of the V/K KIE and the BIE, both of which can be determined experimentally using appropriate competitive experiments with isotopically labeled substrates (Figure 1). For this scheme, assuming the chemical step is irreversible (i.e., k4 and reverse commitment, Cr = 0), the V/K KIE, symbolized as T(V/K), is related to the equilibrium BIE (TKeq) and the intrinsic KIE (Tk3) by Equation 6 (46, 47):
| (6) |
where Tk1 is the intrinsic KIE for binding and Cf is the forward commitment to catalysis, defined as k3/k2. In cases where Cf is low, the Tk1Cf term in Equation 6 can be approximated within experimental error as Cf,4 yielding Equation 7:
| (7) |
Thus, the intrinsic KIE on the chemical step of interest is obtained through the determination of the V/K KIE, the BIE, and the forward commitment.
5′-3H Binding Isotope Effects and Kinetic Isotope Effects on HsPNP
The competitive binding of [5′-3H]inosine and [5′-14C]inosine to HsPNP was measured in the presence of sulfate, a phosphate analogue that precludes phosphorolysis. Normal BIEs of 2% were determined for the native enzyme, His257Phe, and His257Gly (Table 4), indicating that [5′-1H]inosine binds more tightly than [5′-3H]inosine, and therefore, the vibrational environment of the 5′-hydrogen(s) has become less constrained in the Michaelis complex. In contrast, a small but inverse BIE of −2% was found for His257Asp, indicating increased constraint upon the 5′-hydrogen(s) such that the tritiated substrate is slightly preferred in the Michaelis complex. Note that because the labeled substrate exists as a mixture of R- and S-[5′-3H]inosine, the observed isotope effects are averages of each stereoisomer, and therefore, their origin cannot be assigned to a single molecular orbital.
Table 4.
5′-3H Kinetic Isotope Effect and Binding Isotope Effect Data
| Enzyme | V/K KIE | BIE | Cf | Intrinsic KIE |
|---|---|---|---|---|
| Native PNP | 1.054 ± 0.002a | 1.015 ± 0.003 | 0.147a | 1.046 ± 0.004 |
| His257Phe | 0.992 ± 0.003 | 1.022 ± 0.004 | 0.301 ± 0.010 | 0.968 ± 0.005 |
| His257Gly | 0.925 ± 0.005 | 1.024 ± 0.006 | 0.600 ± 0.021 | 0.859 ± 0.007 |
| His257Asp | 1.046 ± 0.004 | 0.980 ± 0.005 | 0.042 ± 0.002 | 1.069 ± 0.007 |
Value reported in reference (5).
Competitive V/K KIEs and forward commitment factors were measured for the three His257 mutants (Table 4), as performed previously with the native enzyme (5). The intrinsic KIEs for all four enzymes were calculated from the V/K KIEs and BIEs by Equation 7. Native HsPNP and the His257Asp mutant gave large normal intrinsic KIEs of 4.6% and 6.9%, respectively, whereas His257Phe and His257Gly exhibited inverse isotope effects of −3.2% and −14.1%, respectively. These results indicate that although the native enzyme retains its preference for the lighter isotopologue throughout catalysis, the mutants undergo a reversal of preference during progress from the Michaelis complex to the transition state.
Although the origin of the 5′-3H isotope effect may be more complicated than can be explained by present theory, two features at this position have been demonstrated in model systems to contribute to remote isotope effects. These factors are 1) polarization of the 5′-hydroxyl group and 2) its orientation relative to one of the C5′–H σ*-orbitals, both of which cause significant changes in the C5′–H bond length, as illustrated by Lewis and Schramm through computational analysis of an isopropanol/formate model (Figure 8a) (24). They demonstrated that as the H-bonding partners are brought closer together, the O–H bond becomes more polarized, resulting in decreased bond order in the adjacent C–H bond. This causes a normal isotope effect whose magnitude increases as the distance between H-bonding partners decreases. Lewis and Schramm further showed that alteration of the conformational orientation of the hydroxyl group (HCOH torsional angle in isopropanol) causes significant changes in C–H bond stretching and bending force constants (Figure 8b). Depending on the dihedral angle, the oxygen’s lone-pair orbitals may hyperconjugate with the C–H antibonding orbitals, giving rise to normal isotope effects, or they may overlap poorly, leading to inverse isotope effects. Thus, contributions from polarization and hydroxyl orientation may work in concert or opposition to yield normal or inverse BIE and KIE.
Figure 8.
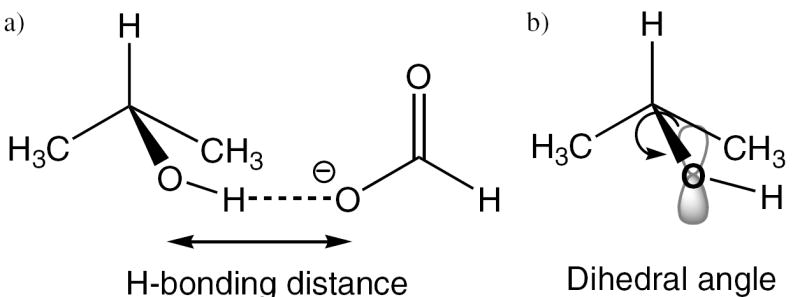
Isopropanol/formate model by Lewis and Schramm (24, 39) to estimate the effects of a) hydrogen-bond distance and b) dihedral angle rotation on the binding isotope effects. Shorter H-bonds cause increased polarization, which enhances hyperconjugation between the oxygen’s lone pair electrons and the antibonding orbital of the adjacent C–H bond. Dihedral angles that increase or decrease overlap of the oxygen’s lone pair orbital with the σ*-orbital give rise to increased or decreased hyperconjugation, respectively, and therefore may result in either normal or inverse isotope effects.
Although the 1.5% BIE found with native HsPNP can be explained by the H-bond to His257, this interaction cannot be invoked for the BIEs of the mutant proteins. His257Gly and His257Phe lack H-bond potential for the 5′-hydroxyl, and although His257Asp retains H-bond capability, it exhibits an inverse BIE. Therefore, the orientation of the 5′-hydroxyl must be a dominant factor in the Michaelis complex. Since the BIEs of native HsPNP, His257Gly, and His257Phe are similar, the dihedral angle between the 5′-HR or 5′-HS and the 5′-O–H bond is proposed to be similar in their respective Michaelis complexes. The generation of the BIE involves increased hyperconjugation relative to unbound inosine in solution. In the case of His257Asp, however, bond polarization is likely the dominant factor, as supported by the ImmH structural data. This would result in poorer n→σ* orbital overlap and the generation of an inverse BIE.
The intrinsic KIEs were larger than the corresponding BIEs, indicating that the V/K KIEs are dominated by transition state chemistry, rather than binding of inosine. This finding is in contrast to TP, where the BIE was found to be equal to the V/K KIE (2). The difference may be related to the SN1-like mechanism involving an oxacarbenium-ion transition state of PNP, while the transition state of TP has SN2 character. These mechanistic distinctions will result in position and H-bonding environment differences for the 5′-hydroxyl.
The intrinsic KIEs for HsPNP and the His257 mutants reveals a dichotomy where enzyme variants capable of H-bonding to the 5′-hydroxyl – native HsPNP and His257Asp – give rise to normal KIEs, while those incapable of H-bonding – His257Gly and His257Phe – result in inverse KIEs. This observation suggests that polarization of the 5′-O–H bond may dominate over other factors. In particular, the dihedral angle of DADMe-ImmH bound at the catalytic site of native HsPNP and all mutants is the same, but the differences in BIE establish differences in vibrational environment and/or bond polarization. Substitution of an aspartate residue, a stronger H-bond acceptor than histidine, results in a larger intrinsic KIE and strengthens this proposal. The inverse intrinsic KIEs observed for the His257Gly and His257Phe mutants can be rationalized by the combined effect of the lack of polarization due to the absence of H-bonding as well as a decrease in electron density at O-5′ during formation of the transition state. As hypothesized in the dynamic mechanism of PNP catalysis (Figure 2), O-5′ is moved toward O-4′ to form an “oxygen stack” that destabilizes electrons on O-4′ and leads to the developing oxacarbenium ion. This donation of electron density from the lone-pair electrons causes a further decrease in hyperconjugation to the 5′-C–H σ*-orbitals, resulting in an inverse KIE.
Conclusions
Accurate resolution of enzymatic transition state structures is important for understanding the reaction mechanism and for the development of transition state analogue inhibitors. Investigation of the interactions between His257 and the 5′-hydroxyl in HsPNP through mutagenesis and subsequent kinetic and structural studies has revealed new information about the remote contributions to catalysis. Further, combined analysis of BIEs and KIEs has enabled the resolution of distortion in the Michaelis complex from intrinsic isotope effects as a result of transition state chemistry. Together, these experiments have provided a more detailed understanding of the enzymatic mechanism (Figure 9). The enzyme approaches the transition state through His257-directed dynamic movement of the 5′-hydroxyl over O-4′ which promotes the departure of hypoxanthine by the movement of electrons toward C-1′. Approach to the dissociative transition state initially proceeds through a point in which the relative electron deficiency of the incipient oxacarbenium ion is localized more at O-4′, and the hypoxanthine is closer to the ribose than the phosphate nucleophile. As the phosphate-ribose distance decreases and the hypoxanthine-ribose distance increases, there is a shift in the positive charge localization to C-1′. These two stages of transition state development are analogous to ImmH and DADMe-ImmH, which are known to mimic early and later transition state structures, respectively. These experiments expand on the nature of intrinsic KIEs and the transition state for PNP. The inhibitory characteristics of the transition state analogues ImmH and DADMe-ImmH are better defined through these results.
Figure 9.
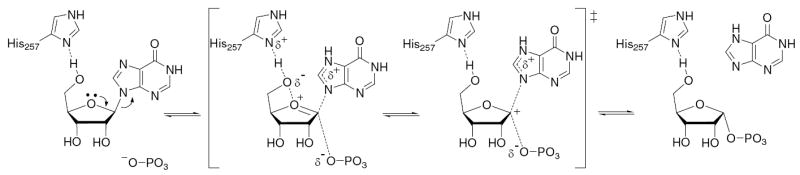
Mechanism of the HsPNP reaction illustrating the role of His257. The H-bond between Nδ of His257 and the 5′-hydroxyl polarizes O-5′, which is steered toward O-4′. This electron density loosens the 4′-oxygen’s lone-pair electrons, which interact with the antibonding orbital of the C-1′–N-9 bond. Further along the reaction coordinate, as the bond to the leaving group is largely broken, significant positive charge density is localized at C-1′. Bond formation to the nucleophile completes the phosphorolysis reaction. N7 of the leaving group has been depicted as being protonated at the transition state, as this has been demonstrated to be a common mechanistic feature in PNP and other nucleoside phosphorylases and hydrolases (37).
Supplementary Material
Calculation of the barrier to 5′-OH rotation in inosine and crystal structures of native and mutant PNPs complexed with DADMe-ImmH and SO4 depicting active site details and closest contacts. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank Drs. Peter Tyler, Richard Furneaux, and Gary B. Evans of Industrial Research, Ltd. (Lower Hutt, NZ) for the synthesis of ImmH and DADMe-ImmH, Dr. Andrzej Lewandowicz for the plasmid encoding native human PNP, and Dr. Suwipa Saen-Oon for computational analysis of the dihedral angles of inosine.
Footnotes
Supported by NIH Research Grant GM41916.
Abbreviations: KIE, kinetic isotope effect; V/K KIE, kinetic isotope effect on the second-order rate constant, kcat/Km; BIE, binding isotope effect; HsPNP, human purine nucleoside phosphorylase; PDB, Protein Data Bank; ImmH, Immucillin-H; DADMe-ImmH, 4′-deaza-1′-aza-2′-deoxy-1′-(9-methylene)-Immucillin-H; RMS, root mean square; TP, thymidine phosphorylase
W. Shi, A. Lewandowicz, P. C. Tyler, R. H. Furneaux, S. C. Almo, and V. L. Schramm, unpublished results.
C-4″ is the carbon linking C-4′ to N-1′. The analogous atom in inosine is the ring oxygen, O-4′.
The unprecedented measurement of a kinetic isotope effect on binding would not significantly change the results and interpretations of the present study. A Tk1 of similar magnitude to the BIEs would result in calculated Tk3 values of 1.044, 0.962, 0.845, and 1.070 for native PNP, His257Phe, His257Gly, and His257Asp, respectively, corresponding to changes of 0.2%, 0.6%, 1.4%, and 0.1%. Thus, for all but His257Gly, which has the largest forward commitment, the assumption of a unity Tk1 is within experimental error.
References
- 1.Northrop DB. The expression of isotope effects on enzyme-catalyzed reactions. Annu Rev Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 2.Birck MR, Schramm VL. Binding causes the remote [5’-3H]thymidine kinetic isotope effect in human thymidine phosphorylase. J Am Chem Soc. 2004;126:6882–6883. doi: 10.1021/ja0492642. [DOI] [PubMed] [Google Scholar]
- 3.LaReau RD, Wan W, Anderson VE. Isotope effects on binding of NAD+ to lactate dehydrogenase. Biochemistry. 1989;28:3619–3624. doi: 10.1021/bi00434a070. [DOI] [PubMed] [Google Scholar]
- 4.Birck MR, Schramm VL. Nucleophilic participation in the transition state for human thymidine phosphorylase. J Am Chem Soc. 2004;126:2447–2453. doi: 10.1021/ja039260h. [DOI] [PubMed] [Google Scholar]
- 5.Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 6.Fedorov A, Shi W, Kicska G, Fedorov E, Tyler PC, Furneaux RH, Hanson JC, Gainsford GJ, Larese JZ, Schramm VL, Almo SC. Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry. 2001;40:853–860. doi: 10.1021/bi002499f. [DOI] [PubMed] [Google Scholar]
- 7.Pugmire MJ, Ealick SE. Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem J. 2002;361:1–25. doi: 10.1042/0264-6021:3610001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nunez S, Antoniou D, Schramm VL, Schwartz SD. Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J Am Chem Soc. 2004;126:15720–15729. doi: 10.1021/ja0457563. [DOI] [PubMed] [Google Scholar]
- 9.McIvor RS, Goddard JM, Simonsen CC, Martin DW., Jr Expression of a cDNA sequence encoding human purine nucleoside phosphorylase in rodent and human cells. Mol Cell Biol. 1985;5:1349–1357. doi: 10.1128/mcb.5.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoeckler JD, Agarwal RP, Agarwal KC, Schmid K, Parks RE., Jr Purine nucleoside phosphorylase from human erythrocytes: physiocochemical properties of the crystalline enzyme. Biochemistry. 1978;17:278–283. doi: 10.1021/bi00595a014. [DOI] [PubMed] [Google Scholar]
- 11.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 12.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 13.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 14.Jones TA. Interactive computer graphics: FRODO. Methods Enzymol. 1985;115:157–171. doi: 10.1016/0076-6879(85)15014-7. [DOI] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- 17.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. 1998;37:8615–8621. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- 18.Kim BK, Cha S, Parks RE., Jr Purine nucleoside phosphorylase from human erythrocytes. I. Purification and properties. J Biol Chem. 1968;243:1763–1770. [PubMed] [Google Scholar]
- 19.Gasteiger EHC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 20.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. 3. Clarendon Press; Oxford: 1986. [Google Scholar]
- 21.Lim M-I, Ren Y-Y, Otter BA, Klein RS. Synthesis of “9-deazaguanosine” and other new pyrrolo[3,2-d]pyrimidine C-nucleosides. J Org Chem. 1983;48:780–788. [Google Scholar]
- 22.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 23.Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. Femtomolar transition state analogue inhibitors of 5’-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli. J Biol Chem. 2005;280:18265–18273. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- 24.Lewis BE, Schramm VL. Binding equilibrium isotope effects for glucose at the catalytic domain of human brain hexokinase. J Am Chem Soc. 2003;125:4785–4798. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 25.Schramm VL. Comparison of initial velocity and binding data for allosteric adenosine monophosphate nucleosidase. J Biol Chem. 1976;251:3417–3424. [PubMed] [Google Scholar]
- 26.Stoeckler JD, Cambor C, Parks RE., Jr Human erythrocytic purine nucleoside phosphorylase: reaction with sugar-modified nucleoside substrates. Biochemistry. 1980;19:102–107. doi: 10.1021/bi00542a016. [DOI] [PubMed] [Google Scholar]
- 27.Erion MD, Takabayashi K, Smith HB, Kessi J, Wagner S, Honger S, Shames SL, Ealick SE. Purine nucleoside phosphorylase. 1. Structure-function studies. Biochemistry. 1997;36:11725–11734. doi: 10.1021/bi961969w. [DOI] [PubMed] [Google Scholar]
- 28.Evans GB, Furneaux RH, Lewandowicz A, Schramm VL, Tyler PC. Synthesis of second-generation transition state analogues of human purine nucleoside phosphorylase. J Med Chem. 2003;46:5271–5276. doi: 10.1021/jm030305z. [DOI] [PubMed] [Google Scholar]
- 29.Lewandowicz A, Shi W, Evans GB, Tyler PC, Furneaux RH, Basso LA, Santos DS, Almo SC, Schramm VL. Over-the-barrier transition state analogues and crystal structure with Mycobacterium tuberculosis purine nucleoside phosphorylase. Biochemistry. 2003;42:6057–6066. doi: 10.1021/bi0343830. [DOI] [PubMed] [Google Scholar]
- 30.Canduri F, dos Santos DM, Silva RG, Mendes MA, Basso LA, Palma MS, de Azevedo WF, Santos DS. Structures of human purine nucleoside phosphorylase complexed with inosine and ddI. Biochem Biophys Res Commun. 2004;313:907–914. doi: 10.1016/j.bbrc.2003.11.179. [DOI] [PubMed] [Google Scholar]
- 31.Rossi C, Picchi MP, Tiezzi E, Corbini G, Corti P. Conformational and dynamic investigation in solution of inosine and its molecular complex, inosiplex, by proton and carbon NMR spectroscopy. Magn Reson Chem. 1990;28:348–354. [Google Scholar]
- 32.Davies DB, Rabczenko A. Assignment and conformational properties of the exocyclic 5’-hydroxymethyl group of nucleosides by nuclear magnetic resonance spectroscopy. J Chem Soc Perkin Trans. 1975;2:1703–1711. [Google Scholar]
- 33.Schweizer MP, Banta EB, Witkowski JT, Robins RK. Determination of pyrimidine nucleoside syn-anti conformational preference in solution by proton and carbon-13 nuclear magnetic resonance. J Am Chem Soc. 1973;95:3770–3778. doi: 10.1021/ja00792a049. [DOI] [PubMed] [Google Scholar]
- 34.Hruska FE, Grey AA, Smith IC. A nuclear magnetic resonance study of the molecular conformation of beta-pseudouridine in aqueous solution. J Am Chem Soc. 1970;92:4088–4094. doi: 10.1021/ja00716a043. [DOI] [PubMed] [Google Scholar]
- 35.Gelbin A, Schneider B, Clowney L, Hsieh S-H, Olson WK, Berman HM. Geometric parameters in nucleic acids: Sugar and phosphate constituents. J Am Chem Soc. 1996;118:519–529. [Google Scholar]
- 36.Sqaillacote M, Sheridan RS, Chapman OL, Anet FAL. Spectroscopic detection of the twist-boat conformation of cyclohexane. A direct measurement of the free energy difference between the chair and the twist-boat. J Am Chem Soc. 1975;97:3245–3246. [Google Scholar]
- 37.Schramm VL. Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch Biochem Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 38.Schramm VL. Enzymatic transition state poise and transition state analogues. Acc Chem Res. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 39.Lewis BE, Schramm VL. Enzymatic binding isotope effects and the interaction of glucose with hexokinase. In: Kohen A, Limbach H-H, editors. Isotope Effects in Chemistry and Biology. CRC Press; Boca Raton, FL: 2005. pp. 1019–1053. [Google Scholar]
- 40.Berti PJ, Blanke SR, Schramm VL. Transition state structure for the hydrolysis of NAD+ catalyzed by diphtheria toxin. J Am Chem Soc. 1997;119:12079–12088. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheuring J, Schramm VL. Kinetic isotope effect characterization of the transition state for oxidized nicotinamide adenine dinucleotide hydrolysis by pertussis toxin. Biochemistry. 1997;36:4526–4534. doi: 10.1021/bi962841h. [DOI] [PubMed] [Google Scholar]
- 42.Parikh SL, Schramm VL. Transition state structure for ADP-ribosylation of eukaryotic elongation factor 2 catalyzed by diphtheria toxin. Biochemistry. 2004;43:1204–1212. doi: 10.1021/bi035907z. [DOI] [PubMed] [Google Scholar]
- 43.Schramm VL. Enzymatic transition-state analysis and transition-state analogs. Methods Enzymol. 1999;308:301–355. doi: 10.1016/s0076-6879(99)08015-5. [DOI] [PubMed] [Google Scholar]
- 44.Kline PC, Schramm VL. Pre-steady-state transition-state analysis of the hydrolytic reaction catalyzed by purine nucleoside phosphorylase. Biochemistry. 1995;34:1153–1162. doi: 10.1021/bi00004a008. [DOI] [PubMed] [Google Scholar]
- 45.Kline PC, Schramm VL. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- 46.Lewis BE, Schramm VL. Enzymatic binding isotope effects and the interaction of glucose with hexokinase. In: Kohen A, Limbach H-H, editors. Isotope Effects in Chemistry and Biology. CRC Press; Boca Raton, FL: 2006. pp. 1019–1053. [Google Scholar]
- 47.Ruszczycky MW, Anderson VE. Interpretation of V/K isotope effects for enzymatic reactions exhibiting multiple isotopically sensitive steps. J Theor Biol. 2006;243:328–342. doi: 10.1016/j.jtbi.2006.06.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Calculation of the barrier to 5′-OH rotation in inosine and crystal structures of native and mutant PNPs complexed with DADMe-ImmH and SO4 depicting active site details and closest contacts. This material is available free of charge via the Internet at http://pubs.acs.org.