

Abstract
Methyl-Coenzyme M reductase (MCR) catalyzes the final step in methane biosynthesis by methanogenic archaea and contains a redox-active nickel tetrahydrocorphin, Coenzyme F430, at its active site. Spectroscopic and computational methods have been used to study a novel form of the Coenzyme, called F330, which is obtained by reducing F430 with sodium borohydride (NaBH4). F330 exhibits a prominent absorption peak at 330 nm, which is blue shifted by 100 nm relative to F430. Mass spectrometric studies demonstrate that the tetrapyrrole ring in F330 has undergone reduction, based on the incorporation of protium (or deuterium), upon treatment of F430 with NaBH4 (or NaBD4). One- and two-dimensional NMR studies show that the site of reduction is the exocyclic ketone group of the tetrahydrocorphin. Resonance Raman studies indicate that elimination of this π-bond increases the overall π-bond order in the conjugative framework. X-ray absorption, magnetic circular dichroism, and computational results show that F330 contains low-spin Ni(II). Thus, conversion of F430 to F330 reduces the hydrocorphin ring but not the metal. Conversely, reduction of F430 with Ti(III) citrate to generate F380 (corresponding to the active MCRred1 state) reduces the Ni(II) to Ni(I), but does not reduce the tetrapyrrole ring system, which is consistent with other studies (Piskorski, R. and Jaun, B. (2003) J. Am. Chem. Soc. 125:13120-5 and Craft, J. L. et al. (2004) J. Biol. Inorg. Chem. 9:77-89). The distinct origins of the absorption band shifts associated with the formation of F330 and F380 are discussed within the framework of our computational results. These studies on the nature of the product(s) of reduction of F430 are of interest in the context of the mechanism of methane formation by MCR and in relation to the chemistry of hydroporphinoid systems in general. The spectroscopic and time dependent DFT calculations add important insight into the electronic structure of the Ni-hydrocorphinate in its Ni(II) and Ni(I) valence states.
Introduction
Methyl-coenzyme M reductase1 (MCR) catalyzes the final step in methane formation from methyl-coenzyme M (methyl-SCoM, 2-(methylthio)ethanesulfonate) and coenzyme B (CoBSH, N-7-mercaptoheptanoylthreonine phosphate) according to Eq. 1 (1). In this reaction, CoBSH serves as the electron donor (2) for the two-electron reduction of methyl-SCoM to produce CH4 and the mixed disulfide CoBS-SCoM. MCR contains Coenzyme F430 (Figure 1), a redox-active nickel tetrahydrocorphin, at its active site (3-5). Coenzyme F430 is the most reduced tetrapyrrole in nature, containing only five double bonds in the macrocycle. X-ray crystallographic studies of MCR revealed that F430 forms the base of a narrow well that accommodates the two substrates and shields the reaction from solvent (6). All methanogens contain F430 (7) and MCR is the only enzyme known to date to use this Coenzyme.
Figure 1.
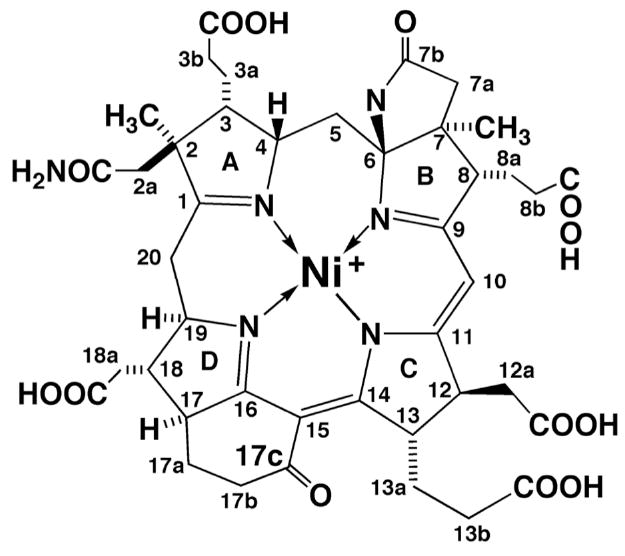
Molecular structure of F430. The formal oxidation state of the Ni ion in F430 is 2+.
| (1) |
F430 requires reductive activation to form a state called MCRred1 that is capable of entering the catalytic cycle. (8). In vitro, MCR is converted to MCRred1 by chemical reduction of the MCRox1 state with Ti(III) citrate at pH 10.0 (8, 9), while, in vivo, the red1 state can be generated by treating cells with 100% H2 prior to harvest (10). MCRred1 has been assigned as a Ni(I) species based on the similarity of its UV-visible and EPR spectra with the Ni(I) species produced when F430 is reduced by Ti(III) citrate (11) or when its pentamethylester is reduced by sodium amalgam (12). While Coenzyme F430 is EPR silent, MCRred1 is characterized by an EPR spectrum with g-values at 2.25, 2.070, and 2.060 (13-15), which is typical of an approximately square planar Ni(I) system with one unpaired electron in the dx2-y2 orbital (11, 16, 17). Coenzyme F430 exhibits a UV-visible spectrum with an absorption peak near 430 nm (431 nm for the free Coenzyme in water (18) and 422 nm for the MCR-bound Coenzyme (19)). Magnetic circular dichroism (MCD) spectroscopic studies clearly demonstrate that at low temperature F430 is a high spin (S = 1) Ni(II) species (20-22) A significant blue-shift of the dominant absorption feature is observed when the Coenzyme is reduced to the Ni(I) state (λmax = 378 nm for the free Coenzyme in water (11) and 388 nm for the MCR-bound Coenzyme (9)). Under the conditions described here, the absorption peak of the Ti(III)-reduced Coenzyme is at 376 nm, so we refer to this state as F380.
Although the assignment of MCRred1 and F380 as Ni(I) species is unambiguous, several results indicated the possibility that the tetrapyrrole ring could undergo reduction. Results of potentiometric titration experiments with Ti(III) citrate revealed that conversion of the Ni(II) state of F430 to F380 involves a 1- to 3-electron reduction (23) The exact number of electrons transferred was ambiguous because Ti(III) citrate was found to be unstable under the titration conditions. Resonance Raman (RR) spectroscopic studies demonstrated that conversion of Ni(II) to the Ni(I) state leads to marked changes in the vibrational spectrum (23). These changes include the disappearance or extensive shift of a band that is sensitive to 14N→15N isotopic substitution, which was interpreted to indicate reduction of a C=N double bond in the corphin ring system. Based on the electrochemical and RR studies coupled to the observation of a ~50 nm blue shift in the UV-visible spectrum upon conversion of F430 to F380, it was proposed that the red1 state contains Ni(I) and a two-electron reduced tetrapyrrole ring (23). However, several recent studies are inconsistent with this proposal. Piskorski and Jaun used the pentamethyl ester form of F430 (F430M) in electrochemical studies to show that conversion of Ni(II)-F430 to F380 is a 1-electron process, thereby indicating only metal-centered reduction (24). Furthermore, recent absorption and magnetic circular dichroism (MCD) spectroscopic and computational studies of Coenzyme F430 in solution and bound to the MCR active site revealed that a metal-centered one-electron reduction of F430 generates the F380 species, characteristic of the active red1 state (21, 25).
In the course of our studies on the reduced states of F430, we discovered that reduction of F430 with sodium borohydride yields a new species with an absorption peak at 326 nm (called F330). The objectives of the work described here were to use spectroscopic and computational studies to better understand the factors that govern metal versus tetrapyrrole ring reduction and to establish whether or not the tetrapyrrole ring is reduced when F430 is converted to F380 and/or F330. Our results indicate that the conversion of F430 to F380 involves a metal-centered one-electron reduction, while the conversion to F330 involves tetrapyrrole ring reduction and alters the metal spin state, but does not affect the metal oxidation state.
Experimental Details
Preparation of Enzyme and Growth Conditions
All manipulations of the enzyme and Coenzyme F430 were performed in a model PC-1-SSG anaerobic chamber (Vacuum Atmospheres Co., Hawthorne, CA) maintained below 5 ppm oxygen. The chamber is equipped with a S2000 Miniature Fiber Optic Spectrometer (Ocean Optics, Inc., Dunedin, FL). Media were prepared as previously described (26). For 15N-labeled F430, Methanothermobacter marburgensis was grown in the presence of 15NH4Cl (Cambridge Isotope Laboratories, Andover, MA) in the growth medium. MCR was purified from Methanothermobacter marburgensis (27) as described earlier (8, 9, 28). Following purification, MCR was concentrated under argon (passed through an Oxisorb (Alltech, Deerfield, IL) column) under 30 PSI pressure in a 50 mL omegacell (Pall, East Hills, NY) with a YM50 50 kDa molecular mass cutoff filter (Millipore, Billerica, MA).
Preparation of F430, F380, and F330
F430 samples were prepared from purified MCR as follows. MCR was titrated with 30% (v/v) HCl to lower the pH below 1.0, which precipitated the protein, releasing F430 into solution. The F430 content was estimated using an extinction coefficient at 430 nm of 23,300 M-1cm-1 (29). Finally, F430 was purified on a Waters C18 μBondapak column (7.8 mm × 30 cm) controlled by an Agilent 1100 HPLC system (Agilent Inc., Palo Alto, CA). The column was equilibrated with water and developed with a 50 min linear gradient; 100 % H2O to 40 % methanol; 2 mL/min. The eluate was monitored at 420 nm, 430 nm and 560 nm. F430 had a retention time of 25 min and was collected directly from the HPLC anaerobically in 9 mL glass serum vials (Alltech, Deerfield, IL) sealed with butyl rubber stoppers (Bellco Inc., Vineland, NJ). Pure F430 was lyophilized and stored at -20 °C.
F380 was prepared chemically, by reducing F430 with Ti(III) citrate at pH 10.0. (11). The F330 form was also prepared chemically, by reducing F430 with either NaBH4 or NaBD4 (as indicated) at pH 10. The formation of the reduced species was confirmed by absorption spectroscopy.
The rate of decay or formation of F430 or one of its reduced derivatives was determined by UV-visible spectroscopy by fitting the data to Equation 2 using Sigma Plot (Systat Software, Inc., Point Richmond, CA), where 1/b is the rate constant, y is the absorption value, y0 is the offset from zero absorption, x is the time, x0 is the time at which the absorption has changed by 50%, and a is the maximum absorption amplitude.
| (2) |
Mass Spectrometry
All mass spectrometry (MS) samples, with the exception of F380, were prepared in a mixture of water (or deuterium oxide):acetonitrile (50:50) + 0.1% concentrated formic acid (Solution A). F330 was loaded onto a 10 μL μ-C18 ZipTip (10μL pipette tips with ~1μL of C18 resin bonded at the bottom) pipette tip (Millipore) as specified by the manufacturer, but with the following modifications: the column was washed five times with 10 μL of water to desalt the sample and eluted from the ZipTip using solution A. F380 samples were studied as prepared according to the Results section. All samples were analyzed on an Applied Biosystems QSTAR LX hybrid quadrupole-TOF LC/MS/MS system equipped with a NanoSpray ion source fitted with a 2 μm 1P-4P coated GlassTip PicoTip emitter (New Objective, Woburn, MA.).
NMR Spectroscopy
All NMR data were acquired on a Bruker Avance DRX 500 MHz NMR instrument (Bruker Biospin Corp., Billerica, MA) equipped with a TXI cryoprobe in the UNL Chemistry Department. In each case, D2O was used as the solvent and the temperature was maintained at 298 K throughout the data acquisition. A one-dimensional (1D) proton NMR spectrum was acquired both before and after each two-dimensional (2D) acquisition to ensure that the sample did not undergo oxidation during the length of the 2D experiment. Furthermore, at the conclusion of the NMR experiment, a UV-visible spectrum was measured to assess the extent of oxidation. In each of these 1D experiments, 4096 scans were performed at a resolution of 64k data points and an acquisition time of 3.17 s. A gradient HMQC experiment was performed with 1024 scans and a resolution of 1024 × 128, with an acquisition time of 0.12 s in F2 (the directly detected axis), and the time data were detected on the proton axis. A gradient COSY experiment was also performed, with 1024 scans and a resolution of 2048 × 128, with an acquisition time of 0.13 s in F2. Gradient HMQC and Gradient COSY techniques are typically used because they increase signal-to-noise relative to the non-gradient methods. Samples were prepared in the anaerobic chamber as indicated (see Figure Legends) and placed in J. Young valve NMR tubes (Norell, Landisville, NJ.) under an atmosphere of argon gas that was passed through an Oxisorb column.
Resonance Raman Spectroscopy
The resonance Raman (RR) measurements were performed on samples of F430, F380, and F330 in tightly sealed 1 mm (internal diameter) capillary tubes. Spectra were obtained at a variety of temperatures ranging from cryogenic (150 K) to ambient in efforts to maximize the S/N ratio. For the cryogenic studies, the capillary tubes were mounted directly onto the cold tip of a closed-cycle liquid He refrigeration system (ADP Cryogenics DE-202).
The RR spectra were acquired with a triple spectrograph (Spex 1877) equipped with a holographically etched 2400 grooves/mm grating in the third stage. Laser excitation at the desired wavelengths was achieved using the discrete outputs of a krypton ion (Coherent Innova 200-K3) or an argon ion (Coherent Innova 400-15 UV) laser. The scattered light was collected in a 90° configuration using a 50 mm f/1.4 Canon camera lens. A UV-enhanced charge-coupled device (CCD) was used as the detector (Princeton Instruments LN/CCD equipped with an EEV1152-UV chip). For F430 and F380, the data acquisition times were ~2 h (60 × 120 s frames) and the laser power at the sample was ~8 mW; for F330 the data acquisition times were ~ 5 h (120 × 150 s frames) and power at the sample was ~13 mW. Cosmic spikes were removed manually prior to the numerical addition of the data sets. The spectral resolution was ~2 cm-1. The frequencies were calibrated using the known vibrational frequencies of indene.
Low-Temperature Absorption and MCD Spectroscopy
Variable-temperature (4 – 100 K) electronic absorption and MCD spectra were collected on a spectropolarimeter (Jasco J-715) in conjunction with a magnetocryostat (Oxford Instruments SM-4000 8T). All MCD spectra were obtained by subtracting the −7T spectrum from the +7T spectrum to eliminate contributions from natural CD.
X-ray Absorption Spectroscopy (XAS)
Ni K-edge XAS data were measured at the Stanford Synchrotron Radiation Laboratory (SSRL) on the 16-pole wiggler beamline 9-3. The monochromator employed dual Si(220) crystals together with a 10 keV cutoff mirror to minimize higher harmonic components. Data were collected in fluorescence mode using a Canberra 30-element Ge-detector. Soller slits and a Co filter were used to minimize contributions from scattered X-rays. X-ray energy calibration data were simultaneously measured on a nickel foil mounted between two ionization chambers positioned after the sample, the first inflection point being assigned to 8331.60 eV. Samples were contained in 10 × 3 × 2 mm mylar cuvettes with one kapton tape window. These were anaerobically loaded using a Vacuum Atmospheres glove box (O2 < 0.5 ppm) and they were immediately frozen and stored in liquid nitrogen until measured. The sample temperature during data collection was maintained at 8 K using an Oxford Instruments CF1208 continuous flow liquid helium cryostat fitted with aluminized mylar windows. Data processing employed the EXAFSPAK suite of programs (30). Each spectrum was at least an average of two scans. The individual scans were compared to ensure that there were no changes due to radiation damage. For each spectrum, a polynomial background function was fitted to the pre-edge and subtracted from the entire spectrum. The data were then normalized to an intensity of 1.0 at 8410 eV.
Computations
Coenzyme Models
As our NMR, MCD, and XAS data indicated that F330 is a diamagnetic species (vide infra), the computational models employed included 4-coordinate Ni(0) and low-spin Ni(II) species. Three possible reduction sites were considered in preliminary density functional theory (DFT) calculations: the metal center (resulting in a Ni(0) species), the C16=N bond associated with ring D, and the peripheral C17c=O carbonyl moiety adjacent to ring D (Figure 1). The coordinates of a previously published, geometry-optimized model of F430 (1) (25) were used as a starting point in the set of computations leading to the F330 models 2, 3, 4a, and 4b (Figure 2). These truncated models of Coenzyme F330 neglect the lactam ring as well as the ring substituents for computational practicality. Previous studies have shown that omitting the lactam ring has minimal effects on the geometry of the hydrocorphin ring (31). Nonetheless, additional computational studies were carried out on the complete Coenzyme using combined quantum mechanical/molecular mechanical (QM/MM) methods. The initial geometry was derived from the crystal structure of 12,13-diepi-F430 pentamethyl ester (32), the hydrocorphin ring was modified to reflect the stereochemistry of Coenzyme F430, and the five ester bonds on the substituents of rings A, B, C, and D were converted to carboxylic acids.
Figure 2.
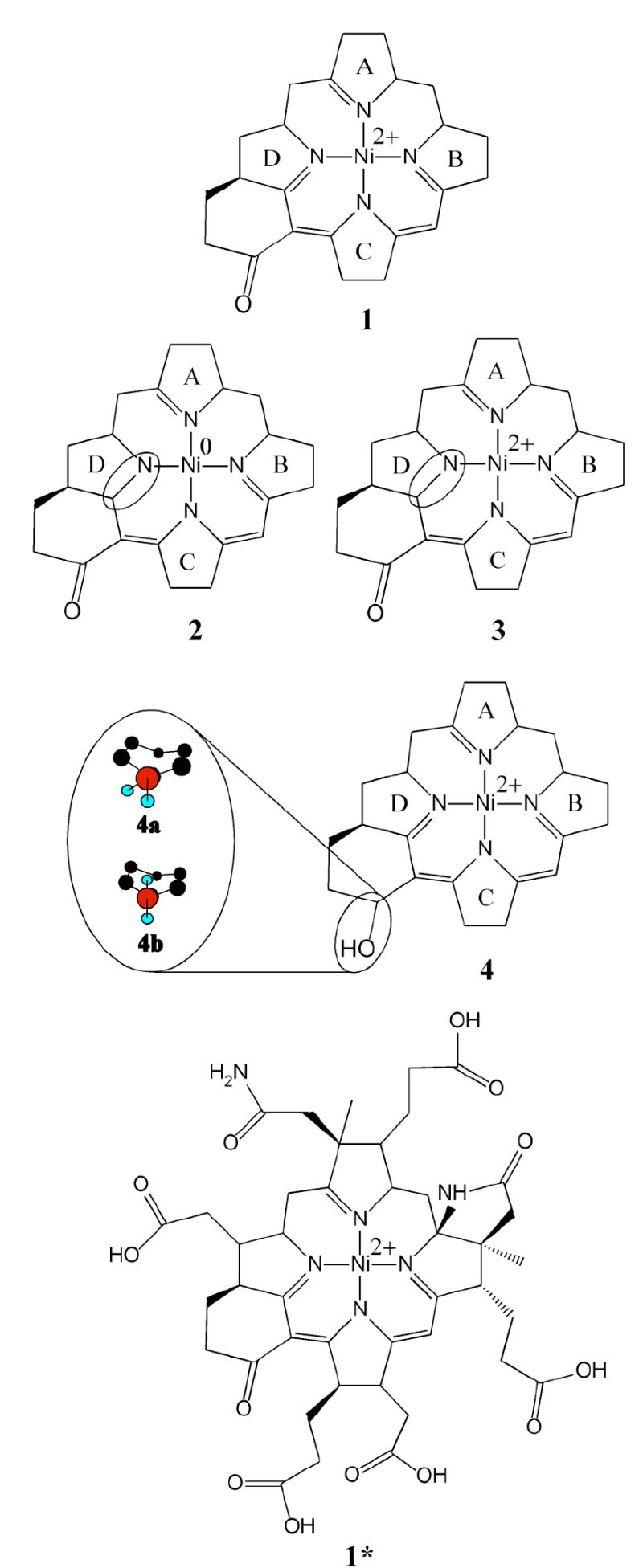
Models of F430 (1 and 1*) and its reduced derivative F330 (2, 3, and 4; hydrocorphin reduction sites are highlighted by an oval) employed for computational studies. The complete Coenzyme model 1* was used as the basis for generating 3* and 4* (not shown). Note that all models contain a low-spin Ni(II) center except for 2, which is a Ni(0) species.
Geometry Optimizations
Pure QM Computations
The Amsterdam Density Functional (ADF) 2004 suite of programs (33-36) was used to refine the atomic positions of models 1, 2, 3, and 4 by DFT energy minimization. These geometry optimizations were performed on a cluster of 20 Intel Xeon processors (ACE computers) using ADF basis set IV, an integration constant of 3.0, and the Vosko-Wilk-Nusair local density approximation (37) (VWN-LDA) with the nonlocal gradient corrections of Becke for exchange (38) and Perdew for correlation (39). Core electrons were frozen through 1s for C, N, and O, and through 2p for Ni.
QM/MM Computations
Based on our results from pure QM optimizations (vide infra), QM/MM computations were performed on three models of the complete Coenzyme; i.e., the Ni(II) F430 model 1* and the Ni(II) F330 models 3* and 4* corresponding to the truncated models 1, 3, and 4a, respectively (Figure 2). The QM region was chosen to include the hydrocorphin core, the lactam ring, and the two methyl groups attached to carbons 2 and 7 (Figure 1), as well as a hydrogen link cap atom in place of a carbon from each side chain. A scaling factor of 1.376 was applied to the six C–C link bonds. All ring substituents were treated at the MM level of theory. The QM/MM geometry optimizations were also performed on a cluster of 20 Intel Xeon processors (ACE computers) using ADF (33-36). For the QM region, ADF basis set IV was employed, along with the VWN-LDA (37) the nonlocal gradient corrections of Becke for exchange (38) and Perdew for correlation (39) and an integration constant of 4.0. The geometry convergence criterion was set at 0.001 Hartree/Å. Core electrons were frozen through 1s for C, N, and O, and through 2p for Ni. The MM region was treated with the AMBER95 force field using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) Hessian update scheme and simple electrostatic coupling to the QM region (40).
TD-DFT Calculations
For time-dependent DFT (TD-DFT) computations, the QM-optimized geometries of 1, 2, 3, 4a, and 4b were used as obtained, while the QM/MM optimized models 1*, 3*, and 4* were suitably truncated. Specifically, in the latter structures the propionate substituents of rings A, B, and C, the acetate substituents of rings C and D, and the acetamide substituents of ring A were replaced by hydrogens. Additionally, the lactam ring was removed, as TD-DFT computations that included the entire QM region failed to converge.
All TD-DFT calculations were performed using the ORCA 2.4.02 software package developed by Dr. Frank Neese (41) Becke’s three-parameter hybrid functional for exchange (42, 43) and the Lee-Yang-Parr functional for correlation (44) (B3LYP/G) were used with the polarized split valence (SV(P)) basis (45) and the SV/C auxiliary basis (46) for all atoms except Ni, for which Ahlrichs’ polarized triple- ζ valence polarization (TZVP) basis (47) was used instead. 50 excited states were calculated for each model with the exception of 2, for which 60 excited states were considered. The effects of solvation on the TD-DFT-computed transition energies and intensities were accounted for by employing the conductor-like screening model (COSMO) with water as the solvent (dielectric constant ε=80.4, refractive index n=1.33) (48). Predicted electronic absorption spectra were generated using the TD-DFT/COSMO results and assuming that each electronic transition gives rise to a Gaussian band with a full width at half maximum of v1/2 = 1700 cm-1. The natures of key electronic transitions were analyzed on the basis of isosurface plots of molecular orbitals (MOs) and electron density difference maps (EDDMs), which were obtained with the gOpenMol program developed by Laaksonen (49) using isodensity values of 0.03 au and 0.003 au, respectively.
Results
Conversion of F430 to F380 and F330 and Stability of F380 and F330
Coenzyme F430 was isolated from purified MCR in order to obtain the pure native Coenzyme and not its 12,13-diepimer. The ratio of absorbance at 430 nm/275 nm was 0.920, confirming that the Coenzyme was indeed in its native state since this ratio is 1.05 for native F430 and 1.2 for the diepimer (50). As described in the Introduction section, previous experimental studies suggested that both the Ni center and the tetrapyrrole ring of Coenzyme F430 can undergo reduction. Reduction of Coenzyme F430 with Ti(III) citrate generates a Ni(I) state, which we call F380, while reduction with sodium borohydride (NaBH4) yields a new species with an absorption peak at 326 nm, termed F330, which has not been previously described (Figure 3).
Figure 3.
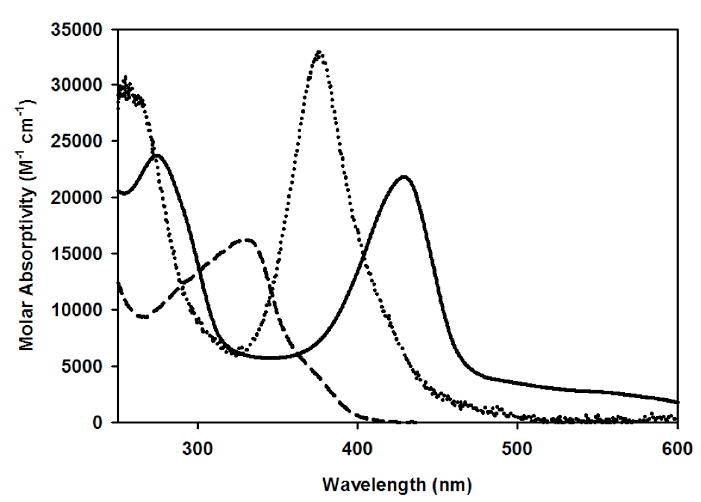
Electronic absorption spectra of F430, F380, and F330 at 300 K. Solid line: spectrum of F430; 20 μM Ni(II)-F430 in 50 mM TAPS pH 10.0. Dotted line: spectrum of F380; 14 μM F430 was converted to F380 in the presence of 70 µM Ti(III)citrate in 25 μM NH4OH at pH 10.5. Dashed line: spectrum of F330; 270 μM F430 was converted to F330 in the presence of 14 mM NaBH4 and 100 mM NH4OH at pH 10.5.
Several spectroscopic methods were used in an attempt to determine whether or not reduction of F430 to F380 and/or F330 involves tetrapyrrole ring reduction. Potentially the most unambiguous methods to address this issue, NMR spectroscopy and mass spectrometry (MS), are also the most challenging because the reduced F430 states are labile and must be maintained intact throughout the time course of the experiment. For two-dimensional NMR studies, this can require that the reduced state be preserved for as long as 36 hours.
Experiments were performed to establish optimal conditions for generating and maintaining F380 and F330. When 26 μM F430 was treated with 0.6 mM Ti(III)citrate (23-fold excess) in 0.5 M TAPS buffer (pH 10.0) at 25 °C, the 432-nm peak due to the Ni(II) F430 state decayed at a rate constant of 0.21 ± 0.05 min-1 concomitant with the development of the 378-nm feature of F380 (k = 0.27 ± 0.05 min-1). Under these conditions, F380 was relatively stable in the anaerobic chamber, converting back to F430 with a rate constant of 0.0082 ± 0.0005 min-1. However, the concentration of Ti(III) citrate used in these experiments was too high for mass spectrometric analysis. Furthermore, incubation of F430 with high concentrations of Ti(III) for extended time periods converts F380 to F330. On the other hand, some Ti(III) was clearly required to prevent oxidation of F380 even in an anaerobic chamber maintained at < 5 ppm O2, as the redox potential for the Ni(II)/(I) couple of F430 is -440 mV (14). Hungate pointed out that, in the absence of a reductant, even a mildly reducing potential of -0.33 V can only be achieved when the oxygen concentration in solution is less than 10-56 molecules per liter (51) Thus, we performed studies to determine both the optimum pH and the minimum amounts of Ti(III) that could be used to maintain F380.
F380 stability is pH dependent (24). When the pH of a solution containing F380 was lowered from pH 10.8 to 9.2, ca. 70% of F380 converted to F430 within 10 minutes (Figure S1, Supplementary Information). This rate of reversion is 20 times faster than the rate of conversion of F380 to F430 at pH 10.8 (Figure S2). The pH-dependent conversion of Ni(I) to Ni(II) is not reversed by adding base; for example, the spectrum of F430, generated by decay of F380 at pH 9.2, does not change when the pH is raised to 10.8 by titrating with base.
Based on the above experiments, the inclusion of Ti(III) citrate and high pH values are required to prevent the reversion to F430. Thus, we maintained the pH between 10.0 and 10.5 and lowered the Ti(III) citrate concentrations as much as possible to maintain F380 during the MS data collection. A 5-fold excess of Ti(III) was sufficient to generate and retain the reduced state of F380 during the mass spectrometric analysis, while a 10-fold excess slightly decreases the signal/noise ratio.
Further precautions included flushing the injection loop with anaerobic water:acetonitrile (50:50) prior to injection and washing the injection needle with the same mixture before introducing the sample into the injection needle. The electronic absorption spectra obtained of a sample before and of a control sample after the MS experiment showed that only 18% loss of F380 had occurred during the mass spectrometric analysis (Figure S3).
While reduction of F430 with Ti(III) citrate generates F380, treatment of F430 with NaBH4 results in an even more dramatic blue shift of the main UV-visible absorption feature from 430 to 330 nm, signaling the formation of F330 (Figure 3). We also optimized conditions for generating and maintaining F330 during the mass spectroscopic and NMR experiments (vide infra).
Mass Spectrometric (MS) Studies
After establishing conditions for reducing and maintaining F380, MS experiments were performed in the positive and negative ion modes (PIM and NIM, respectively) at pH 10.5 where F380 is most stable. The molecular formula of F430 is C42H51N6O13Ni, which corresponds to an exact mass of 905.2868 in full agreement with the mass spectrometric results.
In the PIM, the major peak for the singly charged form of F430 (Figure 4) has an m/z value of 905.3 (29). This corresponds to the expected molecular ion with Ni in the 2+ state and the tetrahydrocorphinoid ligand with a -1 charge (Figure 1). It is difficult to detect the doubly charged form of F430 (Figure 4, inset), which exhibits approximately 3% of the relative intensity of its singly charged counterpart. Presumably, the doubly charged form corresponds to the protonation of one of the nitrogens on the ligand, which has been observed in the doubly charged forms of porphyrins and their analogs (52). Thus, the expected m/z value should be 453.2, i.e., (905.3 + 1)/2, consistent with the experimentally observed value.
Figure 4.
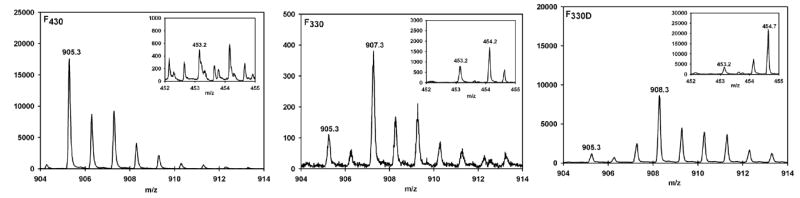
PIM MS spectra of the singly charged F430 (left, 20 μM final concentration), F330 (center) and F330D (right) species. Approximately 80% conversion of F430 to F330 and F330D was achieved by reacting 42 μM F430 with 26 mM NaBH4 or 26 mM NaBD4, respectively, in 60 mM NH4OH. Insets: PIM MS spectral features associated with the doubly charged F430 (left), F330 (center) and F330D (right) species
Mass spectrometry was also used to determine the molecular mass of F380. F380 is difficult to detect in the PIM in either the singly or doubly charged form (not shown), though it is readily detected in the NIM (as described below, Figure 5). Assuming that the ligand remains in the same charged state, the conversion of F430 to F380 by addition of one electron to the Ni ion should generate a neutral molecule, which would not be detected using the PIM. The intensities of the peaks generated from the F380 sample are about 1% of those from the F430 at matched concentrations (not shown). We suspect that those low intensity peaks may actually be due to a small amount of F430 remaining in the F380 sample. If Ti(III) citrate were to reduce a double bond of the tetrapyrrole ring of F430 during formation of F380, the major 905.3 peak would increase by +2 m/z units to 907.3.
Figure 5.
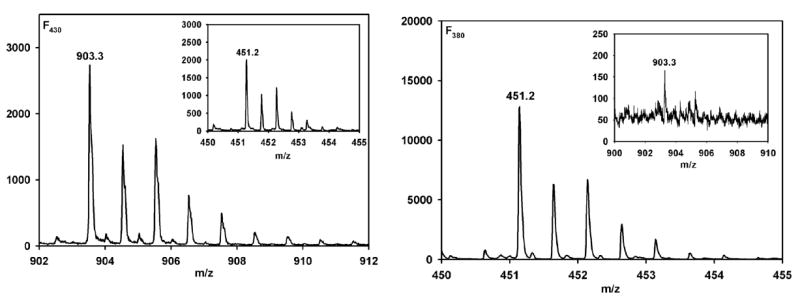
NIM MS spectrum of single and double charged regions for F430 and F380 respectively. (Left) A 20 μM solution (final concentration) of F430 was prepared for analysis in 50% ACN. Inset: Double protonated region for F430. (Right) Conversion of F430 to F380 was achieved by reacting 25 μM F430 with 125 μM Ti(III) citrate in 63 mM NH4OH at pH 10.5, after conversion the volume was raised by 20% by the addition of 100% anaerobic ACN. Inset: Single charged region for F380.
The NIM is ideal for MS analysis of F380 and F430 (Figure 5). In the NIM, the doubly charged forms of both F430 and F380 produce significant contributions to the corresponding spectra. Specifically, in the case of F430 (Figure 5, left), the singly charged and doubly charged species contribute equally to the spectrum, while for F380 (Figure 5, right), the spectrum is dominated by the doubly charged species. This pattern is quite different from that of F430, suggesting F380 is indeed observed in the mass spectrum. For both F430 and F380, the major peaks are observed at 903.3 (905.3-2), as expected for the loss of 2 protons from the carboxyl groups appended to the tetrahydrocorphinoid ring2 and 451.2 (expect 903.3/2 = 451.6) m/z units for the singly and doubly charged species, respectively. That the major form of the F380 sample is the doubly charged species strongly indicates that Figure 5 (right) is the mass spectrum of F380, since one electron reduction of Ni would increase the negative charge on the complex to -2 without affecting the protonation state of the tetrapyrrole ligand (i.e., in F430 and F380, the tetrahydrocorphin would have a net -3 charge and the Ni would be +1). These results provide the most direct evidence to date that conversion of F430 to F380 does not involve reduction of a double bond on the hydrocorphin ring.
While reduction of F430 with Ti(III) citrate to generate F380 causes a significant blue-shift of the dominant absorption feature from 430 to 380 nm, treating F430 with NaBH4 results in an even more dramatic blue-shift of this feature to 330 nm, yielding F330 (Figure 3). To determine if this UV-visible spectral perturbation associated with F430 conversion to F330 (Figure 3) results from metal- or hydrocorphin-centered reduction, we compared the mass spectrum for F430 with those of samples treated with either NaBH4 (Figure 4, center) or NaBD4 (Figure 4, right). We took the same precautions described above for the analysis of F380 except that we were able to remove the reductant (NaBH4), since F330 is more stable than F380. In the PIM MS spectral region associated with the singly protonated species, the major peak shifts by +2 m/z, from 905.3 to 907.3, when F430 is reacted with NaBH4 and by +3 m/z, to 908.3, when F430 is reacted with NaBD4. Similarly, the major peaks associated with the doubly charged products formed upon NaBH4- and NaBD4-treatment of F430 (insets of Figure 4, center and right), which we call F330 and F330D, occur at m/z values of 454.2 and 454.7, respectively. Multiplying these values by two and subtracting one yields m/z ratios of 907.3 for F330 and 908.3 for F330D. The doubly charged forms of F330 and F330D contribute approximately 72-88% of the total intensity, corresponding to an ~25-fold increase in the fraction of doubly charged species formed in these experiments relative to those involving F430 (Figure 4, left).
The +2 unit increase in mass when F430 is treated with NaBH4 and the +3 unit increase upon treatment with NaBD4 in water indicate that two hydrogens are incorporated upon conversion of F430 to F330 and that one of the hydrogens is introduced from the solvent and one is not. This result shows that transfer of one nonexchangeable hydride (or deuteride) and one solvent-introduced proton (or deuteron) occurs when F430 is converted to F330 (or F330D). We also performed reduction of F430 with NaBD4 in D2O, expecting that under these conditions an increase in m/z of 4 units should be observed, since a deuteride would come from NaBD4 and a deuteron from the solvent. However, even when F430 itself is placed in D2O, the peaks at m/z 905.3 are replaced with a heterogeneous mixture of peaks that appear around m/z of 914 (Figure S4), indicating that several protons associated with the Coenzyme can exchange with the deuterons in the solvent, making it difficult to analyze the mass spectrometric data for the reduction with NaBD4. These results strongly indicate that NaBH4 (or NaBD4) reduces a C=N or C=O double bond of the tetrapyrrole ring of F430, which would result in hydride (or deuteride) uptake on carbon and protonation of N or O, respectively.
NMR Studies
One-dimensional (1D) and two-dimensional (2D) NMR spectroscopy was used to obtain unambiguous 1H and 13C spectral assignments for F330 and, thereby, identify the site of hydride transfer to the tetrapyrrole ring during reduction of F430 by NaBH4. All experiments were carried out in a sealed tube using a 1.0 mM sample in D2O, and special attention was paid to ensure the stability of the sample during the experiment (see Materials and Methods). To confirm that F330 had not undergone oxidation (or further reduction) during data collection, the electronic absorption spectrum was measured after the NMR experiment; and indeed this spectrum was found to be almost identical to that collected before the experiment. Consistent with a previous report (53), our NMR data indicate that at room temperature F430 exists as a mixture of low-spin and high-spin Ni(II) species, which results in a paramagnetic shift and broadening of all the resonances. In contrast, F330 exhibits the sharp lines characteristic of a diamagnetic species, which facilitates the assignment of signals. Experiments involving 1H-detected heteronuclear multiple quantum coherence (HMQC) via direct proton-carbon one-bond coupling were used to assign the carbon signals. The HMQC method is extremely useful for assigning geminal proton signals since both 1H signals correlate with a single-carbon frequency. Proton resonance assignments of atoms that are interacting via through-bond scalar coupling were established by homonuclear correlated spectroscopy (COSY). The 1D 1H NMR and 2D HMQC and COSY experiments were performed on samples of F430 reduced with NaBH4 or NaBD4. In the latter case, it is the absence of a particular signal that identifies the site of double-bond reduction. The 1H and 13C signal assignments based on 1D and 2D NMR experiments are listed in Tables 1 and 2 (see Figure 1 for the numbering scheme used). The NMR resonance assignments are consistent with those reported for the two-dimensional NMR analysis of native Coenzyme F430 and its pentamethyl ester derivative (32, 54)
Table 1.
1H-NMR data of F330
| Assignmenta | δ(ppm) | Assignment | δ(ppm) |
|---|---|---|---|
| H3C-C(2) | 1.02 | H′-C(3b) | 2.26 |
| H3C-C(7) | 1.11 | H-C(3b) | 2.42 |
| H′-C(5) | 1.33 | H′-C(8b) | 2.30 |
| H-C(5) | 1.85 | H-C(8b) | 2.51 |
| H′-C(3a) | 1.35 | H′-C(7a) | 2.25 |
| H-C(3a) | 1.65 | H-C(7a) | 2.42 |
| H′-C(17b) | 1.30 | H′-C(2a) | 2.46 |
| H-C(17b) | 1.45 | H-C(2a) | 2.62 |
| H′-C(8a) | 1.37 | H,H-C(12a) | 2.25 |
| H-C(8a) | 1.76 | H-C(17) | 2.52 |
| H′-C(13a) | 1.68 | H-C(8) | 2.63 |
| H-C(13a) | 2.10 | H-C(3) | 2.68 |
| H′-C(17a) | 1.75 | H′-C(20) | 2.75 |
| H-C(17a) | 2.20 | H-C(20) | 2.85 |
| H-C(18) | 1.88 | H-C(12) | 2.92 |
| H′-C(13b) | 1.95 | H-C(13) | 3.38 |
| H-C(13b) | 2.18 | H-C(19) | 3.55 |
| H′-C(18a) | 2.15 | H-C(4) | 4.62 |
| H-C(18a) | 2.46 | H-C(17c) | 4.65 |
see Figure 1 for atom numbering scheme.
Table 2.
13C-NMR data of F330
| Assignmenta | δ(ppm) | Assignment | δ(ppm) |
|---|---|---|---|
| H3C-C(7) | 14 | C(12a) | 40 |
| H3C-C(2) | 19 | C(3) | 42.5 |
| C(3a) | 21.5 | C(2a) | 43.5 |
| C(8a) | 22.5 | C(7a) | 44.5 |
| C(17a) | 25 | C(18) | 46.5 |
| C(17b) | 28 | C(13) | 49 |
| C(13a) | 28 | C(17) | 49.5 |
| C(20) | 29 | C(12) | 50 |
| C(8b) | 34.5 | C(8) | 52 |
| C(3b) | 35 | Q | 63 |
| C(13b) | 35.5 | C(17c) | 61 |
| C(18a) | 36.5 | C(4) | 65 |
| C(5) | 38 | ||
see Figure 1 for atom numbering scheme. In the text, the numbers of these carbon atoms are shown without parentheses, e.g. C(17c) is referred to as C17c.
Assignment of the one- and two-dimensional NMR spectra of F330
The 1H and 13C NMR assignments of F330 are shown in Tables 1 and 2. Signals from exchangeable protons bound to carboxylic acid and amide functionalities are not observed since all spectra were recorded in D2O. Many of the chemical shifts observed in the F330 spectrum are similar to those observed in the 1H NMR spectrum of the pentamethyl ester derivative of F430. The details of the assignments are given in the Supplementary Materials.
A proton signal in the spectrum of F330, which was generated by reduction of F430 with NaBH4, is observed at 4.65 ppm. This signal has no counterpart in the spectrum of F430. Furthermore, this resonance disappears when native F430 is reduced using NaBD4, which is clearly evident from the two-dimensional HMQC spectra (Figures 6 and 7). The 4.65 ppm resonance is assigned as the methine proton (HC-OH) of C17c that arises from reduction of the carbonyl group at C17c of F430 (Figure 1) by NaBH4. The absence of this resonance in the 1H NMR spectra of F330D is diagnostic because NaBD4 transfers a deuteride instead of a hydride, which would generate DC-OH at C17c. This assignment agrees with the secondary alcoholic proton signals reported in the literature (55, 56). The reduction of the >C=O group of F430 at C17C to >HC-OH in F330 or >DC-OH in F330D greatly influenced the chemical shifts of the adjacent protons at C17b. These (-H2C-HC-OH) protons shift upfield by about 1.2 ppm and appear around 1.4 ppm in F330/F330D, whereas for F430 these (H2C-C=O-) protons resonate at 2.5 ppm as reported in the literature (32, 54). Reduction to a secondary alcoholic group relieves the deshielding effect of carbonyl group and therefore causes an upfield shift of C17b from about 39 ppm in F430 to 28 ppm for F330/F330D.
Figure 6.
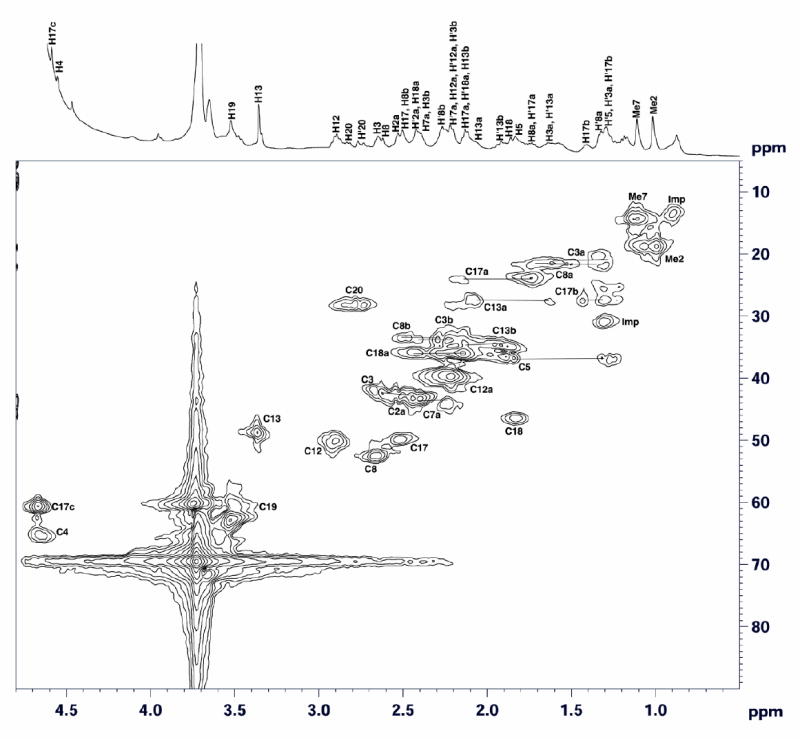
Two-dimensional 1H-13C HMQC spectrum of F330 recorded in D2O at 25°C. Horizontal lines connect signals of geminal protons. Horizontal and vertical axes represent proton and carbon chemical shifts, respectively. The 1H NMR spectrum of F330 is shown at the top.
Figure 7.
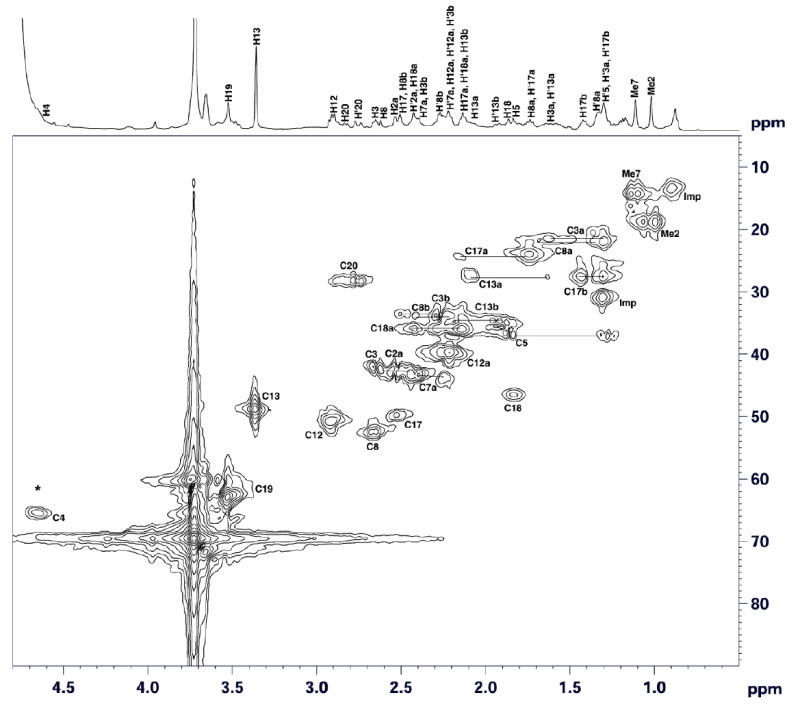
Two-dimensional 1H-13C HMQC spectrum of F330D recorded in D2O at 25°C. Horizontal and vertical axes represent proton and carbon chemical shifts, respectively. * designates the position of the (H)-C17c crosspeak in the 1H-13C HMQC spectrum of F330 (Figure 6). Horizontal lines connect signals of geminal protons. The 1H NMR spectrum of F330D is shown at the top.
The signals for all of the protons of F330 and their directly attached carbons have been assigned based on the 1H-13C HMQC (Figure 6) and 1H-1H COSY (Figure 8) spectra. With respect to the site of hydride transfer in F430 to generate F330, the most important assignment corresponds to the proton signal at 4.65 ppm (Figures 6 and 8), which disappears when the sample is reduced with NaBD4 (Figure 7). In the 1H-13C HMQC spectrum of F330 (Figure 6), this 4.65 ppm proton signal correlates with the carbon signal at 61 ppm, which is assigned to C17C. This methine proton in ring D of F330 arises from the secondary alcoholic proton transferred during the reduction of the cyclic ketone group on ring D of F430. Accordingly, in the 1H-13C HMQC spectrum of the NaBD4-reduced compound, F330D, the crosspeak due to the methine proton disappears (Figure 7). This is the only proton signal present in the 1H-13C HMQC spectrum of F330 that is absent in the spectrum of F330D.
Figure 8.
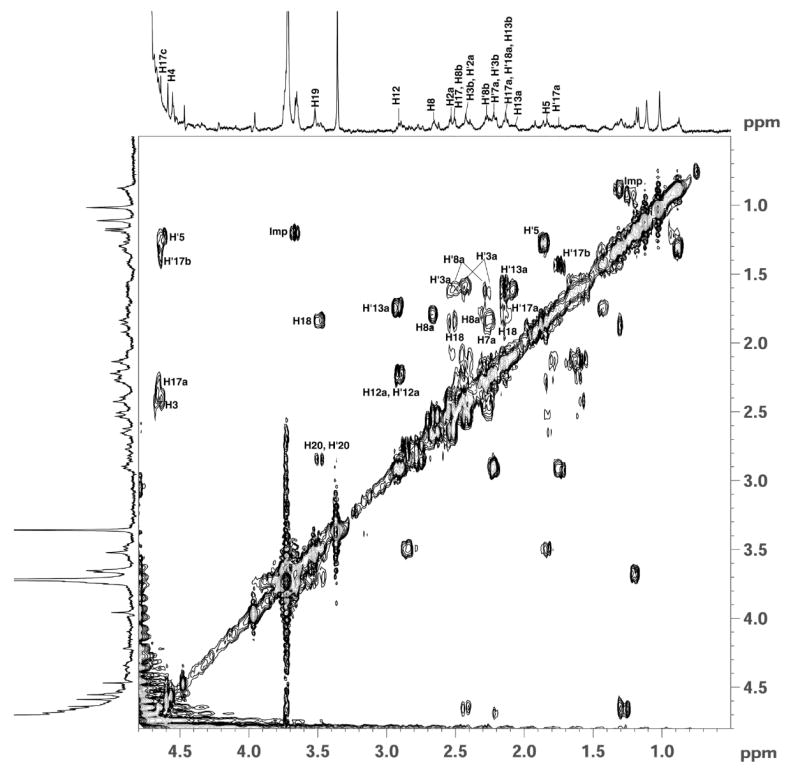
Two-dimensional 1H-1H COSY spectrum of F330 recorded in D2O at 25°C. (Imp= impurity).
This disappearance of the methine proton signal of C17c was also observed in the 2D COSY spectra shown in Figures 8 and S5. Direct scalar connectivities are established from the 2D COSY spectra. The signal at 4.65 ppm from H17c correlates directly with protons at C17b and C17a, which appear at 1.30 and 2.20, respectively. There is an apparent cross-correlation between the protons of C17c and C17 at 2.52 ppm; however, the C17 peak is adjacent to a large water signal decreasing the reliability of this assignment. These cross coupling peaks are not observed in the COSY spectrum of F330D as shown in Figure S5 and in the COSY of F430 reported in the literature (32, 54). The only cross peaks that appear in this region are the ones arising from the 4.62 ppm signal from the methine proton at C4, which correlates with its neighboring protons at C5 and C3. To highlight the couplings observed in the COSY spectra, the C19 methine proton interacts with its adjacent protons at C20 and C18 by showing cross peaks near 2.80 and 1.88 ppm respectively. The proton signal at 2.92 ppm, arising from the methine proton at C12 correlates with protons at C12a and one of the geminal protons of C13a that appear at 2.25 and 1.68 ppm, respectively. Similarly, the methine proton at C8 shows coupling with one of its adjacent protons at C8a. While H’17a shows a crosspeak due to its neighbor H’17b, H5 only shows direct coupling with H’5 that is attached to the same carbon C5. Also, the signals due to protons at positions C2a, C17, C8b, C3b, C7a, C17a, C18a, and C13b exhibit through-bond coupling with protons in their vicinity as shown in the figures (Figures 8 and S5).
Thus, the 1H, COSY, and HMQC spectra are consistent with the hypothesis that formation of F330 involves the reduction of the ketone group at carbon C17c, which is part of the ring attached to ring D of the tetrapyrrole (Figure 1). The complete description of the assignment of signals for all of the protons of F330 and their directly attached carbons based on the 1H-13C HMQC spectrum shown in Figure 7 is presented in the Supplementary Materials (Figures S6 and S7).
Low-temperature Absorption and MCD Studies of F330
The 10 K absorption spectrum of F330 (Figure 9, top) is very similar in general appearance to that obtained at ambient temperature (Figure 3), indicating that the molecular structure of this species is preserved at cryogenic temperatures. The MCD spectrum of F330 is essentially featureless and remains unchanged over a wide range of temperatures (4.5 – 100 K, data not shown). These results conclusively demonstrate that F330 possesses a diamagnetic (S = 0) ground state, consistent with the narrow dispersion and sharpness of all features in the corresponding NMR spectra described above. Therefore, two oxidation states of the nickel ion in F330 appear plausible: low-spin Ni(II) or Ni(0). The latter possibility would seem more probable in light of the reducing conditions required to generate F330. This issue has been addressed by computational studies as described in the next section.
Figure 9.
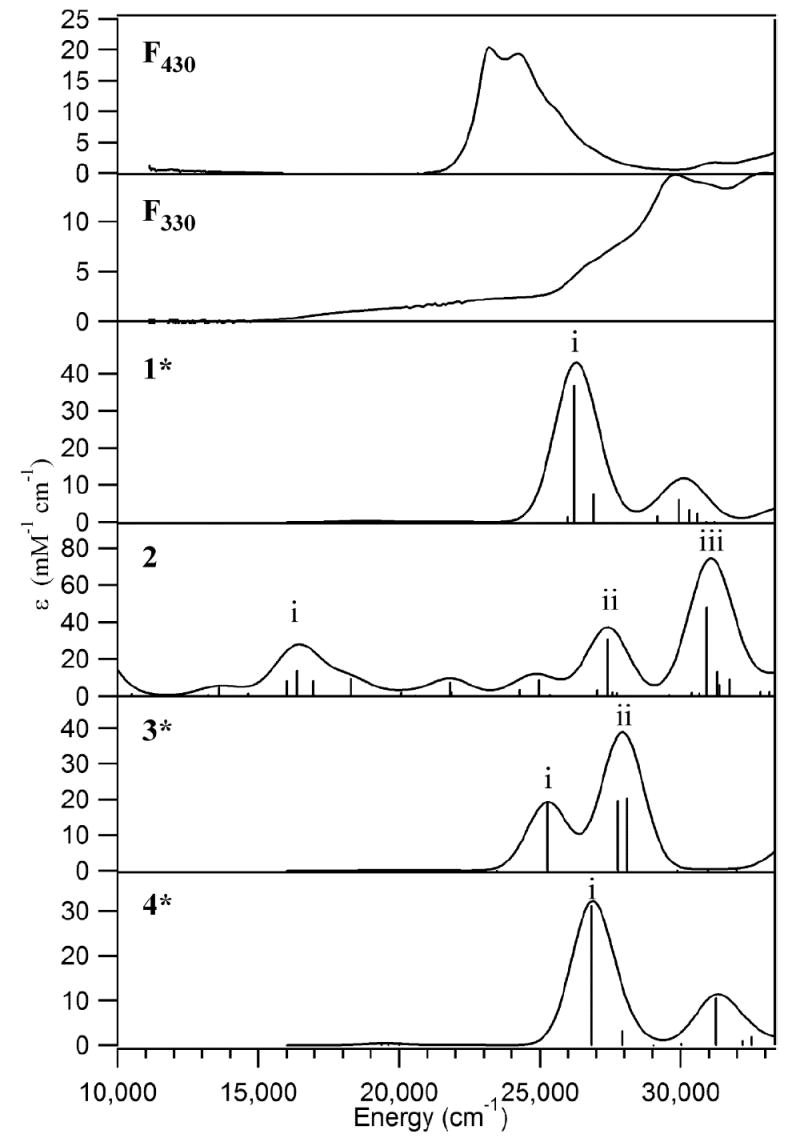
Experimental 10 K absorption spectra of F430 and F330 (top 2 panels) and TD-DFT/COSMO computed spectra for the Coenzyme models 1*, 2, 3*, and 4* (bottom 4 panels).
Computational Studies
To obtain quantitative insight into the electronic structure of F330, we performed computational studies on various Coenzyme models. As revealed by our NMR and variable-temperature MCD data described above, F330 contains a peripheral alcohol group at C17c in the ring adjacent to ring D and a low-spin Ni core, which is either a Ni(0) or a low-spin Ni(II) center. A powerful means for better understanding the electronic structure of F330 and for discriminating among possible Ni oxidation states in F430 and its derivatives is provided by DFT/TD-DFT computations. In this approach, TD-DFT is used to calculate the electronic absorption spectra of viable Coenzyme models (optimized using DFT), which can be compared directly to the experimental absorption spectrum of the species of interest. As we demonstrated in our previous studies of F430 and F380 (25), the TD-DFT predicted (and experimental) absorption spectra of this Coenzyme vary considerably as a function of Ni oxidation state, suggesting that discrimination between low-spin Ni(II) and Ni(0) descriptions for F330 should be relatively straightforward.
Since F430 has been shown to bind two axial H2O ligands at cryogenic temperatures to yield a six-coordinate high-spin (S = 1) Ni(II) species displaying intense temperature-dependent MCD features, a putative Ni(II)F330 species would also be expected to bind two axial water molecules (and thus to become paramagnetic) at low temperatures. While for this reason, and in light of the reducing conditions employed to generate this species, a Ni(0) description for F330 appears more likely, full DFT geometry optimizations were performed on both 4-coordinate Ni(0)- and Ni(II)-containing F330 models. As the changes in the electronic absorption spectrum accompanying a F430 → F330 conversion most likely reflect the reduction of a double bond at one end of the conjugated π system of the hydrocorphin macrocycle, our F330 computational models were generated by adding two electrons and two protons to the C16=N bond associated with ring D or, alternatively, to the peripheral C17c=O carbonyl moiety adjacent to ring D (Figure 2). The DFT-optimized models were then evaluated on the basis of our spectroscopic data for F330 using TD-DFT calculations, previously shown to be well suited for predicting electronic absorption spectra of various F430 models.25 Parallel computations were performed on analogous Ni(II)F430 models that provided a well-defined reference point in these evaluations.
Geometry-Optimized NiF330 Models
Tables 3 and 4 compare key structural parameters for the geometry-optimized models of Ni(II)F430 (1 and 1*), Ni(0)F330 (2), and Ni(II)F330 (3, 3*, 4a, 4b, and 4*) shown in Figure 2. Although an eclipsed H–C17c–O–H configuration was initially chosen for model 4a, the proton of the alcohol moiety moved during the geometry optimization, leading to a dihedral angle τ of 54° in the fully optimized model. In contrast, when the alcoholic proton was positioned in a staggered conformation (τ of 180°), it exhibited only a minimal shift, as revealed by the value of τ = 176° in the optimized model 4b. The partially eclipsed configuration of the alcoholic proton in model 4a is predicted to be more stable than the staggered conformation in 4b by ~5 kJ/mol (Table 3; note that a direct comparison of the computed total energies is only meaningful for different sets of isomers). Due to this energetic preference, only the partially eclipsed geometry of the alcoholic proton was considered in subsequent QM/MM computations.
Table 3.
Key structural parameters for the QM-optimized truncated models of F430 and F330
| Model | 1 | 2 | 3 | 4a | 4b |
|---|---|---|---|---|---|
| Description | Ni(II)F430 | Ni(0)F330 | Ni(II)F330 | Ni(II)F330 | Ni(II)F330 |
| Ni – N (A) (Å) | 2.072 | 2.099 | 2.103 | 2.024 | 2.027 |
| Ni – N (B) (Å) | 1.994 | 1.997 | 1.968 | 1.944 | 1.967 |
| Ni – N (C) (Å) | 1.974 | 2.077 | 1.972 | 1.964 | 1.955 |
| Ni – N (D) (Å) | 1.953 | 2.179 | 2.027 | 1.912 | 1.920 |
| C17c – O (Å) | 1.232 | 1.270 | 1.232 | 1.437 | 1.448 |
| τ (H-C-O-H) | N/A | N/A | N/A | 53.5° | 176.2° |
| Fold anglea | 5.73° | 10.89° | 17.49° | 18.92° | 17.45° |
| Energy (eV)b | -341.29 | -352.83 | -347.71 | -348.45 | -348.39 |
The fold angle is defined as the angle between the mean planes defined by the atoms C15–N–C19–C20–C1–N–C4–C5 and C15–C14–N–C1–C10–C9–N–C6–C5.
Note that a direct comparison of the computed total energies is only meaningful for the isomers 3, 4a, and 4b.
Table 4.
Key structural parameters for the QM/MM-optimized complete models of F430 and F330
| Model | 1* | 3* | 4* |
|---|---|---|---|
| Description | Ni(II)F430 | Ni(II)F330 | Ni(II)F330 |
| Ni – N (A) (Å) | 1.919 | 1.901 | 1.908 |
| Ni – N (B) (Å) | 1.922 | 1.910 | 1.922 |
| Ni – N (C) (Å) | 1.936 | 1.903 | 1.914 |
| Ni – N (D) (Å) | 1.872 | 1.947 | 1.876 |
| C17c – O (Å) | 1.281 | 1.232 | 1.454 |
| O – H (Å) | N/A | N/A | 0.977 |
| τ (H-C-O-H) | N/A | N/A | 53.3° |
| Fold anglea | 60.20° | 61.51° | 56.22° |
| Energy (eV)b | -408.35 | -416.26 | -416.92 |
See Table 3 for a definition of the fold angle.
Note that a direct comparison of the computed total energies is only meaningful for the isomers 3* and 4*.
Excited-State TD-DFT Calculations
Figure 9 compares the experimental absorption spectra of F430 and F330 to the TD-DFT/COSMO computed spectra for the Ni(II)F430 model 1*, the Ni(0)F330 model 2, and the Ni(II)F330 models 3* and 4* (TD-DFT calculated absorption spectra for all computational models considered are presented in Figure S7). Consistent with the computational results obtained in our previous study of a truncated Coenzyme model,25 the TD-DFT results for the four-coordinate Ni(II)F430 model 1* agree reasonably well with our experimental data, predicting a single dominant feature at 26206 cm-1 (382 nm) that clearly corresponds to the prominent absorption band at 23260 cm-1 (430 nm) in the experimental spectrum (Figure 9, top). Note that the transition energies for F430 and the related B12 cofactors are consistently overestimated by the B3LYP TD-DFT method (21, 25, 57-64). Analysis of the calculated MO diagram for 1* (Figure 10, left) reveals that the absorption spectrum is dominated by hydrocorphin-centered π→π* transitions, as the occupied Ni 3d-based MOs are too low in energy to participate in electronic transitions in the UV/vis/near-IR spectral region.
Figure 10.

DFT/COSMO calculated MO diagrams for models 1*, 2, 3*, and 4* (Figure 2, Tables 3 and 4). MOs are labeled according to their principal orbital contributors and are plotted relative to the energy of the corresponding LUMO. The one-electron excitations responsible for the dominant transitions in the TD-DFT calculated absorption spectra shown in Figure 9 are indicated by arrows.
Reduction of the C16=N and C17c=O double bonds of the hydrocorphin macrocycle of 1 to generate models 3* (C=N reduction) and 4* (C=O reduction), respectively, has a small but non-negligible effect on the TD-DFT calculated absorption spectrum (Figure 9). As proposed previously on the basis of qualitative considerations,18 reduction of a C=N (or C=O) double bond at one end of the conjugated π system of the hydrocorphin macrocycle elicits a blue-shift of the dominant UV/visible absorption feature. The TD-DFT calculated blue-shifts of 1719 cm-1 (from 1* to 3*) and 629 cm-1 (from 1* to 4*) predict the trend in the experimentally observed shift of this feature from 23260 to 30300 cm-1 (from 430 to 330 nm) upon F430 → F330 conversion, possibly indicating that F330 may in fact possess a Ni(II) center. Comparison of the calculated bonding description for 1* to those obtained for 3* and 4* (Figure 10) reveals the expected increase in energy separation of the hydrocorphin π-based occupied and π*-based unoccupied MOs involved in the dominant transition upon ring reduction, while the occupied Ni 3d-based MOs remain too low in energy to participate in electronic transitions throughout the UV/vis/near-IR spectral region. The similarities between 3* and 4* in their calculated absorption spectra and MO diagrams signify that spectral predictions alone are insufficient to distinguish between the two possible reduction sites on the hydrocorphyn ring. However, the DFT-predicted stabilization of 4* relative to 3* (as well as of 4a relative to 3, Tables 3 and 4) favors carbonyl reduction over C=N bond reduction, consistent with our NMR results for F330 presented above.
While spectral predictions do not provide a conclusive means by which to distinguish between models 3* and 4*, they are well suited to address the issue of metal- versus tetrapyrrole-based reduction. Metal-centered two-electron reduction of the Ni(II)F330 model 3 to generate the Ni(0)F330 model 2 (Figure 2) gives rise to the appearance of numerous intense features across the entire UV/vis/near-IR spectral region of the TD-DFT/COSMO calculated absorption spectrum (Figure 9). The presence of these low-energy features in the computed absorption spectrum for 2 can be understood in terms of a dramatic destabilization of the occupied Ni 3d-based MOs upon Ni(II) → Ni(0) reduction (Figure 10). In the reduced Ni(0)F330 model, the metal 3d-based MOs lie between the hydrocorphin π-based occupied MOs and the π*-based unoccupied MOs, thereby allowing these MOs to serve as donor orbitals for several low-energy Ni 3d→hydrocorphin π* charge-transfer (CT) transitions. As these features have no experimental counterparts (Figure 9, top), our TD-DFT results strongly suggest that the tetrapyrrole ring is the only site of two-electron reduction upon F430 conversion to F330.
Interestingly, inspection of the calculated MO description for 2 reveals that the two electrons added to Ni(II)F330 do not actually localize on the Ni center alone. Rather, they are almost equally shared between the Ni 3dx2-y2 orbital and a hydrocorphin π*-based orbital; i.e., the LUMO+1 of 2 (MO 117 in Figure 10) contains nearly equal contributions from the Ni 3dx2-y2 and hydrocorphin π* orbitals. This result suggests that the anionic hydrocorphin macrocycle is too strong a donor ligand to stabilize Ni(0), providing a rationale as to why treatment of F430 with NaBH4 results in the reduction of a C=O double bond while preserving the Ni(II) oxidation state.
Axial Ligand Binding Affinities of Ni(II)F430 and Ni(II)F330
Based on our TD-DFT based evaluation of possible F330 models, only a low-spin Ni(II) description appears consistent with our experimental data. However, it is not clear whether such a Ni(II)F330 species would indeed be four-coordinate and diamagnetic (similar to the free Coenzyme F430 at room temperature), as required by our MCD data. To estimate the relative binding affinities for axial H2O ligands of Ni(II)F330 and Ni(II)F430, DFT geometry optimizations were performed on the high-spin bis-aquo analogues of 1 and 4 using a spin-unrestricted formalism. Importantly, the ligand binding affinities of the two Ni(II) species were found to be nearly identical, merely differing by ΔΔH <1 kcal/mol. Thus, in light of the fact that the six-coordinate bis-aquo F430 species predominates only at cryogenic temperatures, our calculations appear qualitatively consistent with F330 being a four-coordinate Ni(II) species; i.e., the actual ΔΔH value might be sufficiently large to ensure that F330 remains four-coordinate at all temperatures, similar to the isoelectronic Co(I)-cobalamin cofactor (65).
X-ray Absorption Studies
To further address the issue of whether or not the Ni oxidation state changes when F430 is reduced to F330, we collected Ni K-edge X-ray absorption near-edge spectroscopic (XANES) data of F330 at pH 2 and pH 11 and of F430 (Figure 11). First derivative spectra are also presented in the lower panel to disclose the edge structure.
Figure 11.
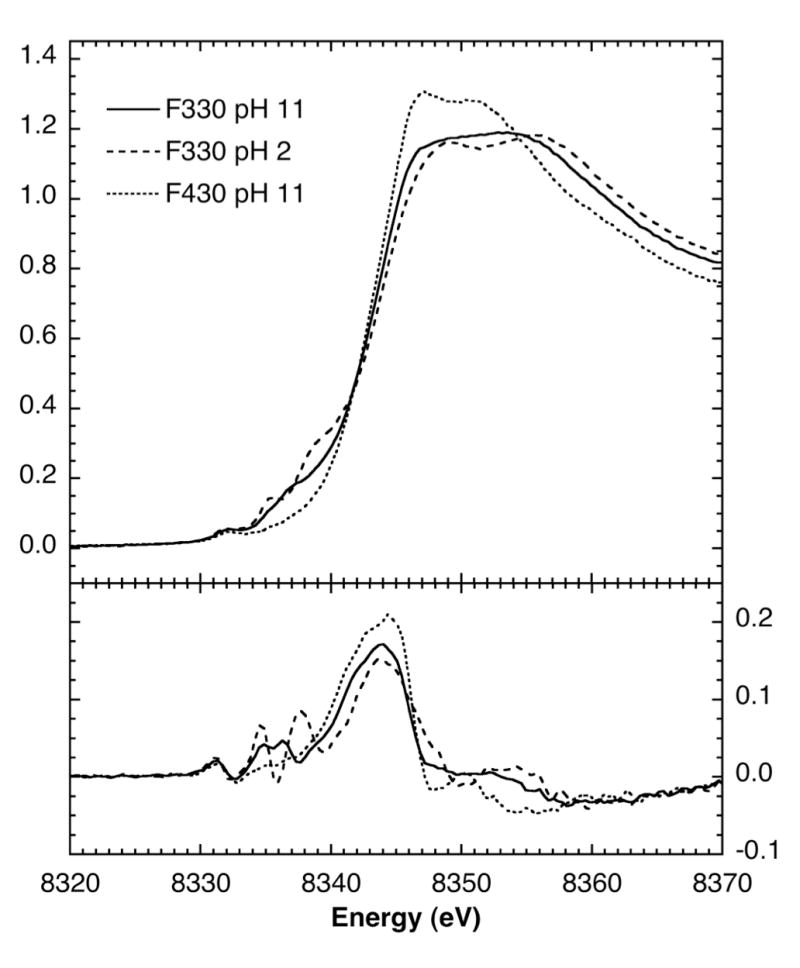
Top: Ni K-edge XANES data of F330 at pH 2 (- - - -) and 11 (———) compared with F430 (-------). Bottom: First derivative spectra. Temperature, 10 K.
The Ni K-edge spectrum of F430 comprises a relatively unstructured broad edge with a major inflexion at 8343.9 eV. A small pre-edge peak at 8332.3 eV can be assigned to a formally forbidden Ni 1s→3d transition. Both of these features correspond to those observed previously for the MCR enzyme (23). The lack of any significant pre-edge structure at ~ 8338 eV suggests that the Ni site is six-coordinate (23, 66) and is consistent with the formation of bis-aquo Ni(II)F430 on freezing.
The spectrum of F330 at pH 11 shows that the energies of the edge inflexion and the Ni 1s→3d band are essentially unchanged. While the intensity of the Ni 1s→3d transition is also unchanged, additional pre-edge structure appears around 8337 eV, which is revealed to comprise two bands by the derivative spectrum. These features are usually assigned to Ni 1s→4p transitions with ligand-to-metal charge-transfer shakedown contributions (67, 68).
At pH 2, the F330 spectrum is more complex. There is apparently a 0.2 eV downshift of the edge inflexion point to 8343.7 eV; however, this is small compared to the ~ 1 eV shift expected for a one-electron reduction. The Ni 1s→3d feature remains at 8332.3 eV, but the additional pre-edge structure around 8337 eV has become even more complex, with two well defined features at 8335.5 eV and ~8339 eV.
These data clearly show that conversion of F430 to F330 is not accompanied by reduction of the central Ni because the energy shifts observed for the Ni K-edge and 1s→3d transitions are rather insignificant. Thus, consistent with the MCD data presented above, our XAS results also demonstrate that F330, like F430, contains a Ni(II) ion. We note that the edge-shift observed on one-electron reduction of Ni compounds can vary considerably with the metal-ligand environment, as the net increase in Ni d-electron density can be moderated through metal-ligand covalency (66).. Although a shift of 1.8 eV has been observed for Ni oxides, a value of slightly less than 1 eV is more typical. In this case, however, we can calibrate the expected shift using Ni K-edge XANES measurements previously reported on the MCR enzyme (23). These data showed an 0.5 eV shift when comparing the spectrum from Ni(II) MCRsilent with that for a 60% Ni(I) MCRred1 sample – the 40% balance presumably being Ni(II) MCRsilent. This implies a ~ 0.8 eV shift for complete Ni(II) → N(I) reduction of the Coenzyme. Similar shifts were observed in studies of MCRred1 by Duin et al (10). Consequently, at least a 1.6 eV shift would be expected for the conversion of Ni(II) → N(0). Therefore, our results clearly rule out the reduction of Ni(II) to Ni(0) during the conversion of F430 to F330 and strongly indicate that the Ni in F430 and both pH states of F330 share the same Ni(II) oxidation state.
A second conclusion is that the appearance of Ni 1s→4p shakedown transitions in the F330 spectra reflect geometric and ligand coordination changes at the Ni site on Coenzyme reduction. These transitions tend to be absent in six-coordinate Ni sites (23) and this is the basis of our suggestion that the F430 in our frozen sample is bis-aquo axially coordinated. By contrast, their presence in F330 suggests a lower ligand coordination number, and possibly a lowering of symmetry, such as movement of the Ni out of the macrocycle plane. Interestingly, the intensities of the Ni 1s→3d transitions in the F330 and F430 spectra are virtually identical; in both cases the corresponding areas are ~2.4 × 10-2 eV. Analysis of model compound data indicates that the intensity of this transition also correlates with the ligand coordination number and geometry (23, 66). A value of 2.4 × 10-2 eV is consistent with either a six-coordinate octahedral or four-coordinate planar Ni site; a five-coordinate bipyramidal Ni is expected to have a significantly more intense transition with a typical area of ~6 × 10-2 eV. Therefore, the most straightforward interpretation of these data is that even in the frozen state F330 has four-coordinate planar geometry, and by inference, no axial ligands, which is consistent with the low-spin assignment based on our NMR, MCD, and computational results.
Finally, the pH sensitivity of the F330 XANES data, in particular in the Ni 1s→4p shakedown region that provides a sensitive probe of ligand-to-metal charge transfer transitions, is consistent with protonation of the macrocycle ligand at low pH. The detailed changes in the F330 spectra are intriguing and presumably reflect subtle changes in the ligand environment and/or conformational changes at the Ni site.
Resonance Raman Studies
RR studies were undertaken to investigate the structural/conformational changes in the macrocycle that accompany conversion of F430 to F330. In principle, the RR studies could also provide an independent probe of reduction of C17c=O group to an alcohol. However, the C=O stretching vibration of F430, which should be observed in 1660-1700-cm-1 region, is not resonance enhanced in the Raman spectrum (23). Accordingly, RR spectroscopy only offers an indirect probe of the effects of carbonyl reduction.
The high-frequency RR spectra of F430, F380, and F330 are compared in Figure 12. The RR scattering characteristics of F430 and F380 have been previously discussed in detail (23) and will not be elaborated on herein. Instead, we focus on the RR features that distinguish F330 from the other forms of the Coenzyme. The RR spectrum of F430 is dominated by a single strong band at 1563 cm-1; all other vibrational features are relatively weak. 15N-labeling studies have shown that the 1563-cm-1 band contains substantial C=N stretching character. The RR spectrum of F380 is much richer than that of F430 and exhibits a number of relatively intense bands in the high-frequency region, including features at 1378, 1490, and 1624 cm-1. None of these bands of F380 corresponds to the 1563-cm-1 band of F430. Inspection of the RR spectra shown in Figure 12 reveals that the vibrational characteristics of F330 are distinct from those of either F430 or F380; however, the spectrum of F330 is qualitatively more similar to that of F430. In particular, the F330 spectrum is dominated by a single strong band at 1585 cm-1, while all other bands are relatively weak.
Figure 12.
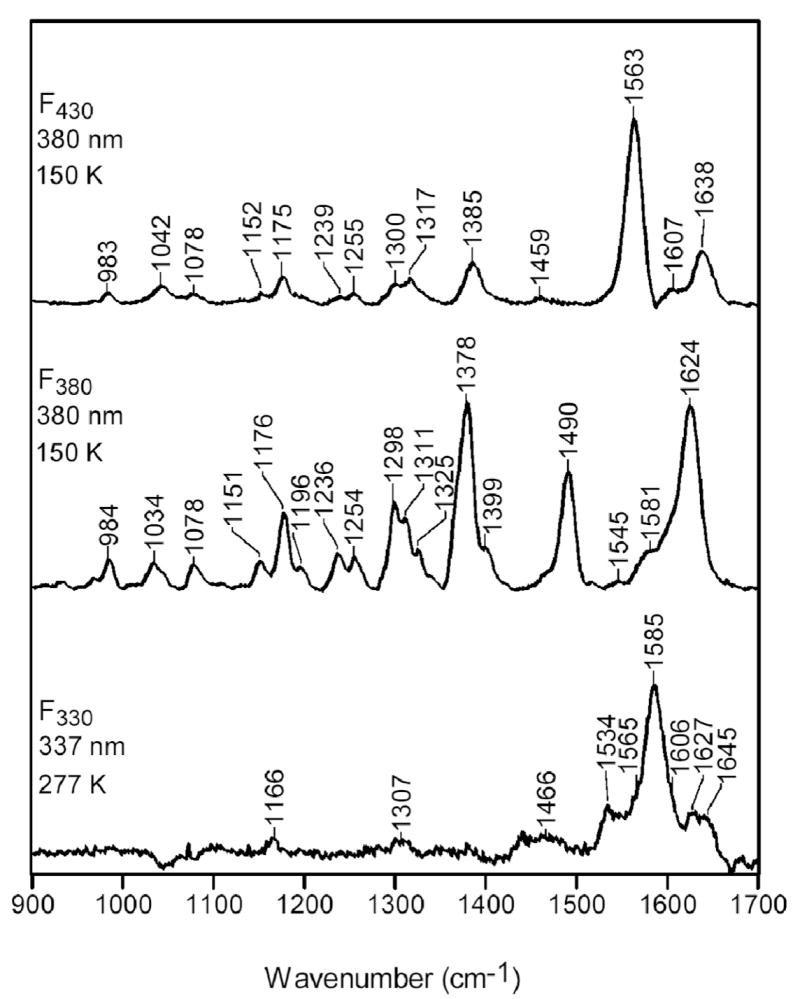
High-frequency RR spectra of F430, F380, and F330.
To further investigate the vibrational characteristics of F330, RR spectra were obtained for the 15N-labeled Coenzyme, as well as F330D containing both natural abundance nitrogen and the 15N label (Figure 13). Our 14N→15N isotopic labeling studies show that the 1585-cm-1 band of F330 downshifts to 1575 cm-1 in 15N-F330. None of the other RR bands of F330 exhibits any discernible isotope shift. The 10-cm-1 14N→15N shift observed for the 1585-cm-1 band of F330 is comparable to the 14-cm-1 shift observed for the 1563-cm-1 band of F430 (23), suggesting that these modes have similar compositions for the two forms of the Coenzyme. In the case of F330D, no 1H→2D shifts can be discerned for any of the RR bands. Likewise, the spectral features observed for 15N-F330D are similar to those of 15N-F330. These results indicate that vibrations involving motions of atoms that are deuterated do not contribute to the resonance-enhanced Raman features observed for F330.
Figure 13.
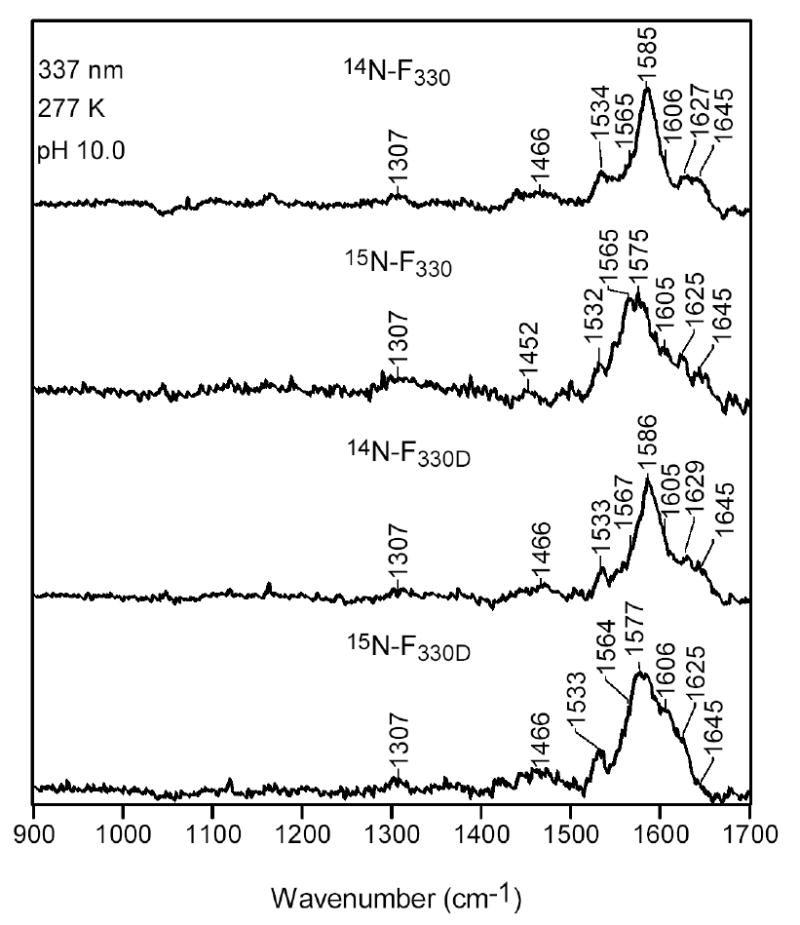
High-frequency RR spectra of F330, 15N-F330, F330D, and 15N-F330D.
The vibrational characteristics of F330 are interesting in light of the other spectroscopic and computational data reported herein, which indicate that conversion of F430 to F330 is due to reduction of the C17c=O group accompanied by conversion of the Ni(II) ion from high- to low-spin. In particular, previous RR studies have shown that the C=N skeletal mode observed at 1563 cm-1 for six-coordinate (high-spin) F430 downshifts to 1532 cm-1 when the Coenzyme is converted to its four-coordinate (low-spin) form (69-71). In contrast, this skeletal vibration of F330, which also contains a four-coordinate, low-spin Ni(II) ion is observed near 1585 cm-1, an upshift of ~22 cm-1 relative to six-coordinate, high-spin F430. Accordingly, the frequency difference between the C=N skeletal mode of the four-coordinate, low-spin forms of F430 versus F330 is greater than 50 cm-1. This observation is qualitatively consistent with the view that reduction of the C17c=O bond increases the π-bond order in the remaining π-framework.
Discussion
Native Coenzyme F430 has been studied by EPR and related advanced EPR (13, 72-75) (76, 77), UV-visible and MCD (21, 25, 73), X-ray absorption (10, 23), NMR (32, 54, 78), and RR (23) spectroscopies, spectroelectrochemistry (24, 79) and X-ray crystallography (6, 80). These spectroscopic properties of MCR and Cofactor F430 have been reviewed (17, 81). The Coenzyme is routinely isolated in a high-spin (S = 1) Ni(II) state that can undergo various reactions. When heated, F430 epimerizes to the 12,13-diepimer; thus, when isolated from the cytoplasm, F430 is isolated as a mixture of native F430 and the diepimer (50, 82). In the studies described here, homogeneous preparations of F430 were obtained from purified MCR, which contains only the native Coenzyme and lacks the diepimer. F430 can be reduced by Ti(III) citrate to form the Ni(I)-bound F380 state or, alternatively, by NaBH4 to generate the F330 state. A major aim of the studies described here was to characterize the novel F330 state using a variety of experimental methods (MS, electronic absorption, MCD, RR, NMR, XAS) and by computational techniques (DFT and QM/MM methods). Another aim was to develop a better understanding of the relatively well-characterized F380 state and to establish conclusively whether the tetrapyrrole ring undergoes reduction upon F430→F380 conversion. An important aspect of these studies is that they provide an understanding of the factors that lead to metal versus tetrapyrrole ring reduction.
Our MS results show unambiguously that reduction of F430 by Ti(III) citrate to generate F380, the form of the Coenzyme in the red1 state of MCR, does not involve reduction of the tetrahydrocorphinoid ring. Earlier, it had been proposed that generation of F380 involves one-electron reduction of the Ni(II) center to Ni(I) along with a two-electron reduction of the tetrapyrrole ring (23) Unambiguous assignment of F380 as a Ni(I) species could be made based on its typical d9 S = ½ EPR spectrum. Tetrapyrrole ring reduction was proposed because the UV-visible spectrum underwent a 40 nm blue shift upon F430→F380 conversion, which is characteristic of a single double-bond reduction within a conjugated π-system. Furthermore, a peak in the RR spectrum of F430 assigned to a mode involving predominant C=N double bond stretching motion either disappears or undergoes a major shift when the Coenzyme is reduced to the F380 state, which would be consistent with reduction of one of the C=N bonds associated with the conjugated tetrapyrrole system (23). However, the MS analysis in the negative ion mode clearly demonstrates that the mass does not change when F430 is reduced to F380 by Ti(III) citrate. Therefore, we can definitively rule out incorporation of hydrogen into the Coenzyme when the F380 state is generated. These results are, therefore, inconsistent with tetrapyrrole ring reduction during formation of the red1 state. Instead, they agree with the conclusions of cyclic voltametric and spectroelectrochemical experiments using the tetramethyl ester of F430 that conversion of F430 to F380 involves one-electron reduction of Ni(II) to Ni(I) without tetrapyrrole ring reduction (24). This conclusion is reinforced by computational results on the F380 state described below.
Although Ti(III) citrate does not reduce the tetrapyrrole ring of F430 during conversion to F380, our MS results clearly demonstrate that treatment of F430 with NaBH4 to form F330 does lead to two-electron reduction of the tetrahydrocorphinoid ring. Conditions were optimized to ensure that the F330 state was maintained throughout the course of the experiment. Mass spectrometric results show that F430 reduction with NaBH4 in H2O increases the mass by two units and reduction with NaBD4 in H2O increases the mass by 3 units. Thus, one of the protiums (or deuteriums) that is transferred is introduced from the solvent (or is exchangeable with the solvent) and the other is not. This result is consistent with transfer of a hydride (or deuteride from NaBD4) and a proton from the solvent, with the hydride (or deuteride) forming a stable C-H (C-D) bond and the proton bound to N or O at an exchangeable position.
Although our MS experiments conclusively demonstrate that the tetrapyrrole ring undergoes reduction upon conversion of F430 to F330, but not to F380, they do not permit determination of the specific site on the tetrahydrocorpinoid ring to which the hydride and proton are transferred. Attempts to further fragment F330 and determine the site(s) containing the increased mass using tandem MS/MS were unsuccessful because the fragmentation cleaved the various groups appended to a stable tetrapyrrole ring system. Thus, NMR, RR, MCD, XAS, and computational approaches were used to identify the site of reduction on the tetrapyrrole ring and to establish the oxidation and spin states of nickel in the core of F330.
NMR experiments, which are severely compromised for studying F380 because of its paramagnetism and rapid decay to Ni(II), are ideal for investigating F330. The 1H and 13C NMR signals of F330 in water/D2O are narrow and well-resolved, indicating that F330 contains low-spin (S = 0) Ni(II) or Ni(0). In contrast, F430 exhibits NMR signals in water/D2O that are fairly broad and un-assignable due to the high-spin (S = 1) Ni(II) central ion in this species. Therefore, the NMR spectra of native F430 were previously collected in F3CCD2OD (TFE) and the pentamethyl ester of F430 was studied in CD2Cl2 or CDCl3, where the Ni(II) ion is low spin (S = 0), so that the signals are narrow enough to permit detailed assignments (32, 54).
While the 1H and 13C chemical shifts for F330 are similar to those of native F430 and its pentamethyl ester, they exhibit some important differences that reveal the site of reduction of the tetrahydrocorphinoid ring during the conversion of F430 to F330. In comparing the 1H-13C HMQC spectra of F330 and F330D, the methine proton signal at 4.65 ppm correlating to the carbon signal at 61 ppm disappears in the spectrum of F330D. This is the only proton signal that is lost in F330D; all of the remaining proton signals in the 1H-13C HMQC spectrum of F330D are the same as those of F330. Therefore, it is clear that the proton signal at 4.65 ppm and the carbon signal at 61 ppm correspond to the site to which the hydride is transferred.
Although, based on RR data, it was previously suggested that the reduction takes place at a C=N double bond (23), the assignments of the 4.51 ppm (H) and 61 ppm (C) signals are unambiguous. Both the 1H and 13C NMR resonances for F330 are in the range for secondary alcoholic groups. Furthermore, there is no proton signal between 2.5-3.5 ppm (for −HC-NH) that correlates to a carbon between 40-50 ppm that selectively appears in the F330 HMQC spectrum and is lost in the spectrum of F330D. Thus, it is clear that no imine (-CH=N-) moieties, e.g. those conjugated to the tetrapyrrole system in rings B and D, undergo reduction when F430 is treated with NaBH4 or NaBD4. Consequently, our 2D-NMR results strongly favor the reduction of the carbonyl group in the exocyclic ring attached to ring D of F430, thereby implying that F430→F330 conversion leads to the formation of a secondary alcoholic group, e.g., HC-OH at C17c (Figure 1).
The RR data are consistent with the NMR analysis and provide additional information about the conjugative network in the corphin ring of F330. A large frequency difference of the C=N skeletal mode for F430 versus F330 is observed. For the four-coordinate, low-spin Ni(II) state of F430 (which predominates at room temperature), this band is observed at 1532 cm-1, while for four-coordinate, low-spin F330 it is upshifted by 50 cm-1. This upfield shift is consistent with an overall increase in π-bond order of the conjugative framework as the number of π –bonds in the conjugated network decreases from five to four (note that the imine in ring A is isolated from the conjugated network, see Figure 1). On the other hand, a 50 cm-1 is quite large and could reflect a conformational difference between F430 and F330 (see below).
The narrow, well-resolved NMR signals of F330 indicate that this species is diamagnetic and thus contains either low-spin Ni(II) or Ni(0). NaBH4 might be expected to reduce the metal to generate Ni(0), since borohydride is typically used to convert cobalamins from Co(III) to Co(I) (83), or to reduce the C=O and C=N double bonds of the macrocycle, since borohydride is routinely used to reduce carbonyl and imine groups (84-87). To unambiguously establish the oxidation and spin states of the nickel in F330, XAS was used. Only minor shifts (<1 eV) and a small decrease in overall intensity are observed in the lower-energy region of the XANES spectra from F430 to F330. As the conversion of Ni(II) to Ni(0) would give rise to at least a 1.6 eV decrease in the edge energy, it is clear that F330 contains a low-spin Ni(II) center. The minor downshifts of the Ni 1s→3d transitions most likely result from changes in the Ni spin state (high spin in F430 vs low spin in F330) and coordination number (6-coordinate in F430 vs 4-coordinate in F330).
Reduction of F430 by NaBH4 leads to a 100 nm blue shift of the dominant feature in the UV-visible spectrum, which is much larger than the 20-30 nm shift that one might expect for reduction of a C=O double bond at the end of a conjugated network. As mentioned above, the computational results predict only an 11 nm shift in the UV-visible spectrum. The UV-visible spectra have been reported for a biosynthetic precursor of F430, 15,17c-seco-F430-17c-acid, in which the carbonyl group at 17c is disconnected from the chromophore because the six-membered ring is not yet closed, and a model compound which has the 5 C=N double bonds of the tetrapyrrole, but lacks the conjugated carbonyl group (88). The biosynthetic precursor and the model compound would be expected to exhibit UV-visible spectra similar to that of F330. Indeed, these complexes display absorption bands at approximately 300 and 350 nm similar to F330. However, the precursor and model complex exhibit an additional, considerably more intense band at 427 nm and 418 nm, respectively, which are shifted by only 3 and 12 nm from the dominant F430 absorption feature. We speculate that reduction of the carbonyl group and the associated change in hybridization at C17 upon conversion of F430 to F330 leads to a major conformational change that isolates the double bond in ring D from the conjugated system or, perhaps causes the Coenzyme to fold in a way that the two double bonds on either side of ring C are co-planar and thus the only ones that are conjugated. This speculative conformational change would also help explain the 50 cm-1 upshift of the 1532 cm-1 band in the RR spectrum.
TD-DFT calculations were used in conjunction with our spectroscopic data to evaluate viable NiF330 models that differ with respect to the metal oxidation state and to help reconcile the large spectral shift of the dominant absorption feature that occurs upon borohydride reduction of F430. The TD-DFT/COSMO computed absorption spectrum for the Ni(0)F330 model 2 (Figure 2) is clearly inconsistent with our spectroscopic data, exhibiting numerous intense features across the entire UV/vis/near-IR spectral region that have no counterparts in the experimental spectrum (Figure 9). In contrast, the TD-DFT/COSMO calculated absorption spectra for our Ni(II)F330 models 3* and 4* agree reasonably well with the experimental spectrum, reproducing the trend for the observed blue-shift of the dominant absorption feature upon conversion of F430 to F330 (Figure 9). Further computational studies will be performed to test the speculation that a folding of the tetrahydrocorphinoid ring in the F330 state causes an additional shift of this feature toward higher energy. In summary, even with the vast array of spectroscopic and computational methods used, the large spectral shift is not fully explained and we offer the testable hypothesis that reduction of the carbonyl group at C17c elicits a change in conformation that disrupts the conjugation in the tetrahydrocorphinoid ring system.
Together, our spectroscopic and computational data provide compelling evidence that F330 possesses a four-coordinate Ni(II) center, presumably because the anionic nature of the hydrocorphin macrocycle prevents stabilization of Ni(0). It is interesting to note that both reduction of Ni(II) to Ni(I) (yielding F380) and hydrocorphin reduction (generating F330) have qualitatively similar effects on the electronic absorption spectrum of F430, in each case leading to a marked shift of the dominant feature at 23260 cm-1 (430 nm) to higher energy. However, the electronic origin of this blue-shift is fundamentally different in these two cases. Upon conversion of F430 to F380, the occupied Ni 3d-based MOs are significantly raised in energy due a substantial decrease in the effective nuclear charge on Ni, thereby shifting between the π-based occupied and π*-based unoccupied frontier orbitals of the hydrocorphin macrocycle (25). Consequently, the energy splitting between these two sets of orbitals increases from F430 to F380, leading to the observed blue-shift of the hydrocorphin-centered π→π* transition that produces the dominant absorption feature (corresponding to the HOMO→LUMO transition (i) of F430, see Figures 9 and 10). Importantly, the close energetic proximity of the Ni 3d-based occupied MOs and the hydrocorphin π*-based unoccupied MOs in F380 also leads to the occurrence of numerous Ni 3d→hydrocorphin π* CT transitions in the vis/near-IR region that are particularly intense in the corresponding MCD spectrum (25). On the other hand, in the case of F330, the shortening of the conjugated π-system due to the ring reduction accompanying F430 → F330 conversion gives rise to a direct increase in the energy separation between the hydrocorphin π-based occupied and π*-based unoccupied orbitals involved in the dominant electronic transition, while the Ni 3d-based MOs remain largely unperturbed and thus too low in energy to participate in electronic transitions in the vis/near-IR spectral region (Figure 10, cf. models 1 and 3*/4*).
Because both Ni(II) and hydrocorphin reduction give rise to substantial blue-shifts of the dominant F430 absorption feature at 22360 cm-1 (430 nm), electronic absorption spectroscopy is not necessarily suited to discriminate between metal-centered and ring-centered reduction in this Coenzyme. Thus, it appeared reasonable to interpret the spectral changes associated with F430 → F380 conversion as indicating a 2-electron reduction of the hydrocorphin ring coupled to Ni(II) → Ni(I) reduction. However, our MS data presented in this paper along with recently published results from electrochemical (24) and combined spectroscopic/computational (25) studies now provide compelling evidence that F430 and F380 merely differ by one electron, both possessing a native-like hydrocorphin macrocycle.
Conclusions
Reduction of F430 with sodium borohydride (NaBH4) elicits a 100 nm blue shift in the absorption spectrum to generate a Coenzyme form that we call F330. To study this novel form of Coenzyme F430, we have used a wide range of spectroscopic methods (UV-visible, NMR, X-ray absorption, MCD, and RR), MS, and computational chemistry. MS studies directly demonstrate reduction of the corphin ring system by identifying the incorporation of one non-exchangeable protium (or deuterium) when F330 is generated with NaBH4 (or NaBD4). One- and two-dimensional NMR studies identify the site of reduction at the exocyclic ketone group adjacent to ring D of the tetrahydrocorphin (C17c according to the labeling of Figure 1). X-ray absorption, MCD, and computational studies reveal that the two-electron reduction of the tetrahydrocorphin ring to form F330 results in a spin-state change in the metal ion from high- to low-spin Ni(II) and rule out the possibility of an associated reduction of the metal ion. Based on RR studies, reduction of the hydrocorphin ring to eliminate the π-bond at C17c increases the overall π-bond order in the conjugative framework. It was proposed that generation of F380 involved both a metal and a ring-centered reduction (23); however, subsequent work provided strong evidence that conversion of F430M to F380 only involved reduction of the metal (24). Recent spectroscopic and computational work (25) and the mass spectrometric results described here demonstrate that reduction of F430 with Ti(III) citrate to generate F380 (corresponding to the active red1 state of MCR), results in reduction of the Ni(II) to Ni(I), but does not result in tetrapyrrole ring reduction. Computational data trace different origins of the significant blue shifts in the UV-visible spectra for both ring reduction (in F330) and metal center reduction (in F380). For F330 formation, it is the shortening of the conjugated π-system relative to F430 that directly increases the energy separation between the hydrocorphin π (HOMO) → π* (LUMO) transition responsible for the dominant electronic transition. On the other hand, for F380, the decrease in the formal oxidation state of Ni (from 2+ to 1+) causes the occupied Ni 3d-based MOs to shift above the hydrocorphin π-based frontier MOs, thereby leading to an increase in the energy of the dominant hydrocorphin-centered π→π* transition.
Supplementary Material
Acknowledgments
We acknowledge the NMR facility in the Chemistry Department at the University of Nebraska with special thanks to Sara Baisaga for her assistance. We thank Haiteng Deng, Jiong Yu, and Ashraf Raza for help with the mass spectrometric studies.
Footnotes
This work was partly supported by a grant from the Department of Energy (SWR, DE-FG03-ER20297), the National Science foundation (TCB, CAREER award MCB-0238530), and the National Institute of General Medical Sciences (DFB, GM-36243). The mass spectrometry instrumentation was purchased with funds from an NIH grant (1P20RR17675) to help support the Instrumentation Core of the Redox Biology Center at UNL. The Advanced Biological and Environmental X-ray Group (ABEX) at Lawrence Berkeley National Laboratory is funded by the Department of Energy, Office of Biological and Environmental Research. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
Abbreviations: MCR, Methyl-coenzyme M reductase; methyl-CoM, methyl-coenzyme M or , 2-(methylthio)ethanesulfonate; CoBSH, coenzyme B or N-7-mercaptoheptanoylthreonine phosphate; RR, Resonance Raman; MCD, magnetic circular dichroism; XAS, X-ray absorption spectroscopy; DFT, density functional theory; TD-DFT, time dependent density functional theory; QM/MM, combined quantum mechanical/molecular mechanical; VWN-LDA, Vosko-Wilk-Nusair local density approximation; BFGS, Broyden-Fletcher-Goldfarb-Shanno; PIM, positive ion mode; NIM, negative ion mode; HQMC, heteronuclear multiple quantum coherence; COSY, homonuclear correlated spectroscopy; XANES, X-ray absorption near-edge spectroscopy.
The formal charge on Ni is +2 and that on the directly bonded nitrogen is -1, so with the loss of two protons from the carboxyl groups, the net charge on the ligand would be -3.
References
- 1.DiMarco AA, Bobik TA, Wolfe RS. Unusual coenzymes of methanogenesis. Ann Rev Biochem. 1990;59:355. doi: 10.1146/annurev.bi.59.070190.002035. [DOI] [PubMed] [Google Scholar]
- 2.Ellermann J, Kobelt A, Pfaltz A, Thauer RK. On the role of N-7-mercaptoheptanoyl-O-phospho-L-threonine (component B) in the enzymatic reduction of methyl-coenzyme M to methane. FEBS Lett. 1987;220:358–62. doi: 10.1016/0014-5793(87)80846-3. [DOI] [PubMed] [Google Scholar]
- 3.Diekert G, Klee B, Thauer RK. Nickel, a component of factor F430 from Methanobacterium thermoautotrophicum. Arch Microbiol. 1980;124:103–106. doi: 10.1007/BF00407036. [DOI] [PubMed] [Google Scholar]
- 4.Diekert G, Jaenchen R, Thauer RK. Biosynthetic evidence for a nickel tetrapyrrole structure of factor F430 from Methanobacterium thermoautotrophicum. FEBS Letters. 1980;119:118–120. doi: 10.1016/0014-5793(80)81011-8. [DOI] [PubMed] [Google Scholar]
- 5.Whitman WB, Wolfe RS. Presence of nickel in factor F430 from Methanobacterium bryantii. Biochem Biophys Res Commun. 1980;92:1196–1201. doi: 10.1016/0006-291x(80)90413-1. [DOI] [PubMed] [Google Scholar]
- 6.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Crystal structure of methyl-Coenzyme M reductase: the key enzyme of biological methane formation. Science. 1997;278:1457–1462. doi: 10.1126/science.278.5342.1457. [DOI] [PubMed] [Google Scholar]
- 7.Diekert G, Konheiser U, Piechulla K, Thauer RK. Nickel requirement and factor F430 content of methanogenic bacteria. J Bacteriol. 1981;148:459–64. doi: 10.1128/jb.148.2.459-464.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goubeaud M, Schreiner G, Thauer RK. Purified methyl-coenzyme-M reductase is activated when the enzyme-bound coenzyme F430 is reduced to the nickel(I) oxidation state by titanium(III) citrate. Eur J Biochem. 1997;243:110–114. doi: 10.1111/j.1432-1033.1997.00110.x. [DOI] [PubMed] [Google Scholar]
- 9.Becker DF, Ragsdale SW. Activation of methyl-SCoM reductase to high specific activity after treatment of whole cells with sodium sulfide. Biochemistry. 1998;37:2639–47. doi: 10.1021/bi972145x. [DOI] [PubMed] [Google Scholar]
- 10.Duin EC, Cosper NJ, Mahlert F, Thauer RK, Scott RA. Coordination and geometry of the nickel atom in active methyl-coenzyme M reductase from Methanothermobacter marburgensis as detected by X-ray absorption spectroscopy. J Biol Inorg Chem. 2003;8:141–8. doi: 10.1007/s00775-002-0399-2. [DOI] [PubMed] [Google Scholar]
- 11.Holliger C, Pierik AJ, Reijerse EJ, Hagen WR. A spectroelectrochemical study of factor F430 Nickel(II/I) from methanogenic bacteria in aqueous solution. J Am Chem Soc. 1993;115:5651–5656. [Google Scholar]
- 12.Jaun B, Pfaltz A. Coenzyme F430 from Methanogenic Bacteria: Reversible One-electron Reduction of F430 Pentamethyl Ester to the Nickel(I) Form. J Chem Soc Chem Commun. 1986:1327–1329. [Google Scholar]
- 13.Telser J, Horng YC, Becker D, Hoffman B, Ragsdale SW. On the assignment of nickel oxidation states of the Ox1 and Ox2 Forms of Methyl-Coenzyme M Reductase. J Am Chem Soc. 2000;122:182–183. [Google Scholar]
- 14.Mahlert F, Grabarse W, Kahnt J, Thauer RK, Duin EC. The nickel enzyme methyl-coenzyme M reductase from methanogenic archaea: in vitro interconversions among the EPR detectable MCR-red1 and MCR-red2 states. J Biol Inorg Chem. 2002;7:101–12. doi: 10.1007/s007750100270. [DOI] [PubMed] [Google Scholar]
- 15.Rospert S, Voges M, Berkessel A, Albracht SPJ, Thauer RK. Substrate-analogue-induced changes in the nickel-EPR spectrum of active methyl-coenzyme-M reductase from Methanobacterium thermoautotrophicum. Eur J Biochem. 1992;210:101–107. doi: 10.1111/j.1432-1033.1992.tb17396.x. [DOI] [PubMed] [Google Scholar]
- 16.Telser J, Fann YC, Renner MW, Fajer J, Wang SK, Zhang H, Scott RA, Hoffman BM. Investigation by EPR and ENDOR spectroscopy of the nickel(I) form of cofactor F-430 of Methanobacterium thermoautotrophicum and of nickel(I) octaethylisobacteriochlorin. J Am Chem Soc. 1997;119:733–743. [Google Scholar]
- 17.Telser J. In: Nickel in F430, in Structure and Bonding. Williams RJP, editor. Springer Verlag: Heidelberg; 1998. pp. 32–63. [Google Scholar]
- 18.Gunsalus RP, WolFe RS. Chromophoric factors F342 and F430 of Methanobacterium thermoautotrophicum. FEMS Microbiol Lett. 1978;3:191–193. [Google Scholar]
- 19.Ellefson WL, Whitman WB, Wolfe RS. Nickel-containing factor F430: chromophore of the methylreductase of Methanobacterium. Proc Natl Acad Sci U S A. 1982;79:3707–10. doi: 10.1073/pnas.79.12.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheesman MR, Ankel-Fuchs D, Thauer RK, Thompson AJ. The magnetic properties of the nickel cofactor F430 in the enzyme methyl-coenzyme M reductase of Methanobacterium thermoautotrophicum. Biochem J. 1989;260:613–616. doi: 10.1042/bj2600613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craft JL, Horng YC, Ragsdale SW, Brunold TC. Nickel Oxidation States of F430 Cofactor in Methyl-Coenzyme M Reductase. J Am Chem Soc. 2004;126:4068–4069. doi: 10.1021/ja038082p. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton CL, Scott RA, Johnson MK. The magnetic and electronic properties of Methanobacterium thermoautotrophicum (Strain ΔH) methyl coenzyme M reductase and its nickel tetrapyrrole cofactor F430: a low temperature magnetic circular dichroism study. J Biol Chem. 1989;264:11605–11613. [PubMed] [Google Scholar]
- 23.Tang Q, Carrington PE, Horng Y-C, Maroney MJ, Ragsdale SW, Bocian DF. X-Ray Absorption and Resonance Raman Studies of Methyl-Coenzyme M Reductase Indicating That Ligand Exchange and Macrocycle Reduction Accompany Reductive Activation. J Am Chem Soc. 2002;124:13242–13256. doi: 10.1021/ja020314h. [DOI] [PubMed] [Google Scholar]
- 24.Piskorski R, Jaun B. Direct determination of the number of electrons needed to reduce coenzyme F430 pentamethyl ester to the Ni(I) species exhibiting the electron paramagnetic resonance and ultraviolet-visible spectra characteristic for the MCR(red1) state of methyl-coenzyme M reductase. J Am Chem Soc. 2003;125:13120–5. doi: 10.1021/ja037862v. [DOI] [PubMed] [Google Scholar]
- 25.Craft JL, Horng YC, Ragsdale SW, Brunold TC. Spectroscopic and computational characterization of the nickel-containing F(430) cofactor of methyl-coenzyme M reductase. J Biol Inorg Chem. 2004;9:77–89. doi: 10.1007/s00775-003-0499-7. [DOI] [PubMed] [Google Scholar]
- 26.Schönheit P, Moll J, Thauer RK. Nickel, cobalt, and molybdenum requirement for growth of Methanobacterium thermoautotrophicum. Archives of Microbiology. 1980;127:59–65. doi: 10.1007/BF00403508. [DOI] [PubMed] [Google Scholar]
- 27.Wasserfallen A, Nolling J, Pfister P, Reeve J, Conway de Macario E. Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov. Int J Syst Evol Microbiol. 2000;50(Pt 1):43–53. doi: 10.1099/00207713-50-1-43. [DOI] [PubMed] [Google Scholar]
- 28.Horng YC, Kunz R, Ragsdale SW. Interaction and reaction of Methyl Coenzyme M Reductase with its potent inhibitor, 3-bromopropane sulfonate. in preparation. [Google Scholar]
- 29.Hausinger RP, Orme-Johnson WH, Walsh C. Nickel tetrapyrrole Cofactor F430: Comparison of the forms bound to Methyl-Coenzyme M Reductase and protein free in cells of Methanobacterium thermoautotrophicum DeltaH. Biochemistry. 1984;23:801–804. [Google Scholar]
- 30.George GN, Pickering I. Laboratory, S S R. Stanford Linear Accelerator Center; Stanford, CA: 2000. [Google Scholar]
- 31.Pelmenschikov V, Blomberg MRA, Siegbahn PEM, Crabtree RH. A Mechanism from Quantum Chemical Studies for Methane formation in Methanogenesis. J Am Chem Soc. 2002;124:4039–4049. doi: 10.1021/ja011664r. [DOI] [PubMed] [Google Scholar]
- 32.Färber G, Keller W, Kratky C, Jaun B, Pfaltz A, Spinner C, Kobelt A, Eschenmoser A. Coenzyme F430 from methanogenic bacteria: complete assignment of configuration based on an X-ray analysis of 12,13-diepi-F430 pentamethyl ester and on NMR spectroscopy. Helv Chim Acta. 1991;74:697–716. [Google Scholar]
- 33.Baerends EJ, Ellis DE, Ros P. Self-consistent molecular Hartree—Fock—Slater calculations I. The computational procedure. Chem Phys. 1973;2:41. [Google Scholar]
- 34.Versluis L, Ziegler T. The Determination of Molecular-Structures by Density Functional Theory - the Evaluation of Analytical Energy Gradients by Numerical-Integration. J Chem Phys. 1988;88:322–328. [Google Scholar]
- 35.Velde GT, Baerends EJ. Numerical-Integration for Polyatomic Systems. J Comp Phys. 1992;99:84–98. [Google Scholar]
- 36.Guerra CF, Snijders JG, te Velde G, Baerends EJ. Towards an order-N DFT method. Theor Chem Acc. 1998;99:391–403. [Google Scholar]
- 37.Vosko SH, Wilk L, Nusair M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin-Density Calculations - a Critical Analysis. Can J Phys. 1980;58:1200–1211. [Google Scholar]
- 38.Becke AD. Density Functional Calculations of Molecular-Bond Energies. J Chem Phys. 1986;84:4524–4529. [Google Scholar]
- 39.Perdew JP. Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 40.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 41.Neese F. ORCA 2.4.02, an ab initio, density functional, and semiempirical program package. Max-Planck-Institut für Bioanorganische Chemie, Mülheim an der Ruhr; Germany: [Google Scholar]
- 42.Becke AD. Density-Functional Thermochemistry .3. The Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 43.Becke AD. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 44.Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti Correlation-Energy formula into a Functional of the Electron-Density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 45.Schafer A, Horn H, Ahlrichs R. Fully Optimized Contracted Gaussian-Basis Sets for Atoms Li to Kr. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 46.Weigend F, Haser M. RI-MP2: first derivatives and global consistency. Theor Chem Acc. 1997;97:331–340. [Google Scholar]
- 47.Schafer A, Huber C, Ahlrichs R. Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 48.Klamt A, Schuurmann G. Cosmo - a New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J Chem Soc - Perkin Trans. 1993:799–805. [Google Scholar]
- 49.Laaksonen L. A graphics program for the analysis and display of molecular dynamics trajectories. J Mol Graphics. 1992;10:33–34. doi: 10.1016/0263-7855(92)80007-z. [DOI] [PubMed] [Google Scholar]
- 50.Shiemke AK, Hamilton CL, Scott RA. Structural heterogeneity and purification of protein-free F430 from the cytoplasm of Methanobacterium thermoautotrophicum. J Biol Chem. 1988;263:5611–6. [PubMed] [Google Scholar]
- 51.Hungate RE. A roll tube method for cultivation of strict anaerobes. In: Norris JR, Ribbons DW, editors. Methods in Microbiology. Chapter IV. Academic Press, Inc; New York: 1969. p. 117. [Google Scholar]
- 52.Gorski A, Vogel E, Sessler JL, Waluk J. Magnetic Circular Dichroism of Octaethylcorrphycene and Its Doubly Protonated and Deprotonated Forms. J Phys Chem A. 2002;106:8139–8145. [Google Scholar]
- 53.Livingston DA, Pfaltz A, Schreiber J, Eschenmoser A, Ankel-Fuchs D, Moll J, Jaenchen R, Thauer RK. Helv Chim Acta. 1984;67:334–351. [Google Scholar]
- 54.Won H, Summers MF, Olson K, Wolfe RS. Two-Dimensional NMR Studies of Native Coenzyme F430. J Am Chem Soc. 1990;112:2178–2184. [Google Scholar]
- 55.Santos CD, Zukerman-Schpector J, Imamura PM. J Braz Chem Soc. 2003;14:998–1004. [Google Scholar]
- 56.Kemp W. Organic Spectroscopy. Macmillan Press; London: 1975. [Google Scholar]
- 57.Stich TA, Seravalli J, Venkateshrao S, Spiro TG, Ragsdale SW, Brunold TC. Spectroscopic Studies of the Corrinoid/Iron-Sulfur Protein from Moorella thermoacetica. J Am Chem Soc. 2006;128:5010–5020. doi: 10.1021/ja054690o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stich TA, Brooks AJ, Buan NR, Brunold TC. Spectroscopic and computational studies of Co3+-corrinoids: spectral and electronic properties of the B12 cofactors and biologically relevant precursors. J Am Chem Soc. 2003;125:5897–914. doi: 10.1021/ja029328d. [DOI] [PubMed] [Google Scholar]
- 59.Stich TA, Buan NR, Brunold TC. Spectroscopic and computational studies of Co2+corrinoids: spectral and electronic properties of the biologically relevant base-on and base-off forms of Co2+cobalamin. J Am Chem Soc. 2004;126:9735–49. doi: 10.1021/ja0481631. [DOI] [PubMed] [Google Scholar]
- 60.Stich TA, Buan NR, Escalante-Semerena JC, Brunold TC. Spectroscopic and computational studies of the ATP:corrinoid adenosyltransferase (CobA) from Salmonella enterica: insights into the mechanism of adenosylcobalamin biosynthesis. J Am Chem Soc. 2005;127:8710–9. doi: 10.1021/ja042142p. [DOI] [PubMed] [Google Scholar]
- 61.Brooks AJ, Vlasie M, Banerjee R, Brunold TC. Spectroscopic and computational studies on the adenosylcobalamin-dependent methylmalonyl-CoA mutase: evaluation of enzymatic contributions to Co-C bond activation in the Co3+ ground state. J Am Chem Soc. 2004;126:8167–80. doi: 10.1021/ja039114b. [DOI] [PubMed] [Google Scholar]
- 62.Casida EM, Jamorski C, Casida KC, Salahub DR. J Chem Phys. 1998;108:4439–4449. [Google Scholar]
- 63.Neese F, Olbrich G. Chem Phys Lett. 2002;362:170–178. [Google Scholar]
- 64.Andruniow T, Kozlowski PM, Zgierski MZ. J Chem Phys. 2001;115:7522–7533. [Google Scholar]
- 65.Liptak MD, Brunold TC. J Am Chem Soc. doi: 10.1021/ja061433q. submitted. [DOI] [PubMed] [Google Scholar]
- 66.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Mascharak PK, Baidya N. X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases. Inorg Chem. 1991;30:920–8. [Google Scholar]
- 67.Smith TA, Penner-Hahn JE, Berding MA, Doniach S, Hodgson KO. J Am Chem Soc. 1985;107:5945–5955. [Google Scholar]
- 68.Bair RA, Goddard WA., III Phys Rev B. 1980;22:2767–2776. [Google Scholar]
- 69.Shiemke AK, Kaplan WA, Hamilton CL, Shelnutt JA, Scott RA. Structural and spectroscopic characterization of exogenous ligand binding to isolated factor F430 and its configurational isomers. J Biol Chem. 1989;264:7276–84. [PubMed] [Google Scholar]
- 70.Shiemke AK, Shelnutt JA, Scott RA. Coordination chemistry of F430. Axial ligation equilibrium between square-planar and bis-aquo species in aqueous solution. J Biol Chem. 1989;264:11236–11245. [PubMed] [Google Scholar]
- 71.Li M. Chemistry. University of Georgia; Athens, GA: 1993. [Google Scholar]
- 72.Goenrich M, Mahlert F, Duin EC, Bauer C, Jaun B, Thauer RK. Probing the reactivity of Ni in the active site of methyl-coenzyme M reductase with substrate analogues. J Biol Inorg Chem. 2004;9:691–705. doi: 10.1007/s00775-004-0552-1. [DOI] [PubMed] [Google Scholar]
- 73.Duin EC, Signor L, Piskorski R, Mahlert F, Clay MD, Goenrich M, Thauer RK, Jaun B, Johnson MK. Spectroscopic investigation of the nickel-containing porphinoid cofactor F(430). Comparison of the free cofactor in the (+)1, (+)2 and (+)3 oxidation states with the cofactor bound to methyl-coenzyme M reductase in the silent, red and ox forms. J Biol Inorg Chem. 2004;9:563–76. doi: 10.1007/s00775-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 74.Harmer J, Finazzo C, Piskorski R, Bauer C, Jaun B, Duin EC, Goenrich M, Thauer RK, Van Doorslaer S, Schweiger A. Spin density and coenzyme M coordination geometry of the ox1 form of methyl-coenzyme M reductase: a pulse EPR study. J Am Chem Soc. 2005;127:17744–55. doi: 10.1021/ja053794w. [DOI] [PubMed] [Google Scholar]
- 75.Telser J, Davydov R, Horng YC, Ragsdale SW, Hoffman BM. Cryoreduction of methyl-coenzyme M reductase: EPR characterization of forms, MCR(ox1) and MCR(red1) J Am Chem Soc. 2001;123:5853–60. doi: 10.1021/ja010428d. [DOI] [PubMed] [Google Scholar]
- 76.Finazzo C, Harmer J, Jaun B, Duin EC, Mahlert F, Thauer RK, Van Doorslaer S, Schweiger A. Characterization of the MCRred2 form of methyl-coenzyme M reductase: a pulse EPR and ENDOR study. J Biol Inorg Chem. 2003;8:586–93. doi: 10.1007/s00775-003-0450-y. [DOI] [PubMed] [Google Scholar]
- 77.Finazzo C, Harmer J, Bauer C, Jaun B, Duin EC, Mahlert F, Goenrich M, Thauer RK, Van Doorslaer S, Schweiger A. Coenzyme B induced coordination of coenzyme M via its thiol group to Ni(I) of F430 in active methyl-coenzyme M reductase. J Am Chem Soc. 2003;125:4988–9. doi: 10.1021/ja0344314. [DOI] [PubMed] [Google Scholar]
- 78.Won H, Olson KD, Hare DR, Wolfe RS, Kratky C, Summers MF. Structural modeling of small molecules by NMR: solution-state structure of 12,13-diepimeric coenzyme F430 and comparison with the x-ray structure of the pentamethyl ester derivative. J Am Chem Soc. 1992;114:6880–92. [Google Scholar]
- 79.Jaun B, Pfaltz A. Coenzyme F430 from methanogenic bacteria: reversible one-electron reduction of F430 pentamethyl ester to the Nickel (I) form. J Chem Soc Chem Commun. 1986;17:1327–1329. [Google Scholar]
- 80.Grabarse WG, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U. On the mechanism of biological methane formation: Structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J Mol Biol. 2001;309:315–330. doi: 10.1006/jmbi.2001.4647. [DOI] [PubMed] [Google Scholar]
- 81.Ragsdale SW. Biochemistry of Methyl-CoM Reductase and Coenzyme F430. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Academic Press; New York: 2003. pp. 205–228. [Google Scholar]
- 82.Pfaltz A, Jaun B, Diekert G, Thauer RK, Eschenmoser A. Helv Chim Acta. 1985;68:1338–1358. [Google Scholar]
- 83.Weissbach H, Peterkofsky A, Redfield BG, Dickerman H. Studies on the Terminal Reaction in the Biosynthesis of Methionine. J Biol Chem. 1963;238:3318–24. [PubMed] [Google Scholar]
- 84.Verdaguer X, Lange UEW, Buchwald SL. Amine Additives Greatly Expand the Scope of Asymmetric Hydrosilylation of Imines. Angew Chem Int Ed Engl. 1998:1103–1107. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1103::AID-ANIE1103>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 85.Brown HC, Hess HM. Selective reductions. XIII. Reaction of 2-cyclopentenones with representative complex hydrides. Aluminum hydride as a selective reagent for the reduction of the carbonyl group in 2-cyclopentenones. J Org Chem. 1969;34:2206–2209. [Google Scholar]
- 86.Johnson MR, Rickborn B. Sodium borohydride reduction of conjugated aldehydes and ketones. J Org Chem. 1970;35:1041–1045. [Google Scholar]
- 87.Deloux L, Srebnik M. Asymmetric boron-catalyzed reactions. Chem Rev. 1993;93:763–784. [Google Scholar]
- 88.Pfaltz A, Kobelt A, Huster R, Thauer RK. Biosynthesis of coenzyme F430 in methanogenic bacteria. Identification of 15,17(3)-seco-F430-17(3)-acid as an intermediate. Eur J Biochem. 1987;170:459–67. doi: 10.1111/j.1432-1033.1987.tb13722.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.