

Abstract
ATP binding cassette (ABC) transporters make up one of the largest superfamilies of proteins known and have been shown to transport substrates ranging from lipids and antibiotics to sugars and amino acids. The dysfunction of ABC transporters has been linked to human pathologies such as cystic fibrosis, hyperinsulinemia, and macular dystrophy. Several bacterial ABC transporters are also necessary for bacterial survival and transport of virulence factors in an infected host. MsbA is a 65kDa protein that forms a functional homodimer consisting of two six-helix transmembrane domains and two approximately 250 amino acid nucleotide binding domains (NBD). The NBDs contain several conserved regions such as the Walker A, LSGGQ and H-motif that bind directly to ATP and align it for hydrolysis. MsbA transports lipid A, its native substrate, across the inner membrane of Gram-negative bacteria. Loss or dysfunction of MsbA results in a toxic accumulation of lipid A inside the cell, leading to cell membrane instability and cell death. Using site-directed spin labeling (SDSL) electron paramagnetic resonance (EPR) spectroscopy, conserved motifs within the MsbA NBD have been evaluated for structure and dynamics upon substrate binding. It has been determined that the LSGGQ NBD consensus sequence is consistent with an α-helical conformation and that these residues maintain extensive tertiary contacts throughout hydrolysis. The dynamics of the LSGGQ and the H-motif region have been studied in the presence of ATP, ADP, and ATP plus vanadate to identify the residues that are directly affected by interactions with the substrate before, after, and during hydrolysis, respectively.
ATP binding cassette (ABC) transporters are one of the largest superfamilies of proteins known (1;2). They are classified by their ability to couple the hydrolysis of ATP to the transport of a variety of substrates either into or out of cells (1;3;4). Defects in endogenous human ABC transporters have been implicated in several pathologies including cystic fibrosis, cholestasis, artherosclerosis, hypoglycemia, hyperbiliruginemia, and macular dystrophy and degeneration diseases (3). Also in humans, ABC transporters such as P-glycoprotein are able to transport chemotherapeutic drugs and lipids resulting in the reduced effectiveness of cancer treatments (5;6). ABC transporters in bacteria are not only essential for survival but are also able to secrete toxins and antimicrobial agents. This ability to expel antimicrobial agents may be involved in the development of multidrug resistance (MDR) (7-13). MDR has become a serious problem facing the medical community today, and its impact can be seen in the treatment of diseases ranging from cancer to human immunodeficiency virus (HIV), to numerous bacterial infections. These facts make ABC transporters an important target in the fight against MDR. By studying these transporters in detail it may be possible to identify new and novel agents to combat infections by pathogens. Bacterial efflux ABC transporters transport a variety of solutes and surface components of the bacterial outer membrane such as capsular polysaccharides, lipopolysaccharides, and teichoic acid. Bacterial ABC transporters have been directly linked to pathogenesis via the export of virulence factors such as hemolysin, heme-binding protein, and alkaline protease, as well as antibiotics, hemes, and siderophores (3;14). ABC transporters may also associate with accessory proteins that mediate the transport of solutes across both the inner and outer membranes simultaneously.
MsbA is a 65 kDa ATPase lipid A transporter found in the inner membranes of Gram-negative bacteria such as E. coli, S. typhimurium, and V. cholerae. Lipid A is a major component of the outer membranes of Gram-negative bacteria and its synthesis and transport is essential for bacterial survival. The functional loss of MsbA results in a toxic accumulation of lipid A within the inner membrane followed by cell death. MsbA is the only known ABC transporter that is essential for bacterial cell viability (15).
ABC transporters contain four domains, two helical membrane spanning domains (MSDs) and two nucleotide binding and hydrolysis domains (NBDs). All four domains, though not always found on the same protein, are conserved in both human and bacterial ABC transporters. The NBDs contain several conserved domains essential for function (Fig 1). The Walker A, LSGGQ, and H-motif bind directly to ATP aligning it for hydrolysis (16-18). The NBDs also contain the Q-loop and Walker B motifs, which coordinate magnesium and water for hydrolysis (3). The energy released through ATP hydrolysis may provide the energy for transport of the lipid A substrate, or to simply reset the MsbA dimer following transport (19). Crystallographic structures suggest that the active conformation of the NBDs consists of the Walker A motif from one NBD and the LSGGQ from the second NBD, forming a “nucleotide-sandwich” (20). Each NBD is able to bind one ATP, and it is proposed that two ATPs are bound to the homodimer to form the catalytically active closed dimer state as demonstrated by the structures of MJ0796 (18), E. coli MalK (21), and Rad50 (22).
Figure 1.
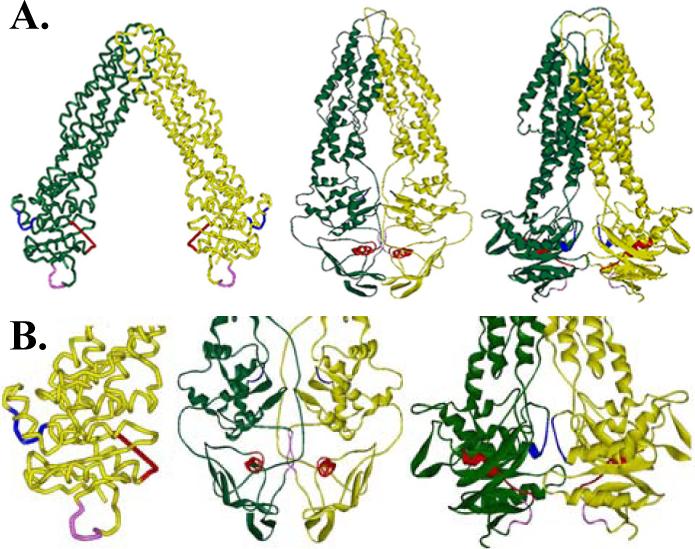
A. Crystal structure of MsbA from E. coli, V. cholera, and S. typhimurium (left to right). B. Enlarged NBD domains from each of the crystal structures are shown with the Walker A (red), LSGGQ (blue), and H-motif (pink) highlighted to demonstrate the differences in these conserved residues between the three structures.
Three crystal structures for MsbA have been solved and provide insight into both its resting and post hydrolysis state conformations (Fig. 1A). Two resting state crystal structures for MsbA have been determined, one from E. coli (23), and a second from V. cholerae (24), however the structures of the protein crystals have significant differences. E. coli MsbA is shown in an open conformation with the transmembrane domains (TMDs) angled away from the axis of symmetry resulting in the periplasmic tips of the helices coming in contact and the cytoplasmic NDBs approximately 40 Å apart. This large gap between the two NBDs has provoked controversy because both NBDs must interact with ATP and likely come together for hydrolysis of ATP (25;26). Also in the E. coli MsbA crystal structure, the NBD conserved Walker A domain is unresolved and the LSGGQ motif appears to be out of position for ATP hydrolysis since it must act in conjunction with the Walker A domain on the opposite monomer in order to bind ATP (Fig. 1B). The second MsbA crystal structure, from V. cholerae, is in a closed conformation with the NBDs in close proximity. (Fig. 1A). However, in this structure, unlike other ABC transporter NBDs that contain 10 β-strands and 6 α-helices tightly packed, this structure contains 6 β-strands and 3 α-helices (3). In addition, this structure is missing several of the dimer contacts observed in other NBDs. The Walker B motif and Q-loop, two additional conserved NBD regions, also do not appear to be in a position to interact with ATP. The third and most recent crystal structure of MsbA is from S. typhimurium (27) and it depicts MsbA in a post hydrolysis state with one ADP and vanadate bound to the NBDs (Fig. 1A). This structure does appear to contain the angled transmembrane domains (TMDs) found in the previous E. coli open conformation state, with an angle of about 30°, however the NBDs are in closer proximity to each other, as expected for the post hydrolysis state.
The SDSL EPR spectroscopy technique has proven to be especially applicable to the study of integral membrane proteins such as MsbA in the solution state and SDSL studies may help to understand the differences between the MsbA crystal structures (28-30). SDSL can also be used to monitor changes that may occur during substrate binding and transport. In this study we evaluate for the first time the NBD LSGGQ and H-motif consensus sequences of E. coli in each of their resting and active state conformations by SDSL EPR spectroscopy.
Experimental Procedures
Materials
2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (MTSL) was purchased from Toronto Research Chemicals (Ontario, Canada), dodecylmaltoside (DM) was purchased from Anatrace (Maumee, OH) and all lipids were purchased from Avanti Polar Lipids (Alabaster, AL).
Methods
Site-directed mutagenesis and protein purification
The gene encoding the E. coli MsbA protein was obtained and inserted into a pET28b vector (Novagen) with an N-terminal 6xHis tag as previously described (28). In addition, both native cysteines (C88 and C315) were previously substituted with serines to create a functional cysteine-less construct. Single cysteines at positions L481, S482, G483, G484, Q485, the LSGGQ, and S540, T541, I542, downstream of the H motif at position 537, were individually introduced into the cysteine-less MsbA plasmid by site-directed mutagenesis using the QuikChange Mutagenesis Kit (Stratagene). Mutant plasmids were sequenced by the MCW Protein and Nucleic Acid Facility (Milwaukee, WI) for verification of the introduced cysteine mutations. The plasmids containing the MsbA gene mutations were transformed into NovaBlue cells (Novagen) and each protein was purified by cobalt affinity chromatography using Talon resin (BD Biosciences Clontech) as described previously (28).
Activity Assays
ATPase activity of the cysteine-less MsbA construct was previously determined to be similar to that of wild type (WT) MsbA (29). All newly introduced cysteine mutants in the cysteine-less background were assayed for ATPase activity in the presence and absence of lipid A using a colorimetric assay to detect the release of Pi following ATP hydrolysis (31). Briefly, the basal level of ATPase activity in each MsbA mutant in detergent micelles was measured followed by activity measurements due to lipid A stimulation in the presence and absence of the spin label MTSL. The ATP activity of all cysteine mutants were able to be stimulated by the addition of lipid A. Lipid A stimulation of ATP activity for each of the cysteine mutants in the LSGGQ region was 5%, 25%, 24%, 5%, and 10%, respectively, and 30%, 10%, and 45% stimulation for the H-motif cysteine mutations at S540, T541, and I542. Activity measurements of MsbA mutants in proteoliposomes were consistent with the activities in detergent. For comparison Lipid A stimulated activity of the cysteine-less background was 45%. Spin labels attached to MsbA within the LSGGQ and in proximity to the H-motif did decrease the lipid A stimulated activity of MsbA by about 45% on average, and the addition of excess ATP, boiled vanadate, and Mg eliminated all ATP hydrolysis activity.
Sample Preparation
MsbA cysteine mutants were labeled with a 10:1 molar ratio of the sulfhydryl-specific spin label 2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (MTSL; Fig. 2) overnight at 4°C in 50mM NaPO4, 0.01% DM, pH 7.0 buffer. Excess label was removed by extensive dialysis against the same buffer, and protein concentrations were determined by the detergent-compatible BCA protein assay (Pierce) using BSA as a protein standard. Synthetic phosphatidylethanolamine (PE), phosphotidylglycerol (PG), and mammalian cardiolipin (CL) were purchased in chloroform and mixed at a molar ratio of 65:25:10 PE:PG:CL to mimic the inner membrane composition of E. coli. The lipid mixture was dried down under nitrogen, desiccated, and then resuspended and solubilized in DM prior to the addition of MsbA at a protein:lipid molar ratio of 1:500. Biobeads (BioRad) were added to the mixture in order to remove the detergent, and the resulting proteoliposome solution was then concentrated by high speed centrifugation (30 min. at 100,000xg). The proteoliposomes were resuspended in the appropriate volume of 50mM NaPO4, pH 7.0 buffer. Proteolipid samples containing substrate typically had final concentrations of 100 μM protein, 40 mM ATP, 2 mM EDTA, 40 mM MgCl2, and 2 mM sodium orthovanadate (Vi), similar to those previously described (29).
Figure 2.
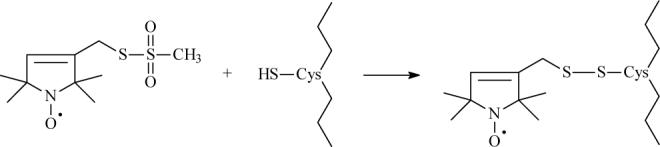
The sulfhydryl-specific spin label MTSL is reacted directly with the introduced cysteine residue to form a covalent disulfide bond between the protein and the spin label.
Electron paramagnetic resonance spectroscopy
X-band continuous wave (CW) EPR spectroscopy was carried out on a Bruker ELEXSYS E500 fitted with a super high Q cavity with samples contained in clay sealed glass capillaries. Power saturation measurements were done on a Varian E-102 Century series spectrometer fitted with a loop-gap resonator (Medical Advances, Milwaukee, WI). Power saturation samples were contained in a gas-permeable TPX capillary for gas equilibration during accessibility measurements. The saturation data were collected and analyzed using a LabView program written by Dr. C. Altenbach (UCLA).
The accessibility of paramagnetic reagents to a spin label can be measured by power saturation experiments. The effective spin-lattice relaxation time, T1, decreases with increasing accessibility, therefore a decrease in the effective T1 results in an increase in the measured power saturation value (P1/2) due to collisions of the spin label with the paramagnetic relaxation reagent. The change in P1/2 relative to a standard measured under N2 (e.g. P1/2(O2) – P1/2(N2) = ΔP1/2(O2)), is directly proportional to the bimolecular collision rate of the spin label with the paramagnetic probe (32). Samples were equilibrated with a stream of either nitrogen or air (20% oxygen) during the power saturation experiments. The P1/2 values for each sample in the presence of nitrogen, air, or 20mM nickel ethylenediaminediacetic acid (NiEDDA) under nitrogen were recorded and calculated as described by Altenbach et al. (33). Proteoliposomes were permeabilized by several freeze thaw cycles to allow NiEDDA to pass through the bilayer.
Results
EPR spectroscopy
SDSL EPR s pectroscopy is a well-established technique to study the dynamics of binding and hydrolysis of ATP during transport within a lipid environment. Spin labels attached to consensus ATP binding and hydrolysis sites within the NBDs are very sensitive to the environment around them. The binding of ATP within the NBDs can result in changes in the motion of the spin label. ATP binding near a spin label, or ATP-induced conformational changes in the protein, will cause either the motion of the spin label to change or force the spin label to adopt a new conformation that can be readily observe d by EPR spectroscopy. It is also possible to determine local secondary structure and changes in the local environment of a spin label due to changes in the accessibility of the spin label to paramagnetic broadening agents.
To determine the changes that occur during ATP binding and hydrolysis, the EPR spectra were recorded at four different stages of the hydrolysis pathway. The four stages are depicted in Figure 3. The first stage is the resting state of MsbA in a synthetic lipid bilayer with no substrate present. The second stage is in the presence of ATP, but the addition of EDTA chelates any magnesium in solution and prevents its hydrolysis and conversion to ADP, locking it in the ATP-bound state for analysis by EPR spectroscopy. The third state studied mimics the high energy intermediate that occurs post hydrolysis where the cleaved inorganic phosphate, which normally rapidly dissociates from the complex, is immediately replaced by vanadate, ultimately preventing the release of ADP. This traps the protein in the high energy intermediate state and allows analysis by EPR methods. The fourth and final stage analyzed is the ADP-bound state induced by the addition of excess ATP in the presence of magnesium, which allows hydrolysis to occur, leaving ADP bound to the protein. The addition of ADP and magnesium directly to the protein was also studied and the EPR spectra were consistent with that of the ATP and magnesium samples.
Figure 3.
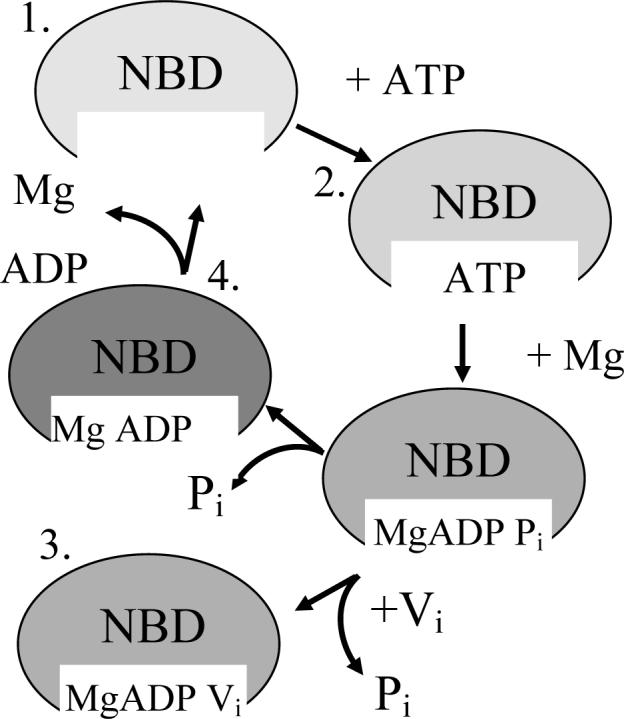
Cartoon of ATP binding and hydrolysis. The MsbA dimer contains two ATP binding sites that must interact to accomplish ATP hydrolysis. The four stages of ATP binding and hydrolysis have each been analyzed and are illustrated here. ATP binding is followed by the addition of MgCl2 immediately followed by sodium orthovanadate (Vi) to first allow hydrolysis of the γ-phosphate from ATP, leading to the ADP and Pi state, which is trapped and mimicked by the replacement of Pi with Vi. The final steps include the release of the Pi, leading to only ADP bound in the pocket, with its complete release reverting the enzyme back to the resting state.
Resting state
Two major motifs were studied: the LSGGQ motif and the H-motif from the NBD (Fig. 1B). Each motif is proposed to be directly involved in ATP binding and hydrolysis. The EPR spectra of the resting state of the labeled mutations in the LSGGQ motif contain two motional components (see arrows in Fig. 4) indicating that each of these sites is involved in extensive tertiary contacts with neighboring side chains. The slow motional component is depicted by the widest peaks from the center and the faster motional component by the narrower peaks observed for sites 481−484. The spectrum of Q485C is unique in that it contains three distinct peaks. Three peaks are typically not seen in EPR spectra of MTSL-labeled sites within α-helical protein segments, indicating that extensive tertiary contacts must exist at this site. Simulations of this spectrum using the Budil and Freed fitting program (34) indicate that it likely arises from only two motional components, with the faster motional component undergoing highly anisotropic motion.
Figure 4.
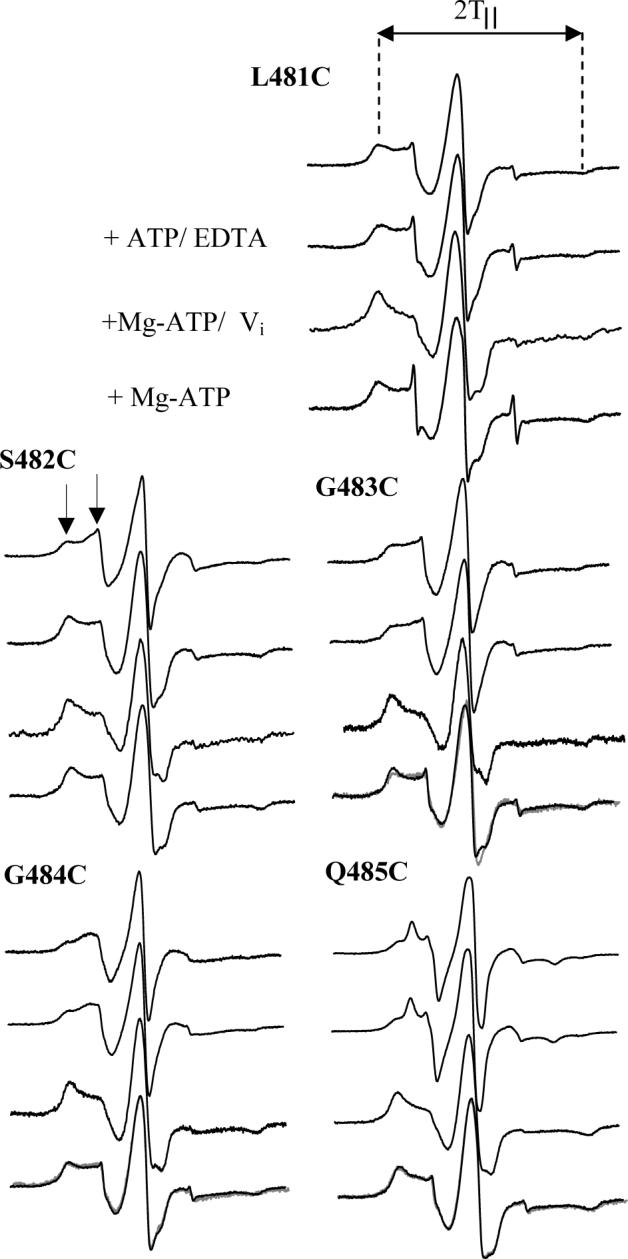
CW X-band EPR spectra of MTSL-labeled MsbA mutants 481−485 reconstituted into membranes. Spectra were recorded at room temperature with a 100G scan width, 10mW microwave power and a typical protein concentration of 100 μM. Spectra without substrate are at the top of each series, with the addition of substrate as indicated. Spectra obtained following addition of magnesium and ATP correspond the Mg-ADP-bound state of MsbA. The gray spectra correspond to the direct addition of excess ADP, as shown for 483 and 484. Arrows denote the two motional components present in the spectrum and outer peak splittings (2T‖) are denoted by dotted lines.
The resting state EPR spectra of the mutations in proximity to the H-motif (540−542) also display two motional components, again a fast motional component with peaks closest to the center and a slower motional component further from the center line (Fig. 5). Interestingly, the spin label at position I542C also displays a spectrum with three distinct peaks in the low-field line resulting from only two motional components, very similar to that observed at Q485C.
Figure 5.
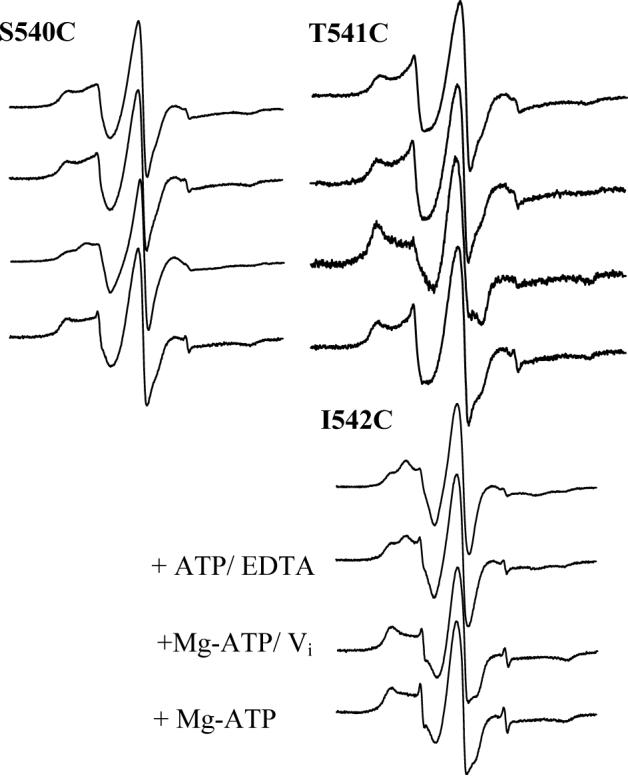
CW X-band EPR spectra of MTSL-labeled MsbA mutants 540−542 reconstituted into membranes. Spectra were recorded under the same conditions as for Fig. 4.
ATP binding and Hydrolysis
Several changes in the motional components of the EPR spectra were observed in both the LSGGQ as well as the H-motif during ATP binding and hydrolysis. The LSGGQ motif contains two types of motional changes. The first type of change was a simple shift in equilibrium from one motional state to another as seen for L481C and S482C (Fig. 4). Specifically, L481C shows no spectral changes in the presence of ATP or ADP (Fig. 4) but does shift from a fast moving component to a slow motional component with an outer peak splitting (2T‖) of 67 G upon vanadate trapping (Table 1). The spin label at position S482C has a shift to the more immobilized component in the presence of ATP, likely due to either direct interaction with ATP or to the formation of new tertiary contacts resulting from a conformational rearrangement within this region of the protein upon binding of ATP. This shift is increased upon binding ATP and vanadate, and remains immobilized in the presence of ADP alone, indicating that this site is directly affected by ATP binding and hydrolysis. The second type of change was the appearance of an additional highly immobilized component at G483C, G484C, and Q485C (Fig. 4). The resting state spectrum of G483C has an outer splitting of 65 G with no observable change in the presence of ATP alone. However; in the presence of ATP and vanadate the splitting widens to 69.5 G, indicative of a highly immobilized component with extensive tertiary contacts with ADP and vanadate or neighboring side chains. In the presence of ADP this immobilization is partially alleviated, but the peaks remain broadened to 67.5 G compared to the 65 G splittings observed for the resting state. For position G484C, the resting state outer splitting of 66 G remains unchanged in the presence of ATP alone, but again broadens to over 68 G in the presence of ATP and vanadate and remains broadened at nearly 68 G in the presence of ADP. The spin label at position Q485C contains three peaks in the resting state with the broadest splitting being 64 G wide, which remains unchanged in the presence of ATP alone but widens in the presence of ATP and vanadate to 67.5 G. Notably, the anisotropic motional component of this spectrum is no longer present, and this two-component spectrum remains unchanged in the presence of ADP following phosphate release. Simulations indicate a high degree of order for these motional components (S > 0.7), but their rotational correlation times remain relatively fast (< 10ns). All of the mutations studied in the LSGGQ motif (481−485) show a shift to the more ordered state when vanadate trapped. ATP does not appear to influence the motional state of most of the mutations with the exception of S482C, believed to be a catalytic residue, where a shift to the immobilized state is observed that remains constant as long as ATP is present (Fig. 4). The spin labels at positions G484C and Q485C also appear to interact with ADP post hydrolysis as well as upon the addition of MgADP.
Table 1.
Outer peak splitting of the LSGGQ and H-motifs (2T∥)
| Resting State | Vanadate Trapped | |
|---|---|---|
| L481C | 66 G | 67 G* |
| S482C | 66.5 G | 67.5 G* |
| G483C | 65 G | 69.5 G |
| G484C | 66 G | 68 G |
| Q485C |
64 G |
67.5 G |
| S540C | 65 G | 68 G |
| T541C | 66 G | 67.5 G |
| I542C | 66 G | 68 G |
Denotes a shift in equilibrium from a fast to a slow moving component
Immediately downstream of the H-motif, similar changes occur at positions 540, 541 and 542 (Fig. 5). Specifically, the spin label at position S540C does not change upon ATP binding, but develops a third intermediate peak in the vanadate trapped state. This intermediate peak disappears in the presence of ADP, resembling both the resting and ATP-bound states. As seen for S540C, the spin label at position T541C remains largely unchanged in the presence of ATP but the spectrum broadens to 67.5 G with a significant increase in the immobilized state in the presence of ATP and vanadate (Fig. 5). After hydrolysis, the spectrum reverts to essentially the same motion observed for the resting and ATP-bound states. The third mutation in the H-motif region, I542C, shows three peaks in the resting state spectrum. Again, no obvious changes were observed in the presence of ATP, but the addition of vanadate causes the three peaks to change to a two component spectrum with the outer peaks nearly 68 G apart. However, unlike the previous two sites studied, the spectrum of I542C does not revert back to the resting and ATP-bound motion in the presence of ADP (Fig. 5).
In summary, only position S482C in the LSGGQ motif showed a change in motion of the spin label upon binding to ATP. This change in the EPR spectrum of S482C is consistent with close proximity to ATP that would be in agreement with previous data indicating that S482 is a catalytic residue. Nevertheless, every site studied in the LSGGQ domain became immobilized in the high-energy vanadate trapped state, showing nearly identical slow motion spectra. The increase in the slow motional component likely indicates close proximity to the penta-coordinate MgADP and vanadate complex but it is also possible that changes in the backbone conformation led to increased tertiary contacts with the label. Interestingly, post hydrolysis, where only ADP was present, sites 481 and 482 reverted back to the ATP-bound motion, whereas the remaining three sites exhibited spectral motion more similar to that seen in the vanadate trapped states, but distinct from either of the previous stages of ATP hydrolysis indicating either interactions with MgADP or possibly a change in the backbone motion due to MgADP binding.
Similarly, each of the sites in proximity to the H-motif showed no change upon ATP binding and large changes upon vanadate trapping. Unlike its two neighboring sites, the spin label at I542C did not revert back to the resting state motion in the post hydrolysis ADP-bound state, but remains in a vanadate-trapped-like conformational state. Because these sites are not thought to directly interact with ATP during hydrolysis it would seem likely that backbone motional changes and/or changes in the tertiary contacts with neighboring side chains are contributing to the motional changes observed upon vanadate trapping and in the presence of MgADP. Overall, our data indicate that site 482 is the only residue within the LSGGQ and H- motifs that is directly affected by ATP binding alone, whereas all of the sites studied showed significant motional effects in the vanadate trapped state. In addition, sites 481, 540, and 541 return to their resting state motion post hydrolysis, however 483, 484, 485, and 542 all retain essentially the same motion post hydrolysis that was observed upon vanadate trapping, indicating these sites retain the same interaction or conformational state even after loss of the hydrolyzed phosphate.
Accessibility Studies
CW power saturation EPR spectroscopy is a proven technique to determine the local environment of introduced spin label side chains within a large protein (33). Studying the local environment of side chains provides information about the local structure. High accessibility to oxygen generally indicates a hydrophobic environment while high accessibility to NiEDDA indicates an aqueous environment. Again, two major regions were studied within the NBD, the LSGGQ and H-motif ATP binding regions. Each spin labeled mutant from L481-Q485 was subjected to power saturation analysis to determine the local environment surrounding each site. Power saturation was done on each mutant in the presence of either nitrogen, air (20% oxygen), or NiEDDA in the presence of nitrogen.
Accessibilities were determined for both the resting and vanadate trapped states (Fig. 6). The region from 481−485 in the resting state appears to have a periodicity of 3.6, consistent with the α-helical conformation proposed in the crystal structures (Fig. 6). The relative accessibility of residues 481−485 in the resting state to both oxygen and NiEDDA are similar for all sites. Power saturation studies show a large decrease in the accessibility to both oxygen and NiEDDA in the vanadate trapped states indicating that ADP and vanadate create significant tertiary contacts within or surrounding the LSGGQ motif and reduce access of the paramagnetic reagents to the spin labeled side chains (Fig. 6).
Figure 6.
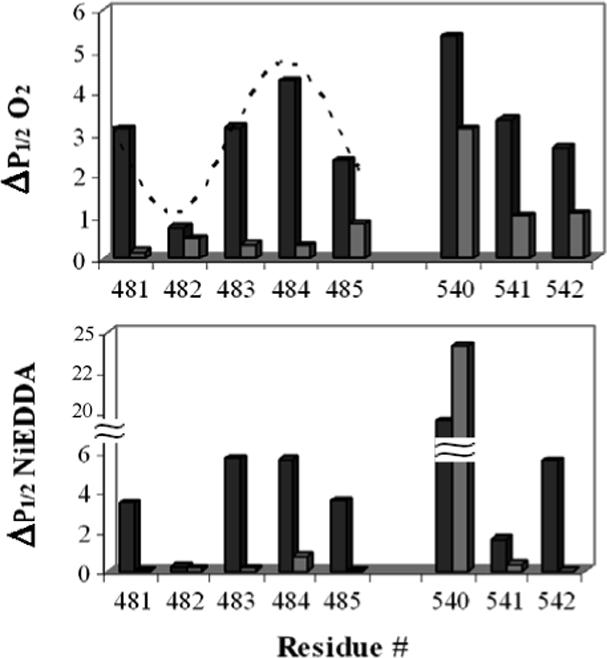
Oxygen and NiEDDA accessibility comparison for both the LSGGQ (481−485) and H-motif (540−542). The top panel shows the oxygen accessibility of the indicated positions in the resting (black bars) and vanadate trapped (grey bars) states. Lower panel shows the accessibility to NiEDDA. The dotted line overlaid on the upper graph is a sine wave with a periodicity of 3.6.
The H-motif was also analyzed by power saturation and a significant decrease in the NiEDDA and oxygen accessibilities was also observed for positions 541 and 542 in the presence of ADP and vanadate (Fig. 6). This dramatic decrease in the accessibility of the spin labels to paramagnetic agents demonstrates that ADP and vanadate block the spin labeled side chains either directly or indirectly through conformational rearrangements of these motifs within the protein structure. Position 540 is slightly different in that the accessibility to oxygen decreases in the presence of vanadate but the accessibility to NiEDDA actually increases (Fig. 6). Although the initial accessibility to NiEDDA is high, the further increase indicates an aqueous pocket forms near this site during ATP hydrolysis.
Discussion
This study directly describes the local structure of the LSGGQ consensus motif in the solution state as an α-helix that becomes less solvent accessible and more tightly packed during ATP binding and hydrolysis. The effects of ATP binding and hydrolysis have been analyzed by EPR spectroscopy to determine which residues are affected by the interaction of MsbA with ATP before, during, and post hydrolysis. It should be mentioned that although ATPase activities for several of these mutants is low when spin labeled, both the ability to be stimulated by lipid A and the observed EPR changes do indicate that ATP hydrolysis does still occur and does represent the native function of MsbA. To confirm that these point mutations do not affect the global structure of MsbA, circular dichroism data also indicate that the overall secondary structure of MsbA is intact in both detergent and liposomes (data not shown). It is likely then that these point mutations and further spin labeling may weaken the interaction of MsbA and ATP thus slowing hydrolysis but not eliminating it entirely. ATP in the presence of EDTA directly affects the motion of the spin label side chain at residue 482 in the LSGGQ region. Positions 481, 483−485, and 540−542 do not appear to be significantly affected by the addition of ATP. This may be due to the lack of magnesium, which may help in the coordination of ATP in the NBD, or indicate only weak interactions at the sites studied that then becomes stronger upon ATP hydrolysis, as demonstrated by the immobilization of the spin label immediately post hydrolysis. Significant changes are observed in the presence of ATP, magnesium, and vanadate resulting in a trapped high energy intermediate state that leads to an EPR shift to an immobilized state in all five LSGGQ residues as well as the 540−542 residues near the H-motif. These results indicate that the LSGGQ and H-motif are directly involved in ATP hydrolysis during transport. It is also interesting to note that positions 483, 484, 485, and 542 also appear to interact with ADP post hydrolysis. In total, these results are consistent with those observed for other ABC transporters where the LSGGQ and H-motifs are directly involved in ATP binding and hydrolysis during transport, such as in the MalK, Rad50, and MJ0796 ATP-bound crystal structures (21;22;26).
The multiple component EPR spectra can be accounted for in two ways. The first possibility is an equilibrium between two conformational states of MsbA, and the second, more likely possibility is the presence of multiple preferred conformational states of the spin label side chains (35) . The shift from a fast motional component of the spectrum to a more immobilized component is likely due to a change in the local conformation of MsbA leading to a shift in the preferred rotameric conformation of the spin label. It is interesting to note that the spin labels at all positions studied show motional components indicative of significant tertiary contacts not typically seen in soluble α-helical structures. Therefore, our data contradict the placement of the LSGGQ region as at least partially water soluble in each of the three MsbA crystal structures (Fig. 1), and suggest that this region is buried within the NBD structure throughout the ATP hydrolysis cycle.
Studies of the paramagnetic accessibility indicate that residue 481 is in a hydrophobic environment and residue 482 has low accessibility to both oxygen and nickel but is slightly hydrophobic and likely packed against the protein backbone. Positions 483 and 484 are accessible to both oxygen and NiEDDA indicating an intermediate environment while 485 is partially protected with moderate accessibility to both oxygen and NiEDDA. These studies are also consistent with the LSGGQ motif being in an α-helical state, which is in general agreement with the previous E. coli and S. typhimurium crystal structures, but differs from the LSGGQ in the V. cholerae crystal structure. Also in the S. typhimurium MsbA structure, the LSGGQ motif appears to be accessible to the solvent in the post hydrolysis vanadate tapped state, which is in stark contrast to our results that indicate that the vanadate trapped state of each of these residues has very little accessibility to the solvent (Fig. 1). In addition, the LSGGQ in the V. cholerae crystal structure has some exposure to the solvent on one monomer but is blocked on the other by the surrounding protein structure. In the E. coli crystal structure, the LSGGQ motif is completely exposed on the outer surface of the protein. Our data indicate that residues 482 and 485 are protected from the solvent but that 481, 483, and 484 are very solvent accessible. This varied accessibility indicates a conformation with at least partial packing against the remaining protein structure as seen in the S. typhimurium and V. cholerae crystal structures. This data is not in agreement with that from the E. coli crystal structure where the LSGGQ is completely exposed and protruding away from the protein backbone. The post hydrolysis crystal structure from S. typhimurium shows that the nucleotide is removed from the LSGGQ motif in the vanadate trapped state. Our data indicate that the LSGGQ must be in contact with vanadate and ADP to account for the large drop in accessibility to both oxygen and NiEDDA.
The H-motif is also depicted in the E. coli crystal structure as a highly exposed loop protruding away from the main protein backbone, however both the V. cholerae and the S. typhimurium MsbA crystal structures depict the H-motif more tightly packed against the protein backbone. The accessibility data presented here indicate that the H-motif is likely packed against the protein backbone since variation occurs in the accessibility of oxygen and NiEDDA. The H-motif must also make extensive contacts with ADP and vanadate or undergo significant structural rearrangement as indicated by the large decrease in the accessibility to oxygen and NiEDDA, as well as decreased mobility, in the vanadate trapped state. Position 540 near the H-motif has high accessibility to both oxygen and NiEDDA. Interestingly in the presence of vanadate this accessibility increases indicating more exposure to the solvent.
Acknowledgements
We thank Kyle Bantz for general assistance on this project and Jimmy Feix for critical reading of the manuscript. This work was supported by NIH grant GM070642.
Abbreviations
- SDSL
site-directed spin labeling
- EPR
electron paramagnetic resonance
- NiEDDA
nickel ethylenediaminediacetic acid
- MTSL
methanethiosulfonate spin label
- DM
dodecylmaltopyranoside
- CW
continuous wave
Footnotes
This work was supported by grant GM070642 from the NIH.
References
- 1.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 2.Dassa E, Hofnung M, Paulsen IT, Saier MH., Jr. The Escherichia coli ABC transporters: an update. Mol. Microbiol. 1999;32:887–889. doi: 10.1046/j.1365-2958.1999.01392.x. [DOI] [PubMed] [Google Scholar]
- 3.Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu. Rev. Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 4.Holland IB, Blight MA. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 1999;293:381–399. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- 5.Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, Haga N. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003;94:15–21. doi: 10.1111/j.1349-7006.2003.tb01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 7.Bolhuis H, van Veen HW, Molenaar D, Poolman B, Driessen AJ, Konings WN. Multidrug resistance in Lactococcus lactis: evidence for ATP-dependent drug extrusion from the inner leaflet of the cytoplasmic membrane. EMBO J. 1996;15:4239–4245. [PMC free article] [PubMed] [Google Scholar]
- 8.Chang G. Multidrug resistance ABC transporters. FEBS Lett. 2003;555:102–105. doi: 10.1016/s0014-5793(03)01085-8. [DOI] [PubMed] [Google Scholar]
- 9.Gros P, Croop J, Housman D. Mammalian multidrug resistance gene: complete cDNA sequence indicates strong homology to bacterial transport proteins. Cell. 1986;47:371–380. doi: 10.1016/0092-8674(86)90594-5. [DOI] [PubMed] [Google Scholar]
- 10.Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr. Relat Cancer. 2003;10:43–73. doi: 10.1677/erc.0.0100043. [DOI] [PubMed] [Google Scholar]
- 11.Levy SB. Active efflux mechanisms for antimicrobial resistance. Antimicrob. Agents Chemother. 1992;36:695–703. doi: 10.1128/aac.36.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu EW, Aires JR, Nikaido H. AcrB multidrug efflux pump of Escherichia coli: composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity. J. Bacteriol. 2003;185:5657–5664. doi: 10.1128/JB.185.19.5657-5664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poole K. Overcoming multidrug resistance in gram-negative bacteria. Curr. Opin. Investig. Drugs. 2003;4:128–139. [PubMed] [Google Scholar]
- 14.Binet R, Letoffe S, Ghigo JM, Delepelaire P, Wandersman C. Protein secretion by Gram-negative bacterial ABC exporters--a review. Gene. 1997;192:7–11. doi: 10.1016/s0378-1119(96)00829-3. [DOI] [PubMed] [Google Scholar]
- 15.Karow M, Georgopoulos C. The essential Escherichia coli msbA gene, a multicopy suppressor of null mutations in the htrB gene, is related to the universally conserved family of ATP-dependent translocators. Mol. Microbiol. 1993;7:69–79. doi: 10.1111/j.1365-2958.1993.tb01098.x. [DOI] [PubMed] [Google Scholar]
- 16.Austermuhle MI, Hall JA, Klug CS, Davidson AL. Maltose-binding protein is open in the catalytic transition state for ATP hydrolysis during maltose transport. J. Biol. Chem. 2004;279:28243–28250. doi: 10.1074/jbc.M403508200. [DOI] [PubMed] [Google Scholar]
- 17.Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure (Camb.) 2001;9:571–586. doi: 10.1016/s0969-2126(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 18.Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF. The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J. Biol. Chem. 2001;276:32313–32321. doi: 10.1074/jbc.M100758200. [DOI] [PubMed] [Google Scholar]
- 19.Lu G, Westbrooks JM, Davidson AL, Chen J. ATP hydrolysis is required to reset the ATP-binding cassette dimer into the resting-state conformation. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17969–17974. doi: 10.1073/pnas.0506039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Lu G, Lin J, Davidson AL, Quiocho FA. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 2003;12:651–661. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- 23.Chang G, Roth CB. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science. 2001;293:1793–1800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- 24.Chang G. Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J. Mol. Biol. 2003;330:419–430. doi: 10.1016/s0022-2836(03)00587-4. [DOI] [PubMed] [Google Scholar]
- 25.Higgins CF, Linton KJ. Structural biology. The xyz of ABC transporters. Science. 2001;293:1782–1784. doi: 10.1126/science.1065588. [DOI] [PubMed] [Google Scholar]
- 26.Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ. Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 2002;277:21111–21114. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reyes CL, Chang G. Structure of the ABC transporter MsbA in complex with ADP-vanadate and lipopolysaccharide. Science. 2005;308:1028–1031. doi: 10.1126/science.1107733. [DOI] [PubMed] [Google Scholar]
- 28.Buchaklian AH, Funk AL, Klug CS. Resting state conformation of the MsbA homodimer as studied by site-directed spin labeling. Biochemistry. 2004;43:8600–8606. doi: 10.1021/bi0497751. [DOI] [PubMed] [Google Scholar]
- 29.Buchaklian AH, Klug CS. Characterization of the Walker A motif of MsbA using site-directed spin labeling electron paramagnetic resonance spectroscopy. Biochemistry. 2005;44:5503–5509. doi: 10.1021/bi047568v. [DOI] [PubMed] [Google Scholar]
- 30.Dong JH, Yang GY, Mchaourab HS. Structural basis of energy transduction in the transport cycle of MsbA. Science. 2005;308:1023–1028. doi: 10.1126/science.1106592. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Romo P, Sanchez-Nieto S, Gavilanes-Ruiz M. A modified colorimetric method for the determination of orthophosphate in the presence of high ATP concentrations. Anal. Biochem. 1992;200:235–238. doi: 10.1016/0003-2697(92)90458-j. [DOI] [PubMed] [Google Scholar]
- 32.Altenbach C, Flitsch SL, Khorana HG, Hubbell WL. Structural studies on transmembrane proteins. 2. Spin labeling of bacteriorhodopsin mutants at unique cysteines. Biochemistry. 1989;28:7806–7812. doi: 10.1021/bi00445a042. [DOI] [PubMed] [Google Scholar]
- 33.Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc. Natl. Acad. Sci. U. S. A. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-Least-Squares Analysis of Slow-Motion EPR Spectra in One and Two Dimensions Using a Modified Levenberg-Marquardt Algorithm. Journal of Magnetic Resonance. 1996;A120:155–189. [Google Scholar]
- 35.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]