

Abstract
AhpC and AhpF from Salmonella typhimurium undergo a series of electron transfers to catalyze the pyridine nucleotide-dependent reduction of hydroperoxide substrates. AhpC, the peroxide-reducing (peroxiredoxin) component of this alkyl hydroperoxidase system, is an important scavenger of endogenous hydrogen peroxide in bacteria and acts through a reactive, peroxidatic cysteine, Cys46, and a second cysteine, Cys165, that forms an active site disulfide bond. AhpF, a separate disulfide reductase protein, regenerates AhpC every catalytic cycle via electrons from NADH which are transferred to AhpC through a tightly-bound flavin and two disulfide centers, Cys345-Cys348 and Cys129-Cys132, through putative large domain movements. In order to assess cysteine reactivity and inter-domain interactions in both proteins, a comprehensive set of single and double cysteine mutants (replacing cysteine with serine) of both proteins were prepared. Based on 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) and AhpC reactivity with multiple mutants of AhpF, the thiolate of Cys129 in the N-terminal domain of AhpF initiates attack on Cys165 of the intersubunit disulfide bond within AhpC for electron transfer between proteins. Cys348 of AhpF has also been identified as the nucleophile attacking the Cys129 sulfur of the N-terminal disulfide bond to initiate electron transfer between these two redox centers. These findings support the modular architecture of AhpF and its need for domain rotations for function, and emphasize the importance of Cys165 in the reductive reactivation of AhpC. In addition, two new constructs have been generated, an AhpF-AhpC complex and a “twisted” form of AhpF, in which redox centers are locked together by stable disulfide bonds which mimic catalytic intermediates.
Keywords: peroxiredoxins, peroxidases, flavoproteins, disulfide redox centers, antioxidants
INTRODUCTION
Findings of the past decade have revealed that cysteine-based peroxidases, including non-selenium-containing glutathione peroxidase homologues and peroxiredoxins (Prxs)1, serve not only as detoxifying systems but also as regulators of signal transduction pathways and peroxide responses (2-5). In many eubacteria, a major player in hydrogen peroxide scavenging is the alkyl hydroperoxide reductase protein AhpC (6, 7). Physical and functional properties of this Prx are relatively well characterized and have led to some astonishing discoveries about this group of enzymes (8). These enzymes catalyze peroxide reduction through a catalytic Cys (C46 in Salmonella typhimurium AhpC). Upon oxidation to a sulfenic acid (R-SOH) by hydrogen peroxide, organic hydroperoxides or peroxynitrite, C46 becomes reoriented through local unfolding of about 10 residues in the active site loop and helix region to reach across and form a disulfide bond with the resolving Cys (C165 in Salmonella typhimurium AhpC) on another subunit (1, 3, 9, 10). These reversibly disulfide-bonded dimers, with two symmetrical active sites per dimer, are thus the minimal unit of activity. However, these dimers also undergo additional oligomerization to form striking toroid-shaped decameric (α2)5 complexes. Decamer formation is promoted to a much lesser extent in the oxidized protein than in the reduced form; our studies have also shown that the decamerization of reduced AhpC contributes significantly to the very high efficiency of peroxide reduction catalyzed by these enzymes (10, 11). This feature of redox-dependent oligomerization which regulates enzyme activity is relevant, perhaps to varying degrees, to the very large group of related “typical 2-Cys” Prxs of higher organisms, as well (12, 13). In addition, even further aggregation of eukaryotic Prx decamers under conditions of oxidative stress may trigger cellular responses and/or switch on a chaperone activity that may help protect cellular proteins (14-17).
In the catalytic cycle of AhpC, only the peroxidatic Cys is required in the peroxide reduction step; this Cys is conserved among all Prxs and is the only participating protein thiol group in Prxs which employ a “1-Cys” mechanism. Most of the Prxs, including AhpC, also possess a conserved, second “resolving” Cys that forms a disulfide bond with the sulfenic acid form of the peroxidatic Cys prior to reactivation of the protein by reduction. In the vast majority of Prxs this resolving Cys is located within the C-terminal tail that reaches across the interface to form the intersubunit disulfide bond of the oxidized Prxs (i.e., the “typical” 2-Cys Prxs, Fig. 1, site 4). In other families of Prxs, the so-called “atypical 2-Cys” Prxs, the resolving Cys is positioned in one of three distinct sites (in helix α2, helix α3 or the loop between strand β7 and helix α5 for BCP, Tpx and Prx5 classes of Prxs, respectively). In these proteins, the resolving Cys forms an intrasubunit disulfide bond with the peroxidatic Cys upon oxidation (13). The enzymatic function of the resolving Cys is certainly in part to protect the enzyme from losing activity due to further oxidation of the sulfenic acid at the active site. By its participation in the disulfide bond with the peroxidatic Cys, it also plays a role in reduction; given the nature of dithiol-disulfide interchange, it must act either as the point of initial thiolate attack by the reductant or as the leaving group during this first step (18). The structural arrangement of the disulfide bond in typical 2-Cys Prxs suggests that the resolving Cys, C165 in AhpC, is most accessible and likely to be the point of attack by the dithiol-containing reductant (10, 19). One study has investigated this point experimentally. In tryparedoxin peroxidase (another typical 2-Cys Prx), this resolving Cys sulfur of the disulfide bond was verified as the point of attack using a mutant version of the reductant, tryparedoxin, lacking the second Cys of the CXXC motif, and mass spectrometric analyses of the disulfide-containing peptide that was generated (20).
Figure 1.
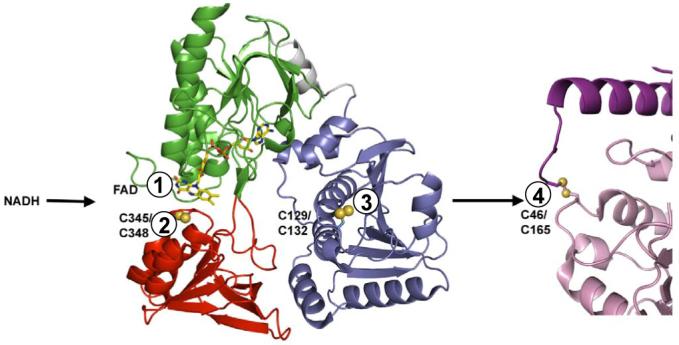
Electron transfer centers in the AhpF-AhpC enzyme system. Shown by numbers for the sequential transfers of reducing equivalents are the redox centers in AhpF (1 is FAD, 2 is C345-C348, and 3 is C129-C132) and AhpC (4 is the C46-C165 intersubunit disulfide center) which mediate transfer of electrons between NADH and the hydroperoxide substrates directly reduced by C46 in AhpC. Structures shown are a monomeric depiction of dimeric AhpF from pdb file 1hyu (NAD/SS domain in red, FAD domain in green, and N-terminal domain in blue) (21), and about half of a symmetrical head-to-tail dimer from the decameric structure of oxidized AhpC, pdb file 1yep (10, 11).
True AhpC-like proteins from bacteria are reduced efficiently not by thioredoxin (Trx)1, the generalized reductant of many typical and atypical 2-Cys Prxs, but by a specialized flavoprotein, AhpF, that replaces both Trx reductase (TrxR) and Trx as the reductase “system”. AhpF possesses three domains in total; two of these in the C-terminal region of AhpF, which correspond to homologous domains in bacterial TrxR (34% identity), are depicted in green (the flavin-binding, or FAD, domain) and red (the pyridine nucleotide-binding and redox disulfide-containing, or NAD/SS, domain) in Fig. 1. These domains, through putative large domain movements, shuttle electron pairs from NADH, through FAD and the TrxR-like redox-active disulfide center (C345,C348 in S. typhimurium AhpF), to the third, Trx-like domain at the N-terminus of AhpF (blue domain in Fig. 1). Not only does the C129,C132 disulfide center of this domain carry out a Trx-like function in mediating electron transfer from the TrxR-like part of AhpF to its protein disulfide substrate, AhpC, it also exhibits a “double Trx” fold, with two repeats of the fold in tandem but intimately associated into a single domain (21). A number of approaches, including the engineering of truncated and chimeric constructs of AhpF, have verified its functional analogy to a TrxR-plus-Trx fusion protein (8, 22-24). Furthermore, these studies, in addition to the generation of heterodimeric mutant constructs (8, 25), have demonstrated that electron transfer through AhpF is an intrasubunit process where the “distal” electron acceptor, the dithiol center in the N-terminal domain (C129,C132), is the direct reductant of AhpC (Fig. 1). Less clear has been the specific role of each of the cysteinyl residues of AhpF in each step of the electron transfer process, although analogies to the TrxR and Trx system, and the modeling of a “rotated” conformation of the AhpF structure determined by X-ray crystallography, have suggested that C348 and C129 participate in a disulfide-bonded intermediate and that C129 would be the likely attacking thiolate in reducing AhpC (8, 21).
The goal of this investigation was to experimentally address the identity and reactivity of the actual attacking and resolving cysteinyl residues in each protein, AhpF and AhpC, in the course of the multiple dithiol-disulfide electron transfer steps occurring through these proteins. In addition, the intent was to generate disulfide-linked complexes mimicking catalysis for crystallographic studies of distinct, functional conformations of these two proteins. Our approaches included complementary protein chemical and kinetic methods to study a comprehensive series of cysteine mutants of AhpF and AhpC. Portions of these results have been presented in abbreviated form (26, 27).
EXPERIMENTAL PROCEDURES
Materials
NADH was purchased from Boehringer Mannheim. Sigma was the supplier of FAD, 3-acetylpyridine adenine dinucleotide (AcPyAD+), molecular biology grade ammonium sulfate, Tris, protocatechuic acid, and protocatechuate-3,4-dioxygenase. TPCK-treated trypsin was purchased from Worthington Biochemicals. Difco bacteriological media, sodium dithionite, and organic solvents were from Fisher. Other reagents and buffer components including 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), sodium dodecyl sulfate (SDS), 2-mercaptoethanol, 1,4-dithiothreitol (DTT) and ultrapure urea were purchased from Research Organics, Inc. Ultrapure guanidine hydrochloride was from Gibco BRL. Restriction enzymes, T4 DNA ligase, calf-intestinal alkaline phosphatase, and other DNA-modifying enzymes were obtained from New England Biolabs or Promega. [α-35S]dATP for DNA sequence analysis was purchased from New England Nuclear. The standard buffer used in most experiments was 50 mM potassium phosphate with 1 mM ethylenediamine tetraacetic acid (EDTA) at pH 7.0 unless otherwise noted.
Mutagenesis, Expression and Purification of Mutant Enzymes
C345S and C348S mutants of AhpF were generated by mutagenesis and purified as described previously (22). In order to replace C129 and C132 of AhpF with Ser or Ala residues, two mutagenic oligonucleotides were designed: 5′-TTCACTCTCC(T/G)CCCATAACTGCCCGGACGTAG-3′and 5′-TTCACTCTCCTGCCATAAC(T/G)CCCCGGACGTAG-3′ (oligonucleotide synthesis was performed in the DNA Synthesis core Laboratory of the Comprehensive Cancer Center of Wake Forest University, supported in part by NIH Grant CA 12197). For mutagenesis, single stranded template was generated using Stratagene R408 helper phage from the phagemid vector pBlF2 as described earlier [entire coding region for AhpF cloned into pBluescriptII SK(+)] (22). Following mutagenesis with the Sculptor in vitro mutagenesis kit from Amersham, mutants were screened and appropriate mutations identified by sequencing of single-stranded DNA using the Sequenase Version 2 sequencing kit (United States Biochemicals). The mutant-containing plasmids were digested with EagI-EcoRI, and the 867 bp fragments were subcloned into the EagI-EcoRI sites of pAF1 (a derivative of the expression vector pOXO4 containing ahpF) (28). For generation of the four double mutants, these same EagI-EcoRI fragments were subcloned into these sites in the C345S- or C348S-expressing mutant forms of pAF1. Mutants C129S/C345S and C132S/C345S were expressed in the presence of 2 mM fresh DTT and purified and stored at -20 °C in the presence of 5 mM DTT to prevent premature formation of disulfide bonds. The DTT was removed before use by gel filtration over a 30 mL G-25 column (Pharmacia) into our standard purification buffer, 25 mM potassium phosphate, with 1 mM EDTA, at pH 7.0. A similar subcloning strategy was used to generate the mutant of AhpF lacking all four Cys residues of interest, the C129S/C132S/C345S/C348S mutant (4C→S for short2), using previously-generated double mutants of AhpF (22).
The N-terminal domain fragment of AhpF, comprising residues 1-202, was mutated to generate C132S as described above for intact AhpF and was expressed and purified as previously described (24). The expression and purification of the recombinant AhpF and AhpC proteins were performed as described previously (29, 30).
Equipment for Spectral and Anaerobic Experiments
A thermostatted Milton Roy Spectronic 3000 diode array spectrophotometer with 0.35 nm resolution was used to collect data for all absorbance spectra, some enzymatic assays and anaerobic titration experiments. Several spectral assays were carried out using a thermostatted Gilford 220 recording spectrophotometer with a Beckman DU monochromator and a Kipp and Zonen (Delft, The Netherlands) chart recorder. For faster spectroscopic rates, a thermostatted Applied Photophysics DX.17MV stopped-flow spectrofluorometer was used. Fluorescence spectra were recorded using a SLM Aminco Bowman Series 2 luminescence spectrophotometer.
Spectral Properties of AhpF
Fluorescence intensity measurements were compared each time with that of an 8.8 μM FAD standard solution (A450 = 0.1); the concentrations of wild-type and mutant proteins ranged between 7.6 and 9.0 μM, corresponding to an A450 of 0.1. The molar extinction coefficients of the protein-bound FAD at 447 or 449 nm were determined by release of the flavin cofactor with 4 M guanidine hydrochloride (GuHCl) and quantitation compared to the corresponding free FAD. Extinction coefficients used for all reduced and oxidized pyridine nucleotides, free FAD, and 2-nitro-5-thiobenzoate (TNB) were those previously reported (29).
Thiol Quantitation Assays for Free Thiol Groups
Protein was incubated with 150 μM DTNB in standard phosphate buffer. The amount of TNB generated was measured at 412 nm until it reached a plateau, at 5-20 min for most mutants, but closer to 1 hr for the native C345S mutant of AhpF. Denaturing conditions, when needed, were achieved by adding an equivalent volume of 8 M GuHCl adjusted with potassium phosphate to pH 7.0. To assess the amount of TNB remaining bound to the protein after DTNB reaction with AhpF double cysteine mutants, excess DTNB was removed by extensive concentration and redilution using an Apollo ultrafiltration unit (30 kDa cutoff) from Orbital Biosciences (Topsfield, MA), then the protein-linked TNB was released by the addition of a 100-fold excess of DTT. For non-reducing SDS-polyacrylamide gel electrophoresis, a 100-fold excess of N-ethylmaleimide was added to the sample buffer to prevent scrambling of thiols.
NADH-Dependent DTNB Reductase Assays
Turnover assays with DTNB and NADH as the oxidizing and reducing substrates, respectively, were conducted as described previously (29) in buffer containing 50 mM Tris-HCl at pH 8.0, 0.5 mM EDTA, and 100 mM ammonium sulfate. The latter component was previously demonstrated to have a moderate activating effect on AhpF when included in peroxidase, DTNB reductase and transhydrogenase assays (29).
Kinetics of Thiol Modification by DTNB with AhpF Mutants
The release of TNB upon treatment of mutants with DTNB was monitored using continuous absorbance measurements at 412 nm in 25 mM potassium phosphate, 1 mM EDTA (pH 7.0) at 25 °C. DTNB was varied between 70 μM and 310 μM whereas the AhpF concentration was 7 μM. This resulted in a total absorbance change of 0.2 at 412 nm for two thiol groups. The absorbance data were fitted using SigmaPlot (Jandel Scientific, Inc.) to a single exponential equation, y=a(1-e-kx), or to a double exponential equation, y=a(1-e-kx)+ b(1-e-mx), where k and m are the rate constants and a and b correspond to the associated amplitudes. In C129S/C345S, increased amounts of DTNB resulted in decreased formation of the interdomain disulfide bond as judged by non-reducing SDS polyacrylamide electrophoresis. Thus, data from C129S/C345S were fitted to y=a(1-e-kx) + b(1-e-mx) + c(1-e-0.00104), where 0.00104 s-1 is the constant intramolecular rate of disulfide bond formation estimated from repeated fitting of the overall data and c is the fraction of this species formed estimated from nonreducing SDS gels.
Large amounts of interdomain disulfide bond-containing C132S/C345S AhpF protein for further characterization were obtained by titrating in 0.25 equivalents of DTNB relative to protein until a final 10-fold excess was reached. At least three washes by a 30 kDa cutoff Apollo concentrator removed the free DTNB and TNB. The extent to which the interdomain disulfide bond had formed was evaluated on nonreducing SDS polyacrylamide gels.
Generation of an Interprotein Mixed Disulfide Bond Between AhpF and AhpC
For oxidation of the thiol to sulfenic acid in the C165S mutant, 20 nmols of the protein was preincubated with one equiv of H2O2 in an anaerobic cuvette following repeated evacuation and nitrogen flushing for 20 min. Mutant AhpF proteins (20 nmol) were added anaerobically to the sidearm of the cuvette, and the solution (0.6 mL total volume) was made anaerobic for an additional 15 min. Oxidized C165S was mixed with each of the single mutants of AhpF, and a 100 μl sample was taken at 5 min and again after 120 min. Samples from the two time points were added immediately to 4x sample buffer lacking 2-mercaptoethanol for analysis by SDS-polyacrylamide gel electrophoresis (10% acrylamide).
The TNB conjugates of C46S and C165S were generated by incubating 100 nmols of each of the mutants with a 2-fold excess of DTNB, and removal of excess TNB in a Centricon CM-30 with four washes in standard phosphate buffer. Twenty nmol of the single mutant AhpC-TNB conjugates was mixed with an equimolar amount of the AhpF single mutants in a total volume of 100 μL with standard phosphate buffer at room temperature for 5 min. Sample buffer containing 2% SDS and lacking 2-mercaptoethanol was added before analyses by SDS-polyacrylamide gel elecrophoresis (10% acrylamide).
To purify large amounts of the F:C129-S-S-C165:C2 interprotein disulfide-containing complex, the TNB conjugate of C46S AhpC (50 nmol) was incubated with a two-fold molar excess of C132S AhpF (100 nmol) for two hours and loaded on a Phenyl Sepharose 6 Fast flow column equilibrated with 20% ammonium sulfate in standard phosphate buffer. The F:C129-S-S-C165:C complex was eluted with a linear gradient from 20% to 0% ammonium sulfate. SDS-polyacrylamide gel analyses of the eluted fractions allowed for the identification of those containing the pure complex and indicated that free C132S AhpF remained bound to the column under these conditions. Pure fractions were pooled and stored at -20 °C until needed. A similar complex between C129 of the N-terminal domain of AhpF and C165 of AhpC was generated in a similar way using the C132S mutant of the separately-expressed N-terminal domain (24).
RESULTS
AhpF Single Cysteine Mutants: DTNB Reactivity and Turnover Assays
For independent assessment of the reactivity of each cysteine thiol toward DTNB, each member of the cysteine pair in the two CXXC redox centers in AhpF was individually mutated to serine to prevent disulfide bond formation; an additional mutant lacking all four catalytic cysteine residues was also constructed as a control (4C→S). Properties of the C345S and C348S mutants of AhpF were described previously (22). With only one disulfide center intact in each case, none of the four single mutants of AhpF possesses measurable activity in reducing the AhpC disulfide bond and supporting turnover with peroxides.
Reaction of all four single mutants of AhpF with DTNB demonstrated the reactivity of about one free thiol in each mutant protein in its native state, confirming the disruption of one disulfide redox center and corresponding generation of one new, accessible, free thiol per mutant (Table 1); two additional thiols were detected (for a total of three free thiols per mutant protein) in all proteins after denaturation with GuHCl and, based on the result with 4C→S AhpF, represent the two inaccessible, non-functional cysteine residues, C476 and C489, at the C-terminus of AhpF as previously demonstrated by site-directed mutagenesis (1).
Table 1.
DTNB reactivity and turnover rates of wild type and single cysteine mutant AhpF proteins
| AhpF (wt)a | C129S | C132S | C345S | C348S | 4C→S | |
|---|---|---|---|---|---|---|
| Native thiol contentb | 0.06 ± 0.02 | 1.20 ± 0.03 | 0.85 ± 0.01 | 1.08 ± 0.03 | 0.76 ± 0.03 | 0.10 ± 0.01 |
| Additional thiol content post denaturationc | 1.93 ± 0.02 | 2.23 ± 0.04 | 1.98 ± 0.02 | 2.07 ± 0.17 | 2.05 ±0.04 | 1.95 ± 0.03 |
| Rate of reaction with DTNBd | N.R.e | 66 | 4050 | <0.75 | 66 | N.R. |
| NADH:DTNB oxidoreductase activityf | 1 150 ± 160 | 18.3 ± 4.9 | 40.2 ± 7.0 | <0.1 | <0.1 | <0.1 |
Thiol content data from previous work (1); oxidized wild-type AhpF, as purified.
Expressed per subunit for untreated (naturally oxidized) proteins; assays were performed in 50 mM potassium phosphate buffer, pH 7.0 with 1 mM EDTA using 9-13 nmol of protein and a 10-fold excess or greater of DTNB in a total volume of 0.45 mL at 25°C. Values shown for thiol contents are the average of at least five measurements.
Expressed per subunit; values shown for thiol contents are the average of at least four measurements following the addition of 0.45 mL 8 M GuHCl to native thiol content samples.
Expressed as M-1 s-1; assays were performed with 200-700 μM DTNB as described in “b”.
N.R. = no reaction
Expressed as μM NADH reduced min-1 μM-1 enzyme. Assays were performed with 10-200 pmol mutant or 0.7-1.5 pmol wild type AhpF proteins with 500 μM DTNB and 200 μM NADH, in 50 mM Tris-HCl buffer, pH 8.0, with 0.5 mM EDTA and 100 mM ammonium sulfate (8-11 replicates). Because of a low, interfering cysteine-independent rate of DTNB reduction, values were those calculated after subtracting the “background” rate obtained with protein pretreated with a 50-fold excess of N-ethylmaleimide for 1 hr, then washed free of the reagent using a Centricon CM-30 ultrafiltration device.
Second order rates of reaction toward DTNB were assessed directly in the individual mutants, giving site-specific information addressing a combination of accessibility and aspects of the protein microenvironment that, among other things, promote the nucleophilicity of the Cys residue of interest. Given that one Cys per mutant protein is reactive under these conditions (C132 of C129S, C129 of C132S, C345 of C348S and C348 of C345S), the order of reactivity for the four catalytic Cys residues in the respective mutant proteins is C129>C345≈C132>C348, with C129 by far the most reactive residue toward DTNB by over 60 fold (Table 1). Interestingly, rates of reaction of DTNB with C46 and C165 in C165S and C46S mutants of AhpC, respectively, were similar to that of the most reactive cysteine identified in AhpF, at 1100-1500 M-1 s-1 (30).
The formation of mixed disulfides with TNB at the mutated redox centers of AhpF may partially mimic the corresponding normal disulfide bonds in AhpF (see below). With the two disulfide centers intact, AhpF exhibits a robust NADH:DTNB oxidoreductase activity (29). We thus asked whether or not the mixed disulfides formed on the mutant proteins could behave similarly to those in the wild-type protein by conducting turnover assays in the presence of NADH. A low rate of turnover was surprisingly exhibited by the 4C→S control protein that was similar in magnitude to the rate observed with the C345S and C348S mutants; because of this, results in Table 1 are shown after subtracting a “background” rate obtained for each mutant after blocking free thiol groups with N-ethylmaleimide. Using this assay, TNB linked to either C129 or C132 in the N-terminal domain was released by electron transfer through the protein from NADH, resulting in a detectable NADH:DTNB oxidoreductase activity greater than the low rate exhibited by the 4C→S mutant. While there is a two-fold greater activity when the TNB is linked to C129 (in C132S) than when the linkage is through C132 (in C129S), both of these rates are far lower than the >28-fold higher activity of the wild type protein. Thus, the TNB-mixed disulfide at the N-terminus is not a very good mimic of the wild type protein as a substrate for the C-terminal disulfide center, probably due to the presence of the relatively bulky TNB group. AhpF without an intact C-terminal disulfide redox center (i.e. in C345S or C348S mutants) is completely inactive for electron transfer to disulfide-containing species including DTNB.
AhpF Double Cysteine Mutants: Stabilization of a Charge Transfer Absorbing Species of AhpF
To further address the role of each cysteinyl group in the intramolecular electron transfer of AhpF and to generate interdomain, disulfide-linked species for crystallographic studies, four double mutants, C129S/C345S, C129S/C348S, C132S/C345S and C132S/C348S, were expressed and purified to homogeneity. Functionally, these mutants have no disulfide-dithiol-mediated enzymatic activity (i.e. catalytic AhpC or DTNB reduction) due to the lack of either intact disulfide redox center; direct flavin-mediated transhydrogenase (hydride transfers to and from pyridine nucleotides) and oxidase activities are retained in these mutants, however, indicating that they are folded properly and binding the flavin in a functional way (Supporting Information, Table S2).
For the most part, absorbance and fluorescence spectra of single and double cysteine mutants of AhpF, primarily reflecting the bound FAD, were quite similar to those of the wild-type protein (see Tables S1 and S2 in Supporting Information for detailed spectral information). Exceptions to this are all the mutants containing a C348S mutation. This single mutant (22), as well as the C129S/C348S and C132S/C348S double mutants, exhibit the typical two broad flavin absorbance peaks, but these are both blue shifted relative to wild-type AhpF and the other mutants, and the two peaks exhibit a lower extinction coefficient for the ∼374 nm peak relative to the ∼445 nm peak (with a difference of 10-20 %; Fig. 2A). In addition, the fluorescence of the flavin is quenched by 18 to 46% in these mutants relative to wild-type AhpF, suggesting that the new Ser at position 348 is both near the flavin and quenching its fluorescence (see Supporting Information and reference 22).
Figure 2.

Comparison of the mutant AhpF visible absorbance spectra and the thiolate → FAD charge transfer absorbance interaction elicited by addition of ammonium ion and Tris to C345S mutants. The proteins whose spectra are shown in panel A were in standard phosphate buffer at neutral pH and room temperature; spectra shown are for wild type AhpF (solid line), C132S/C348S (dashed line), and C129S/C348S (dottedline) proteins. In panel B, 53 μM of the C132S/C345S mutant of AhpF in 0.5 M Tris-HCl at pH 8.0 was titrated with a solution containing 3 M (NH4)2SO4 in the same Tris buffer. Wild type AhpF in standard phosphate buffer (dashed line) is shown for comparison. Spectra (alternating solid and dotted lines) of C132S/C345S AhpF have been adjusted for volume, and contain the following concentrations of (NH4)2SO4 in order of increasing 532 nm and decreasing 450 nm absorbance: 0, 29.7, 58.8, 87.4, 115, 170, 222, 312, 413, 580, 794, 1026, and 1585 mM. Inset, plot of A560 as a function of ammonium sulfate concentration (avoiding the relatively noisy spectral data around the ∼532 nm peak).
One unusual spectral finding arose for another of the mutants, C132S/C345S, during purification; upon changing buffer conditions from the standard potassium phosphate buffer to a buffer containing Tris and ammonium sulfate at pH 8.0, a deep brick-red color appeared. Such an absorbance spectrum indicates the presence of a thiolate-FAD charge transfer interaction centered around 530 nm based on studies of related flavoprotein disulfide reductases including thioredoxin reductase (31, 32). This charge transfer interaction, although weaker, was also identified in the C129S/C345S mutant, but not in C132S/C348S or C129S/C348S mutant proteins (Fig. 2B), again indicating that the thiolate of C348, but not C345, is located in close proximity to FAD. Although previous attempts to identify the charge transfer species in the corresponding single cysteine mutant, C345S, in the presence of ammonium ion did not show evidence for the charge transfer band (22), revisiting this mutant by exchanging the buffer into one containing Tris and ammonium sulfate did indeed result in the appearance of a weak charge transfer species (not shown). The structural and chemical effects promoting the visible charge transfer interaction were fully reversible; no charge transfer species was detected after dialysis to remove the amine-containing buffer or upon lowering the pH (to pH < 5.5) to protonate the active site thiolate (see Supporting Information for more details). Although it may seem surprising that the intensity of the charge transfer absorbance due to interaction between Cys348 and FAD would vary due to mutations in the distal N-terminal domain, this variation may reflect the relative stabilization of different conformations of AhpF in the various mutants as described in more detail in the discussion and observed previously with various mutant forms of TrxR (33).
AhpF Double Cysteine Mutants: DTNB Reactivity
We hypothesized that at least one, and perhaps more, of the four double cysteine mutants of AhpF would stably form an intramolecular disulfide bond between the two redox centers upon oxidation. Addition of the oxidant DTNB to these mutants could potentially result in the formation of two different products depending on relative rates of reaction of the different Cys residues with DTNB and with the mixed disulfides formed with TNB. As depicted in Figure 3A, both of the cysteines could form mixed disulfide bonds with TNB (species C). On the other hand, if the initial reaction with DTNB were efficiently followed by attack of the other Cys residue on the nascent mixed disulfide bond with TNB, an “intercenter” or interdomain disulfide bond would be formed, covalently linking the N-terminal domain with the C-terminal NAD/SS domain (species D). In both of these cases, two equivalents of chromophoric TNB would be released; however, the resulting products could be readily distinguished by the difference in covalently-linkedTNB content (species D having zero and species C having two bound TNB moieties). As expected, the reaction of a 10-fold excess of DTNB over AhpF mutants resulted in the release of approximately two TNB anions in all four AhpF mutants (Table 2). Thus, both remaining cysteine residues of the two redox centers were able to participate in the reaction in each case. After the removal of excess DTNB, the proteins were exposed to the disulfide reductant dithiothreitol (DTT). Both C132S/C348S and C129S/C348S released approximately two equivalents of TNB, supporting the formation of species C after DTNB treatment. C132S/C345S, however, did not release any significant amount of TNB, suggesting that a stable intramolecular, interdomain disulfide bond was formed between the C-terminal C348 and N-terminal C129 residues (species D). The release of 0.83 equivalents of TNB in the fourth mutant, C129S/C345S, suggests that DTNB treatment resulted in the formation of a mixture of species C and D.
Figure 3.


Reaction scheme for AhpF double Cys mutants with DTNB (A), and gel-based analysis of the products obtained after DTNB treatment (B). Panel A depicts the common first step (species A reacting with DTNB to give species B) and the divergent pathways that could lead to the formation of two different products (species C and D). The bi-lobed structure represents the NAD/SS and FAD domains of AhpF (residues 210-521), and the smaller structure the N-terminal domain (1-196), with a linker region (thick black line, residues 197-209) joining it to the FAD domain. Note that two of the rate constants associated with these reactions, k1 and k2, are second order rate constants dependent on the DTNB concentration, while k3 is a first order rate constant (independent of DTNB concentration). Actual measured rates constants are presented in Table 2. Panel B shows the products of each of the double mutants of AhpF following reaction with a 10-fold excess of (140 μM) DTNB as analyzed on a 10% SDS polyacrylamide gel. Samples (10 μg) were boiled in SDS sample buffer either containing (reducing samples, lanes 1-5) or lacking (nonreducing samples, lanes 6-9) 2-mercaptoethanol. The presence of an “interdomain” disulfide bond as depicted for species D (panel A) results in decreased mobility of the proteins (lanes 7 and 9, exhibiting an apparent molecular weight of 68 kDa) that is alleviated by reduction (lanes 3 and 5). Lane 1 contains broad molecular weight markers from Bio-Rad (Hercules, CA) with the indicated Mr values to the left. Proteins loaded were C129S/C348S (lanes 2 and 6), C129S/C345S (lanes 3 and 7), C132S/C348S (lanes 4 and 8), and C132S/C345S (lanes 5 and 9).
Table 2.
Reaction rates and thiol quantitation of AhpF double cysteine mutants with DTNB
| C132S/C348S | C132S/C345S | C129S/C348S | C129S/C345S | ||
|---|---|---|---|---|---|
| Observed ratesa | k1 (M-1s-1) | 6200 ± 500 | 1200 ± 50 | 46 ± 2.6 | 110 ± 5.8 |
| k2 (M-1s-1) | 40 ± 2.4 | 9.8 ± 0.3 | |||
| k3 (s-1) | 0.0010 | ||||
| TNB released upon DTNB reaction b | 2.10 ± 0.04 | 2.10 ± 0.10 | 1.97 ± 0.10 | 1.92 ± 0.11 | |
| TNB released after DTT treatment of oxidized proteinc | 1.91 ± 0.15 | 0.05 ± 0.03 | 2.17 ± 0.15 | 0.83 ± 0.15 | |
| Products detected on geld | C | D | C | C, D |
Reaction rates of AhpF mutants with DTNB in standard phosphate buffer. Rate of TNB release was observed at 412 nm. AhpF at 7.7 μM was mixed with 77-310 μM DTNB.
Equivalents of TNB released upon reaction of the mutants with 10-fold excess DTNB.
Equivalents of TNB released after removal of excess DTNB by ultrafiltration and subsequent addition of DTT.
Analyses of the protein products on nonreducing SDS-PAGE gels matched the above results. In the absence of reductant, TNB-modified proteins migrated with an apparent Mr of 56 kDa (similar to the migration of the reduced mutants), whereas the putative interdomain disulfide-linked species migrated substantially more slowly, giving an apparent Mr of 68 kDa (Fig. 3B). The slower migration of the intrachain, disulfide-bonded forms of the AhpF mutants was surprising, since the presence of such a disulfide bond typically increases the mobility of these more compact forms. Nonetheless, in this case, species D as analyzed by spectral analyses clearly correlated with formation of this upper band on nonreducing SDS-PAGE. Neither C132S/C348S nor C129S/C348S formed any of the upper band, whereas oxidized C132S/C345S was converted to this form in >90% yield and C129S/C345S was a mixture of the two forms. Mapping and identification of the disulfide bond-containing tryptic peptide by HPLC and ESI-MS analysis confirmed the location of the disulfide bridge between C129 and C348 in the oxidized C132S/C345S mutant (see Fig. S1 in Supporting Information).
To further investigate the rates of reaction of each of the Cys residues with DTNB and/or mixed disulfides with TNB formed as intermediates, we analyzed the kinetics of the spectral changes that ensued following addition of DTNB to each of the double cysteine mutants. For the two mutants that form only TNB mixed disulfides at each center, the reactivities of C132S/C348S and C129S/C348S nicely match those expected based on each of the corresponding single mutants [6200 and 40 M-1 s-1 for C129 and C345 of C132S/C348S (Table 2), similar to 4050 and 66 M-1 s-1 for C132S and C348S, respectively (Table 1); and a single rate of 46 M-1 s-1 for C132 and C345 of C129S/C348S (Table 2), similar to 66 M-1 s-1 for both C129S and C348S single mutants (Table 1)]. The mutant which efficiently formed an interdomain disulfide bond according to the lack of TNB in the final product, C132S/C345S, possesses one rapidly reacting and one very slowly reacting Cys residue (differing by more than 3 orders of magnitude), C129 and C348, respectively, according to the DTNB reactivity of the corresponding single mutants (Table 1). As only one DTNB concentration-dependent rate (1200 M-1 s-1) was observed for the release of both TNB moieties from this mutant (Table 2), we attribute this to the facile reaction of C129 with DTNB followed by a very fast and irreversible attack of C348 on this mixed disulfide bond to release the second equivalent of TNB (at > 0.9 s-1, based on the fastest rate of DTNB reduction observed in these experiments, at 310 μM DTNB; data not shown).
As suggested by mixed product formation with DTNB, the most complex reaction was that of the C129S/C345S mutant. The fastest rate observed (110 M-1 s-1) corresponds well with the expected rate of reaction of C132 with DTNB based on the corresponding single mutant (66 M-1 s-1) (Tables 2 and 1, respectively), forming intermediate species “B” (Fig. 3A) which could then partition to species C and D. Indeed, based on both the TNB content of the products (Table 2) and the SDS polyacrylamide gel showing the relative distribution of products (Fig. 3B), rates to form species C and D from the intermediate B must be quite similar as both products are observed. Note, however, that the rate of formation of species C (resulting in the release of the second TNB) should be a second order rate dependent on the concentration of DTNB, while the rate of formation of species D (also releasing a second TNB) should be a first order rate independent of DTNB concentration. Accordingly, gel analyses of products formed upon addition of increasing concentrations of DTNB favored formation of C over D (D present at approximately 40%, 35% and 30% for DTNB concentrations of 140, 210 and 280 μM, respectively; data not shown). From these results and repeated fitting of the datasets to the appropriate first and second order rates and amplitudes, all three rate constants for the full reaction were obtained, corresponding to the partitioning of species B at a rate of 9.8 M-1 s-1 to form species C (dependent on DTNB concentration), and at a rate of 0.001 s-1 to form species D (independent of DTNB concentration).
Conclusions from these studies were thus that C348 must be present to form an interdomain disulfide bond (species D) efficiently enough to compete with the modification of the protein by reaction with two molecules of DTNB, and that C132S/C345S forms the interdomain disulfide bond in >90% yield and at a rate that is at least three orders of magnitude faster than C129S/C345S (>0.9 s-1 versus 0.001 s-1). These results strongly suggest that the oxidized C132S/C345S mutant best mimics the catalytic intermediate generated during electron transfer between the two disulfide/dithiol redox centers of wild-type AhpF.
The C132S/C345S Mutant of AhpF With a Disulfide Bond Tethering Two Domains is Conformationally “Locked” and Cannot Engage in Electron Transfer
Based on the crystal structure and our knowledge of the sequence of electron transfers among AhpF redox centers (Fig. 1), major conformational changes in AhpF are needed in order for electrons to be transferred among the three redox centers (21, 24). The physical connection of C129 in the N-terminal domain of AhpF via an interdomain disulfide bond to C348 located in the the C-terminal NAD/SS domain, as approximated in a model of AhpF (21) based on a similar “tethered” construct between thioredoxin reductase and thioredoxin (34), should not allow this disulfide bond to come into proximity of the flavin. This construct should therefore be resistant to reduction by NADH. In accordance with this expectation, only about one equivalent of NADH is oxidized upon addition to the C132S/C345S mutant whether or not the latter was pretreated with DTNB to generate the disulfide bond between the two remaining reactive Cys residues (see Fig. S2 of Supporting Information). Not surprisingly, also, the relatively compact and less flexible structure of the oxidized (disulfide-bonded) C132S/C345S mutant renders it highly resistant to proteolytic digestion relative to the reduced protein (see Fig. S3 of Supporting Information).
Formation of Covalent Complexes Between AhpF and AhpC Mutants
Having established that C348 in AhpF attacks C129 in the N-terminus, we next turned to the determination of which Cys residues are directly involved in electron transfer between AhpF and AhpC based on biochemical analyses. As with the two disulfide-dithiol redox centers in AhpF, formation of a stabilized disulfide bond between the two proteins is expected to be possible if one Cys residue in each redox center is mutated. To promote disulfide bond formation between the two thiol-containing proteins, DTNB was again used as the oxidant, although in this case the individual proteins were pretreated to form mixed disulfides with TNB before being mixed to assess interaction between the redox centers in the two proteins (vide infra).
In these experiments, another option for Cys oxidation, relating to catalysis, was also important to explore; C46 of AhpC is readily oxidized to the sulfenic acid, Cys-SOH, upon treatment with one equivalent of H2O2, and this species is stable in the absence of C165 (in the C165S mutant) and under anaerobic conditions (30, 35). The ability of AhpF to recycle the C165S mutant of AhpC during catalytic assays, albeit relatively poorly, indicates that a functional interaction can take place between the two proteins even in the absence of C165 on AhpC. Earlier studies, including functional assays with the separately-expressed N-terminal domain of AhpF, established that electron flow to AhpC occurred through direct interaction of the C129,C132 redox center of AhpF with AhpC (8, 24). In accordance with our expectations, then, Cys residues in the N-terminal domain of AhpF (from the C132S and C129S mutants) were able to form a covalent link with oxidized C46 on C165S AhpC after mixing the two in a 1:1 ratio, and the Cys residues in the other disulfide-dithiol redox center, C345 and C348, were not (data not shown). After both 5 and 120 min time points, all of the C132S mutant of AhpF incubated with oxidized C165S AhpC had been converted to the higher molecular weight species as assessed on SDS polyacrylamide gels, whereas C129S under the same conditions gave a shift of only about 40% of the protein to the higher molecular weight complex, even after 120 min incubation.
The above experiments were informative regarding the physical ability for AhpC and AhpF to interact via an interprotein disulfide bond, but the ability of AhpF to reduce Cys-SOH on AhpC is only relevant to catalysis by the C165S mutant of AhpC, a mutant that is clearly crippled for reductive recycling due to its lack of C165 (30). Oxidation of each of the Cys residues in AhpC mutants to their TNB-mixed disulfides, on the other hand, better mimics normal catalysis in presenting a disulfide-linked species to AhpF for attack. In fact, such a strategy was previously used whereby an engineered construct with a fluorescein derivative attached to AhpC via a disulfide bond was presented as a substrate to AhpF or the truncated version (the separately-expressed N-terminal domain). Release of the fluorescein from C165 of the C46S mutant of AhpC was quiterapid, at ∼1.5 × 106 M-1 s-1, suggesting that AhpF can tolerate some degree of extra “bulkiness” in the disulfide center it attacks (24).
On incubating TNB conjugates of C165S and C46S AhpC in the present study with each of the four single AhpF mutants in an equimolar mixture, only the sample generated by incubation of C132S AhpF with TNB-linked C46S AhpC contained a significant amount of higher molecular weight complex (Fig. 4, lane 3). The lack of a significant amount of free AhpC or AhpF protein species in this sample analyzed by SDS polyacrylamide gels indicated that the reaction went essentially to completion. As a control, the predenatured proteins did not form the disulfide complex after mixing (Fig. 4, lane 10). This result indicates that proper folding of each protein is necessary for formation of this complex, designated “F:C129-S-S-C165:C” in the ensuing results and discussion (to emphasize the Cys residues involved in the interprotein disulfide bond).
Figure 4.

Complex formation between single cysteine mutants of AhpF and TNB-conjugates of AhpC mutants. TNB conjugates of C46S AhpC (lanes 2-5 and 10) or C165S AhpC (lanes 6-9), prepared by preincubation with excess DTNB and isolation by ultrafiltration, were incubated for 5 min with equimolar amounts of AhpF mutants C129S (lanes 2 and 6), C132S (lanes 3, 7 and 10), C345S (lanes 4 and 8), or C348S (lanes 5 and 9) prior to separation on a non-reducing 10% SDS polyacrylamide gel. Proteins in lane 10 were denatured in sample buffer before mixing. Lane 1 contains low molecular weight markers from Bio-Rad (Hercules, CA) with the indicated Mr values to the left.
Spectrophotometric analyses following the rate of TNB release, carried out in the presence of excess C132S AhpF, also demonstrated full release of TNB from the conjugate with C46S AhpC which occurred with a second order rate constant of 1.6 × 106 M-1 s-1. This rapid rate of disulfide bond formation suggests that this F:C129-S-S-C165:C complex represents a reasonable model for a catalytically-relevant interprotein disulfide-containing AhpF-AhpC intermediate, and that C129 on AhpF is the attacking residue, and C165 on AhpC the site of attack on the C46-C165 disulfide bond, in the wild-type proteins undergoing catalysis. On the other hand, only a slight amount of higher molecular weight complex was generated on mixing the TNB-conjugate of C165S AhpC with C132S AhpF (Fig. 4, lane 6), and the rate of this reaction is indeed much slower, with a second order rate constant of 15 M-1 s-1. As with the results with the Cys-SOH form of C165S AhpC, AhpF C-terminal single cysteine mutants C345S and C348S showed no sign of complex formation with the TNB-conjugate of either C46S or C165S mutants of AhpC, either by gel analysis (Fig. 4, lanes 3, 4, 7 and 8) or by spectral analysis.
Biochemical Analyses of F:C129-S-S-C165:C
Electrospray ionization mass spectrometry analyses confirmed that the observed high molecular weight species of C132S AhpF and C46S AhpC on the gel (Fig. 4, lane 3) was indeed a 1:1 covalent complex between the AhpC and AhpF mutants (see Supporting Information for details). For additional biochemical analyses, a large amount of the F:C129-S-S-C165:C complex was prepared by addition of a two-fold excess of C132S AhpF to the TNB-linked C46S mutant of AhpC until all of the AhpC had formed complex as judged by nonreducing SDS polyacrylamide gels. The excess C132S AhpF was then removed by chromatography.
As with the interdomain disulfide-linked C132S/C345S mutant of AhpF, we conducted NADH titration experiments to determine whether or not the interprotein disulfide bond was in communication with the other redox centers of AhpF. For wild type AhpF and also the TNB-linked form of the C132S mutant of AhpF, anaerobic titration with NADH results in the consumption of 2.5 - 2.7 equivalents of NADH, one for each disulfide redox center and about 0.5 to 0.7 for the flavin, which is not completely reduced by NADH [(1) and Supporting Information, Fig. S4]. On the other hand, NADH titration of untreated C132S AhpF and the F:C129-S-S-C165:C complex resulted in oxidation of only 1.5 equivalents of NADH (Supporting Information, Fig. S4) and did not reduce the complex into separable AhpF and AhpC proteins as demonstrated on nonreducing SDS polyacrylamide gels (data not shown). Thus, attachment of AhpC to the N-terminal domain of AhpF through a disulfide bond linkage does not allow for the domain rotations in AhpF required to bring the N-terminal and C-terminal disulfide/dithiol redox centers together for electron transfer.
DISCUSSION
In the studies presented here, we have generated single and double mutants of AhpF where at least one of the two disulfide redox centers has been converted to a thiol(ate) center through mutation. By assessing the reactivity of these mutants with DTNB and TNB-linked AhpC mutants as oxidizing substrates, we have learned a great deal about the individual roles of cysteinyl residues in the electron transfer processes through AhpF and on to the AhpC substrate. First and foremost, it is quite clear that C129 in the N-terminal domain plays two critical roles in these transfers; it is the attacking nucleophile during thiol-disulfide interchange to transfer electrons from AhpF to AhpC, and it is the point of attack in the disulfide form for transfer of electrons into the N-terminal domain (NTD) from the C345-C348 redox center in AhpF (Fig. 5B). In both single and double mutants of AhpF, reactivity with DTNB supports C129 as the most accessible and reactive cysteinyl residue in AhpF, although it is likely that its reactivity is enhanced when C132 is also present (Tables 1 and 2, and see below). Experiments assessing the ability of AhpF mutants to form disulfide-linked complexes with AhpC support not only the direct role for C129 of AhpF in the attack on the AhpC disulfide bond, but also C165 in AhpC as the disulfide-bonded residue attacked by the C129 thiolate (Fig. 4). Interestingly, formation of a complex between AhpF and AhpC (F:C129-S-S-C165:C) occurs quite rapidly, at 1.6 × 106 M-1 s-1, even though in these studies the C129 in the AhpF mutant employed is not activated by the missing C132 and the TNB-linked disulfide bond with C165 of AhpC is only a partial mimic of the native disulfide bond. This rate is only about 8-fold slower than the kcat/Km value for the catalytic efficiency of this process for the wild type enzymes (36).
Figure 5.


Two putative conformational states of AhpF (panel A) and schematic representation of the successive electron transfers through the redox centers of AhpF and AhpC (panel B). In panel A, conformation I represents the solved crystal structure of AhpF (pdb 1hyu), colored as in Fig. 1 (21). In this conformation, the C345-C348 disulfide center is in close proximity to FAD. Conformation II is the modeled alternative conformation, based on the solved structure of the complex between thioredoxin and thioredoxin reductase from E. coli (pdb 1f8m) (34). Both the NAD/SS (red) and N-terminal domain (blue) have been rotated, as described in the Discussion and in the original report of this model (21). In panel B, successive electron transfers between the flavin (top, green) and disulfide (red and blue) redox centers of AhpF and AhpC (light and dark purple representing the two subunits) are depicted, along with the conformational changes that bring successive domains into proximity for electron transfer (right side). Protonation states of the thiols as depicted here are not necessarily accurate.
Exploring further the kinetic data collected herein on the cysteinyl reactivity of C129 in AhpF, it is clear that the reactivity depends moderately on context, and that it is far more reactive toward disulfide bonds generated with C165 of AhpC than with the small molecule disulfide bond of DTNB. Looking first at conditions where a single step of C129 thiolate attack on DTNB can be measured, the C132S mutant in the presence of 500 μM DTNB reacts at 120 min-1 (Table 1); for comparison, this step in the two corresponding double mutants occurs at 36 min-1 (for C132S/C345S) and 186 min-1 (for C132S/C348S) (Table 2). Turnover with 500 μM DTNB and saturating amounts of NADH, as catalyzed by the C132S mutant, also occurs at about 40 min-1 although there are some differences in the buffer conditions used for the turnover assays. Where the disulfide redox center of the N-terminal domain is intact (i.e. in wild type AhpF), turnover with DTNB is approximately 30-fold higher than with C132S (Table 1); this may reflect additional reactivity of C129 due to the presence of the closely interacting C132 residue that apparently shares a proton with C129 in the solved crystal structures of this domain (21, 37), and also a more facile reduction by the NAD/SS domain of the native N-terminal disulfide center relative to the TNB-linked center of the C132S mutant. Finally, the reaction of C132S-containing AhpF mutants with DTNB is much slower, by about 3 orders of magnitude, than the reaction of C132S with the AhpC mutant containing TNB-conjugated C165 (1.6 × 106 M-1 s-1 for complex formation versus 1.3 × 103 M-1 s-1 for C132S turnover with DTNB, Table 1). This clearly reflects the contribution of AhpC-AhpF protein interactions and the ability of AhpF to distinguish its natural oxidized partner from other disulfide-bonded species.
Comparing the C129-C132 center within the NTD of AhpF with its functional and structural relative, Trx, the pattern most typically seen with these CXXC motif-containing, Trx fold proteins is maintained in AhpF in that the more N-terminal cysteinyl residue is the nucleophilic attacking residue toward disulfide-bonded substrates, and this more N-terminal residue is also the point of attack by its reductant, TrxR or the TrxR-like portion of AhpF (38). Relative to Trx, the NTD of AhpF is larger, containing two continguous Trx folds, but just the one redox active disulfide within the second of these folds. Interestingly, the putative active site acid-base catalyst near the C129-C132 redox center of AhpF, E86, is contributed by the first Trx fold to the redox center in the intimately-associated second fold and is oriented in a mirror image active site arrangement when compared with its functional counterpart in E. coli Trx, D26 (21).
Another major finding from the present study is that C348 of AhpF can form a thiolate → FAD charge transfer absorbance band in the presence of amine-containing buffers and high concentrations of ammonium sulfate in all three of the mutants where C345 has been changed to serine, and that C348 is also the “interchange thiol” that attacks the disulfide-containing redox center in the N-terminal domain of AhpF. C348 was also shown in the crystal structure to be marginally closer than C345 to the isoalloxazine ring of AhpF, although both are >5 Å away due to the interference of a bound chloride ion in this crystallized form of AhpF (pdb identifier 1hyu) (21). More clarity on this point comes from the crystal structure, reported by Bieger and Essen (39), of the C-terminal fragment of the nearly identical E. coli AhpF, where no chloride ion is interfering; in this structure (pdb 1fl2), C345γS is 3.0 Å from the flavin C4a position and C348γS is 4.6 Å away. In the homologous protein, TrxR, the sulfur of the side chain corresponding to C348 in AhpF, C138γS, is 3.1 Å away from the C4a position of FAD, and C135γS is ∼5.0 Å away (40).
For TrxR, several biochemical studies, including fluorescence quenching of the flavin by a Ser residue replacing the C138 (similar to our results with C348S herein) and formation of a thiolate q→ FAD charge transfer interaction in a C135S mutant at high ammonium ion concentration, have also demonstrated the proximity of C138 to the flavin in TrxR (33, 41, 42). Importantly, TrxR and AhpF differ from most other flavoprotein disulfide reductases (e.g. glutathione reductase and lipoamide dehydrogenase) in having this flavin-interacting disulfide bond located in a sort of “tilted” fashion with respect to the flavin (40), rather than oriented with only one of the cysteinyl residues “toward” the flavin (the more C-terminal one of the CXXXXC motif), and the other cysteine directed away from the flavin and participating in interactions with the disulfide/thiol-containing substrate, as for glutathione reductase and lipoamide dehyhdrogenase (43, 44). This difference in architecture is also linked with the putative large domain rotations in the NAD(P)/SS domains (relative to the flavin domains) of TrxR and AhpF that alternately swing the disulfide center and the nicotinamide ring of the pyridine nucleotide into positions near the flavin (Fig. 5A), unlike the more static flavin-disulfide centers of the other flavoproteins. These two conformations, best characterized in TrxR (34, 40), allow for the flavin-to-disulfide (C135-C138 in TrxR or C345-C348 in AhpF) interaction on the one hand (conformation I in Fig. 5A), or the pyridine nucleotide-flavin interaction on the other (conformation II in Fig. 5A). In AhpF, a model of conformation II was generated based on the conformation I crystal structure (1hyu) and on the orientation of the Trx and TrxR domains in the crystallographically-characterized “twisted” version of TrxR (pdb identifier 1f6m) (34). The magnitude and direction of rotation of the NAD/SS domain required to generate this conformation in AhpF was quite similar to that of TrxR; compared to the crystal structure, this conformation has the NAD/SS domain rotated 57° (21). A further complexity in AhpF is the presence of the NTD fused to the flavin domain through a flexible linker; to generate the modeled alternative conformation, this domain also had to be rotated 100° and shifted 8 Å, along with an unraveling of one turn of the flexible linker. It is important to note that conformation II, which should correspond to species “D”, the interdomain disulfide bond-linked AhpF depicted in Fig. 3, has very well-defined and restricted locations of all the domains relative to one another (Fig. 5A); conformation I shown in the figure represents the structure as crystallized, but the NTD is not significantly interacting with the other two domains and should be quite capable of large motions in solution even where the NAD/SS and FAD domains are maintaining the flavin-close-to-disulfide conformation; it is in this putative conformation I (or family of conformations) where the NTD is expected to present its accessible dithiol center (C129-C132) to its substrate protein, AhpC, for reduction (Fig. 5B).
While many of the structural and functional features of AhpF clearly place it in the family of bacterial TrxR-like flavoprotein disulfide reductases, there are several notable distinctions between AhpF and TrxR. In the DTNB reactivity study by Veine et al. (42), similar to ours, using single cysteine mutants of TrxR and Trx, all of the rates that they obtained, except for their C138S mutant, were qualitatively in the same order as, though apparently quite a bit slower than, the rates we observed with AhpF mutants. In the TrxR/Trx system, C138 of TrxR is more reactive toward DTNB than is C135 of TrxR or C35 of Trx, whereas its counterpart in AhpF, C348, is essentially unreactive in the C345S mutant (Table 1). In the reaction of DTNB with the double mutants of AhpF, however, C348 plays a critical role in attack on the NTD disulfide center, efficiently forming a disulfide bond with C129 in the C345S/C132S mutant. For AhpF, where the C345,C348 center transfers electrons to an attached disulfide-containing domain (C129,C132 of the NTD), C348 may not become particularly reactive until the true disulfide substrate, the NTD, approaches, a phenomenon referred to as substrate-assisted catalysis. Interestingly, the TrxR-like C-terminal portion of AhpF is a good reductant of the separately-expressed NTD, but not of Trx, whereas TrxR can reduce both the NTD and Trx (23, 24). This suggests that AhpF domain interactions have evolved a degree of specificity toward one another and have functionally diverged from the TrxR and Trx ancestral proteins. Based on kcat measured for the AhpF-AhpC interaction (36), these electron transfers within AhpF must proceed at rates at least as fast as 237 s-1 at 25 °C, rather faster than the kcat of about 33 s-1 observed for TrxR turnover with Trx at the same temperature (45).
Finally, we showed herein that, in the “forward” direction of AhpF reducing a disulfide in AhpC, there is essentially complete specificity for C129 in AhpF as the attacking residue and C165 in AhpC as the point of initial attack (Fig. 4); in the reverse direction, this specificity is not maintained for the AhpF NTD residues (see Table S3 in Supporting Information). In TrxR, three out of the four possible mixed disulfide-containing species between TrxR and Trx could be formed using TNB linkage to either TrxR or Trx mutants; the inability to form this species for the C138S and C32S species of TrxR and Trx, respectively, was attributed to the extremely slow rate of formation that permitted side reactions to occur, as well (42). Considering other “typical 2-Cys” peroxiredoxins that are closely related to AhpC, the only system studied in depth for cysteine reactivity between a Trx-like electron donor and the Prx was tryparedoxin and tryparedoxin peroxidase from Trypanosoma brucei brucei; in this case, a similar specificity for the N-terminal cysteine within the CXXC motif of the donor (tryparedoxin) attacking the resolving Cys (comparable to C165 of AhpC) of the Prx was also observed by mass spectrometry, this time using a wild type version of the Prx and mutation only of the electron donor (20). When a similar experiment was tried with AhpF mutants and wild type AhpC, no stable disulfide-linked complex was observed (data not shown). Together, these experiments definitively identify a critical role for the conserved resolving Cys of typical 2-Cys Prxs as the point of attack in the reduction and re-activation of these proteins during catalysis.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Lois LaPrade and C. Michael Reynolds for their technical assistance.
Footnotes
This study was supported by a grant from the National Institutes of Health to L.B.P. (RO1 GM50389) and an Established Investigatorship from the American Heart Association to L.B.P. (0140175N).
- Prx
- peroxiredoxin, Trx, thioredoxin
- TrxR
- thioredoxin reductase, AhpC, alkyl hydroperoxide reductase C component (peroxidase)
- AhpF
- alkyl hydroperoxide reductase F component (flavoprotein reductase)
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid)
- TNB
- 2-nitro-5-thiobenzoic acid
- SDS
- sodium dodecyl sulfate
- DTT
- 1,4-dithiothreitol
- GuHCl
- guanidine hydrochloride
- EDTA
- ethylenediamine tetraacetic acid
- AcPyAD+
- 3-acetylpyridine adenine dinucleotide
- NAD/SS
- the pyridine nucleotide-binding and C345-C348-containing domain of AhpF, residues 328-449
- NTD
- the N-terminal domain of AhpF, residues 1-202
4C→S represents the AhpF mutant lacking all four redox-active half-cystines, C129S/C132S/C345S/C348S; F:Cys129-S-S-Cys165:C denotes the product from the reaction of the AhpC C165-TNB conjugate (made with the C46S mutant) with AhpF C132S, containing the reactive C129 residue.
REFERENCES
- 1.Poole LB. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 2. Cystine disulfides involved in catalysis of peroxide reduction. Biochemistry. 1996;35:65–75. doi: 10.1021/bi951888k. [DOI] [PubMed] [Google Scholar]
- 2.Toledano MB, Delaunay A, Monceau L, Tacnet F. Microbial H2O2 sensors as archetypical redox signaling modules. Trends Biochem Sci. 2004;29:351–357. doi: 10.1016/j.tibs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 4.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci U S A. 2005;102:8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poole LB. Bacterial peroxiredoxins. In: Torres M, Fukuto JM, Forman HJ, editors. Signal Transduction by Reactive Oxygen and Nitrogen Species: Pathways and Chemical Principles. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2003. pp. 80–101. [Google Scholar]
- 8.Poole LB. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch Biochem Biophys. 2005;433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 10.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 11.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood ZA, Schröder E, Harris JR, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 13.Karplus PA, Hall A. Structural Survey of the Peroxiredoxins. In: Flohé L, Harris JR, editors. Peroxiredoxin Systems. Springer; New York: 2007. in press. [Google Scholar]
- 14.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, Jönsson TJ, Poole LB, Heintz NH. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meissner U, Schroder E, Scheffler D, Martin AG, Harris JR. Formation, TEM study and 3D reconstruction of the human erythrocyte peroxiredoxin-2 dodecahedral higher-order assembly. Micron. 2007;38:29–39. doi: 10.1016/j.micron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Moon JC, Hah YS, Kim WY, Jung BG, Jang HH, Lee JR, Kim SY, Lee YM, Jeon MG, Kim CW, Cho MJ, Lee SY. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem. 2005;280:28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- 17.Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW, Choi YO, Kim WY, Kang JS, Cheong GW, Yun DJ, Rhee SG, Cho MJ, Lee SY. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Whitesides GM, Houk J, Patterson MAK. Activation parameters for thiolate disulfide interchange reactions in aqueous-solution. J Org Chem. 1983;48:112–115. [Google Scholar]
- 19.Hofmann B, Hecht H-J, Flohé L. Peroxiredoxins. Biol. Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 20.Budde H, Flohé L, Hofmann B, Nimtz M. Verification of the interaction of a tryparedoxin peroxidase with tryparedoxin by ESI-MS/MS. Biol Chem. 2003;384:1305–1309. doi: 10.1515/BC.2003.146. [DOI] [PubMed] [Google Scholar]
- 21.Wood ZA, Poole LB, Karplus PA. Structure of intact AhpF reveals a mirrored thioredoxin-like active site and implies large domain rotations during catalysis. Biochemistry. 2001;40:3900–3911. doi: 10.1021/bi002765p. [DOI] [PubMed] [Google Scholar]
- 22.Li Calzi M, Poole LB. Requirement for the two AhpF cystine disulfide centers in catalysis of peroxide reduction by alkyl hydroperoxide reductase. Biochemistry. 1997;36:13357–13364. doi: 10.1021/bi9713660. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds CM, Poole LB. Attachment of the N-terminal domain of Salmonella typhimurium AhpF to Escherichia coli thioredoxin reductase confers AhpC reductase activity but does not affect thioredoxin reductase activity. Biochemistry. 2000;39:8859–8869. doi: 10.1021/bi000826d. [DOI] [PubMed] [Google Scholar]
- 24.Poole LB, Godzik A, Nayeem A, Schmitt JD. AhpF can be dissected into two functional units: tandem repeats of two thioredoxin-like folds in the N-terminus mediate electron transfer from the thioredoxin reductase-like C-terminus to AhpC. Biochemistry. 2000;39:6602–6615. doi: 10.1021/bi000405w. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds CM, Poole LB. Activity of one of two engineered heterodimers of AhpF, the NADH:peroxiredoxin oxidoreductase from Salmonella typhimurium, reveals intrasubunit electron transfer between domains. Biochemistry. 2001;40:3912–3919. doi: 10.1021/bi002766h. [DOI] [PubMed] [Google Scholar]
- 26.Jönsson T, Poole LB. Intramolecular electron transfer pathway in mutants of AhpF, a flavoprotein disulfide reductase. In: Chapman S, Perham R, Scrutton N, editors. Flavins and Flavoproteins 2002. Agency for Scientific Publications; Berlin: 2002. pp. 691–696. [Google Scholar]
- 27.Poole LB. Flavin-linked redox components required for AhpC reduction in alkyl hydroperoxide reductase systems. In: Ghisla S, Kroneck P, Macheroux P, Sund H, editors. Flavins and Flavoproteins 1999. Agency for Scientific Publications; Berlin: 1999. pp. 195–202. [Google Scholar]
- 28.Parsonage D, Miller H, Ross RP, Claiborne A. Purification and analysis of streptococcal NADH peroxidase expressed in Escherichia coli. J Biol Chem. 1993;268:3161–3167. [PubMed] [Google Scholar]
- 29.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 30.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 31.Prongay AJ, Engelke DR, Williams CH., Jr. Characterization of two active site mutations of thioredoxin reductase from Escherichia coli. J Biol Chem. 1989;264:2656–2664. [PubMed] [Google Scholar]
- 32.Williams CH., Jr. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase -- a family of flavoenzyme transhydrogenases. In: Müller F, editor. Chemistry and Biochemistry of Flavoenzymes. CRC Press, Inc.; Boca Raton: 1992. pp. 121–211. [Google Scholar]
- 33.Mulrooney SB, Williams CH., Jr. Evidence for two conformational states of thioredoxin reductase from Escherichia coli: use of intrinsic and extrinsic quenchers of flavin fluorescence as probes to observe domain rotation. Protein Sci. 1997;6:2188–2195. doi: 10.1002/pro.5560061013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lennon BW, Williams CH, Jr., Ludwig ML. Twists in catalysis: alternating conformations of Escherichia coli thioredoxin reductase. Science. 2000;289:1190–1194. doi: 10.1126/science.289.5482.1190. [DOI] [PubMed] [Google Scholar]
- 35.Poole LB, Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Methods Enzymol. 2002;348:122–136. doi: 10.1016/s0076-6879(02)48632-6. [DOI] [PubMed] [Google Scholar]
- 36.Poole LB, Higuchi M, Shimada M, Calzi ML, Kamio Y. Streptococcus mutans H2O2-forming NADH oxidase is an alkyl hydroperoxide reductase protein. Free Radic Biol Med. 2000;28:108–120. doi: 10.1016/s0891-5849(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 37.Roberts BR, Wood ZA, Jönsson TJ, Poole LB, Karplus PA. Oxidized and synchrotron cleaved structures of the disulfide redox center in the N-terminal domain of Salmonella typhimurium AhpF. Protein Sci. 2005;14:2414–2420. doi: 10.1110/ps.051459705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kallis GB, Holmgren A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J Biol Chem. 1980;255:10261–10265. [PubMed] [Google Scholar]
- 39.Bieger B, Essen LO. Crystal structure of the catalytic core component of the alkylhydroperoxide reductase AhpF from Escherichia coli. J Mol Biol. 2001;307:1–8. doi: 10.1006/jmbi.2000.4441. [DOI] [PubMed] [Google Scholar]
- 40.Waksman G, Krishna TS, Williams CH, Jr., Kuriyan J. Crystal structure of Escherichia coli thioredoxin reductase refined at 2 Å resolution. Implications for a large conformational change during catalysis. J Mol Biol. 1994;236:800–816. [PubMed] [Google Scholar]
- 41.Prongay AJ, Williams CH., Jr. Evidence for direct interaction between cysteine 138 and the flavin in thioredoxin reductase. A study using flavin analogs. J Biol Chem. 1990;265:18968–18975. [PubMed] [Google Scholar]
- 42.Veine DM, Mulrooney SB, Wang PF, Williams CH., Jr. Formation and properties of mixed disulfides between thioredoxin reductase from Escherichia coli and thioredoxin: evidence that cysteine-138 functions to initiate dithiol-disulfide interchange and to accept the reducing equivalent from reduced flavin. Protein Sci. 1998;7:1441–1450. doi: 10.1002/pro.5560070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karplus PA, Schulz GE. Refined structure of glutathione reductase at 1.54 Å resolution. J Mol Biol. 1987;195:701–729. doi: 10.1016/0022-2836(87)90191-4. [DOI] [PubMed] [Google Scholar]
- 44.Mattevi A, Schierbeek AJ, Hol WG. Refined crystal structure of lipoamide dehydrogenase from Azotobacter vinelandii at 2.2 Å resolution. A comparison with the structure of glutathione reductase. J Mol Biol. 1991;220:975–994. doi: 10.1016/0022-2836(91)90367-f. [DOI] [PubMed] [Google Scholar]
- 45.Lennon BW, Williams CH., Jr. Effect of pyridine nucleotide on the oxidative half-reaction of Escherichia coli thioredoxin reductase. Biochemistry. 1995;34:3670–3677. doi: 10.1021/bi00011a023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.