

Abstract

We report the catalytic asymmetric allylation of ketones under highly concentrated reaction conditions with a catalyst generated from titanium tetraisopropoxide and BINOL (1:2 ratio) in the presence of isopropanol. This catalyst promotes the addition of tetraallylstannane to a variety of ketones to produce tertiary homoallylic alcohols in excellent yield (80–99%) with high enantioselectivities (79–95%). The resulting homoallylic alcohols can also be epoxidized in situ using tert-butyl hydroperoxide (TBHP) to afford cyclic epoxy alcohols in high yield (84–87%).
Synthetic organic chemistry has advanced to the level that most complex natural products can now be prepared.1–3 As a result, a focus of organic synthesis is how natural products can be made in a truly practical manner.4–6 One of the challenges facing chemists, therefore, is the development of transformations that are not only efficient, selective, and high yielding, but that are also more environmentally benign.7,8
Common measures of the ‘greenness’ of reactions are the E factor9 and the volume productivity.10 The E factor is defined as the ratio of weight waste to weight product, and the volume productivity is grams product per liter of reaction medium. The E factor for many pharmaceuticals has been estimated to exceed 100.11 The largest contributors to the magnitude of E factors are organic solvents, many of which are ecologically harmful and require costly remediation.
One approach to reducing a reaction’s E factor and, therefore, its adverse impact on the environment is to conduct them under solvent-free or highly concentrated conditions.11–14 Advantages of solvent-free or highly concentrated reactions include cost savings, reduced energy consumption, decreased reaction times, and a considerable reduction in reactor size and, therefore, capital investment. These attributes have inspired a substantial research effort directed toward the development of solvent-free reactions.10–14
Despite the importance for solvent-free and highly concentrated processes, relatively few studies concerning concentrated catalytic asymmetric reactions have been reported.15–21 This is not surprising, given that catalyst enantioselectivity and efficiency can be highly sensitive to concentration and reaction medium.22 Although the challenges of solvent-free and highly concentrated asymmetric catalysis are significant, they are outweighted by the potential environmental and economical benefits.
In response to these challenges, a few research groups have examined catalytic asymmetric reactions under solvent-free and highly concentrated conditions.15–21 Examples of highly enantioselective catalysts that exhibit good substrate generality under solvent-free conditions include Jacobsen’s salen derivatives for the kinetic resolution of racemic epoxides, desymmetrization of meso epoxides,23–26 and the hetero-Diels-Alder reaction.27 Other examples include Ding’s hetero-Diels-Alder28 and carbonyl-ene17 reactions and the Hoveyda/Schrock enantioselective ring closing metathesis route to cyclic amines.29 We recently reported the asymmetric addition of alkyl and functionalized alkyl groups to ketones under solvent-free conditions.16 Under these conditions, catalyst loadings were reduced by 4–40 fold, while maintaining high levels of enantioselectivity (>90% ee).16
Herein, we report a related effort to develop a more efficient process for the asymmetric allylation of ketones. In contrast to our asymmetric allylation of ketones employing dichloromethane solvent, optimization of the reaction under highly concentrated conditions resulted in the identificaiton of a new titanium catalyst.
We recently introduced a highly enantioselective catalyst for the asymmetric allylation of ketones under standard solvent conditions.30 While new catalysts are emerging for this difficult transformation,31–40 development of efficient and general catalyst remains challenging. Our original catalyst was generated by combining 30 mol % titanium tetraisopropoxide, 30 mol % BINOL, and isopropanol (20 equiv with respect to ketone substrate). The resulting catalyst promotes addition of tetraallylstannane to a variety of ketones in CH2Cl2 with high enantioselectivities (Scheme 1).30
Scheme 1.
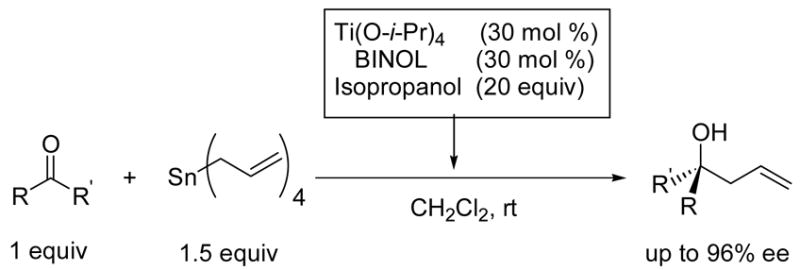
Catalytic Asymmetric Allylation of Ketones in CH2Cl2 Solvent.
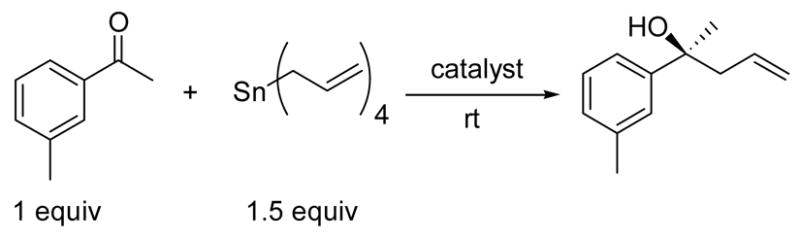
Combining 3-methylacetophenone with 1.5 equiv of tetraallylstannane, 20 equiv of IPA, and 20 mol % catalyst (Ti : BINOL, 1 : 1 ratio) in CH2Cl2, the corresponding homoallylic alcohol was produced in 82% yield and 96% ee (Table 1, entry 1). Decreasing the amount of IPA from 20 to 3 equiv resulted in only a 2% decrease in the enantioselectivity (Table 1, entry 2). At lower catalyst loadings (10 and 5 mol %), the difference in enantioselectivity with 20 vs. 3 equiv of IPA increased (entries 3 vs. 4 and 5 vs. 6).
Table 1.
Comparison of the Asymmetric Allylation of 3-Methylacetophenone Under Standard and Highly Concentrated Reaction Conditions.
 | |||||
|---|---|---|---|---|---|
| entry | solvent | Ti(O -i-Pr)4 (mol %) | BINOL (mol %) | IPA (equiv) | % ee (% yield) |
| 1 | 20 | 20 | 20 | 96 (82) | |
| 2 | 20 | 20 | 3 | 94 (91) | |
| 3 | CH2Cl2 | 10 | 10 | 20 | 88 (94) |
| 4 | 10 | 10 | 3 | 81 (86) | |
| 5 | 5 | 5 | 20 | 69 (93) | |
| 6 | 5 | 5 | 3 | 61 (89) | |
| 7 | 20 | 20 | 20 | 85 (91) | |
| 8 | concentrated | 10 | 10 | 3 | 81 (88) |
| 9 | 5 | 5 | 3 | 79 (84) | |
In an effort to reduce the amount of solvent employed, the allylation in entry 1 (96% ee) was repeated in the absence of dichloromethane. Thus, employing 20 mol % catalyst and 20 equiv of IPA, the enantioselectivity of the product dropped to 85% (Table 1, entries 1 vs. 7). Surprisingly, decreasing the amount of IPA from 20 to 3 equiv at 10 and 5 mol % catalyst produced a smaller change in the enantioselectivity of the product (entries 8 and 9). Reducing the amount of IPA and catalyst loading had only a marginal affect on product yield and enantioselectivity under highly concentrated conditions (Table 1, entries 7–9). In sharp contrast, using less catalyst in CH2Cl2 solution resulted in a 33% decrease in the enantioselectivity (entries 2 and 6). Using less than 3 equiv of IPA under concentrated conditions resulted in lower enantioselectivities.
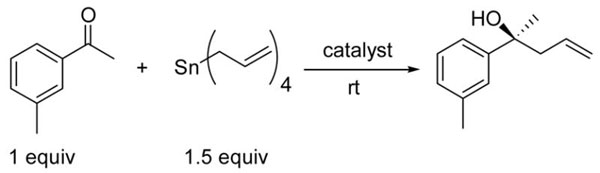
In the allylation of aldehydes, it is known that the ratio of titanium to BINOL can impact the ee of the secondary homoallylic alcohol product.41–43 Based on this observation and the results in Table 1, we examined the impact of the Ti : BINOL ratio on the ketone allylation under concentrated conditions (3 equiv IPA, no CH2Cl2). Thus, reaction of 3-methylacetophenone, tetraallylstannane (1.5 equiv), and 10 mol % catalyst (Ti : BINOL ratio of 1 : 1) resulted in product with 88% ee (Table 2, entry 1). We were pleased to find that doubling the mol % BINOL, while maintaining the mol % titanium tetraisopropoxide, resulted in an increase in ee to 95% (Table 2, entry 2). Based on this result, we focused on catalyst optimization maintaining a 1 : 2 ratio of Ti : BINOL. Reducing the mol % of titanium to 5, 2, and 1 caused a loss of enantioselectivity, which was dramatic below 5 mol % (Table 2, entries 3–5).
Table 2.
Optimization of Ti(O-i-Pr)4 : BINOL Ratio and Catalyst Mol % Under Highly Concentrated Conditions.
 | ||||
|---|---|---|---|---|
| entry | Ti(O -i-Pr)4 (mol % ) | BINOL (mol %) | IPA (equiv) | %ee (% yield) |
| 1 | 10 | 10 | 3 | 88 (88) |
| 2 | 10 | 20 | 3 | 95 (93) |
| 3 | 5 | 10 | 3 | 90 (90) |
| 4 | 2 | 4 | 3 | 55 (97) |
| 5 | 1 | 2 | 3 | 28 (95) |
After optimizing the reaction under concentrated conditions, with a 1 : 2 ratio of Ti : BINOL and 3 equiv of IPA, we investigated the substrate scope of this new catalyst system (Table 3). Only a marginal electronic influence was found with substituted acetophenone derivatives, as 3-(trifluoromethyl) and 4-methoxyacetophenone underwent allylation with similar enantioselectivities at 10 mol % catalyst (89% and 88%, entries 3 and 5). For comparison purposes, 3-methylacetophenone underwent allylation at 10 mol % catalyst, generating the product in 95% ee (Table 3, entry 1). Decreasing catalyst loading to 5 mol % for these substrates resulted in a decrease in enantioselectivity to 90%, 79%, and 82% (entries 2, 4, and 6, respectively). The enone possessing the exocyclic double bond proved to be an excellent substrate, providing enantioselectivities of 91% and 85% at 10 and 5 mol % catalyst (entries 7 and 8, respectively).
Table 3.
Yields and Enantioselectivities for the Asymmetric Allylation of Ketones Under Highly Concentrated Conditions with 1:2 Ti : BINOL Ratio.
| entry | substrate | Ti:BINOL (mol %) | Sn(allyl)4 (equiv) | IPA (equiv) | % ee (% yield) |
|---|---|---|---|---|---|
| 1 |
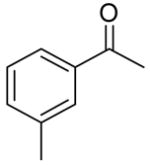
|
10:20 | 1.5 | 3 | 95 (93) |
| 2 | 5:10 | 1.5 | 3 | 90 (90) | |
| 3 |
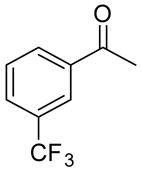
|
10:20 | 1.5 | 3 | 89 (87) |
| 4 | 5:10 | 1.5 | 3 | 79 (85) | |
| 5 |
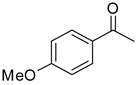
|
10:20 | 1.5 | 3 | 88 (99) |
| 6 | 5:10 | 1.5 | 3 | 82 (92) | |
| 7 |
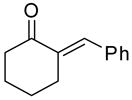
|
10:20 | 1.5 | 3 | 91 (96) |
| 8 | 5:10 | 1.5 | 3 | 85 (99) | |
| 9 |
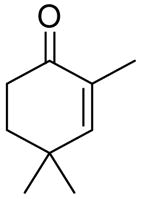
|
10:20 | 1.5 | 3 | 91 (80) |
| 10 | 5:10 | 1.5 | 3 | 86 (85) | |
| 11 |
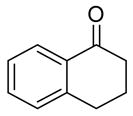
|
10:20 | 1.5 | 3 | 87 (99) |
| 12 | 5:10 | 1.5 | 3 | 79 (99) |
Reaction of the α,β-unsaturated enone produced the corresponding homoallylic alcohol in 91% and 86% ee at 10 and 5 mol % catalyst (entries 9 and 10). The cyclic ketone α-tetralone underwent addition with slightly lower enantioselectivity (87% and 79%, entries 11 and 12). The results listed in Table 3 demonstrate the potential range of substrates that can be transformed into homoallylic alcohols with high levels of enantioselectivity using this new catalyst under highly concentrated conditions. It is noteworthy that these highly concentrated conditions allow for a 3-fold reduction in titanium and a 1/3 reduction in BINOL loading, with a significant decrease in the reaction volume.
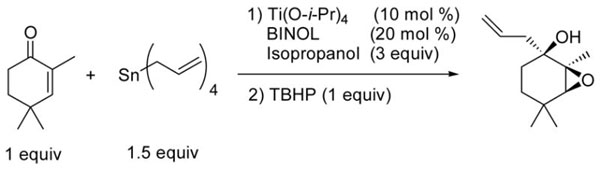
Another method to increase the synthetic efficiency of a transformation is via tandem reactions, which enable rapid increases in molecular complexity with minimal isolation and purification.44 With this aim in mind, we set out to conduct a tandem asymmetric allylation of enones followed by a chemo- and diastereoselective directed epoxidation of the allylic double bond under concentrated conditions. Our approach involves exploitation of the titanium catalyst employed in the asymmetric allylation to conduct the diastereoselective epoxidation reaction. Thus, after the ketone allylation was complete, 1 equiv of anhydrous tert-butyl hydroperoxide (TBHP, 5.5 M) was added to the reaction mixture (Table 4).30
Table 4.
Highly Concentrated Tandem Asymmetric Allylation/Diastereoselective Epoxidation of Cyclic Enones.
 | |||
|---|---|---|---|
| Entry | substrate | product | % ee (% yield) |
| 1 |
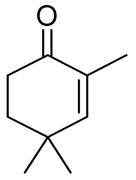
|
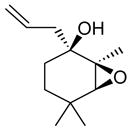
|
91 (84) |
| 2 |
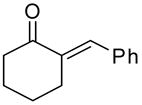
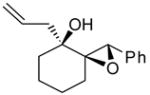
|
|
91 (87) |
| 3 |
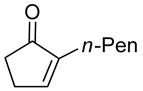
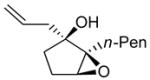
|
|
90 (85) |
The epoxidation proceeded readily at room temperature to afford the syn epoxy alcohols in 84 – 87% yield (Table 4). No erosion in ee during the epoxidation was observed, and only a single diastereomer was detected in each case by 1H NMR spectroscopy and GC analysis. The tandem asymmetric allylation/diastereoselective epoxidation reaction is operationally simple and circumvents the need to isolate and purify the intermediate tertiary allylic alcohols. Starting from achiral precursors, this one-pot sequence results in the generation of three contiguous stereocenters with excellent enantio- and diastereoselectivity and with high yields.
In summary, we have developed a new catalyst for the asymmetric allylation of ketones that is readily prepared from titanium tetraisopropoxide and BINOL (1 : 2 ratio) in the presence of isopropanol. While the role of the isopropanol remains elusive, it is necessary to obtain high enantioselectivity. This catalyst functions under concentrated reaction conditions, providing tertiary homoallylic alcohols with high levels of enantioselectivity. Furthermore, catalyst loadings as low as 5 mol % give high enantioselectivity, representing a significant reduction from our original system, which required 30 mol % catalyst and 20 equivalents of IPA to obtain high enantioselectivity.
The resultant enantioenriched products are particularly useful intermediates in synthesis, because the double bonds can be differentially functionalized. This was illustrated in the tandem asymmetric allylation/diastereoselective epoxidation reaction that chemoselectively oxidizes the more electron-rich allylic double bond, producing the syn epoxy alcohols in high yields and diastereoselectivity. While our new approach is more environmentally friendly, we are currently searching for allyl sources that do not involve tin.
Supplementary Material
Procedures and full characterization of new compounds (PDF) is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the NIH (National Institute of General Medical Sciences, GM58101).
References
- 1.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; New York: 1989. [Google Scholar]
- 2.Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. VCH; Weinheim, Germany: 1996. [Google Scholar]
- 3.Nicolaou KC, Snyder SA. Classics in Total Synthesis II: More Targets, Strategies, Methods. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- 4.Smith AB, III, Beauchamp TJ, LaMarche MJ, Kaufman MD, Qiu Y, Arimoto H, Jones DR, Kobayashi K. J Am Chem Soc. 2000;122:8654–8664. [Google Scholar]
- 5.Smith AB, III, Freeze BS, Xian M, Hirose T. Org Lett. 2005;7:1825–1828. doi: 10.1021/ol050455z. [DOI] [PubMed] [Google Scholar]
- 6.Mickel SJ, Niederer D, Daeffler R, Osmani A, Kuesters E, Schmid E, Schaer K, Gamboni R, Chen W, Loeser E, Kinder FR, Jr, Konigsberger K, Prasad K, Ramsey TM, Repic O, Wang R-M, Florence G, Lyothier I, Paterson I. Org Proc Res Dev. 2004;8:122–130. [Google Scholar]
- 7.DeSimone JM. Science. 2002;297:799–803. doi: 10.1126/science.1069622. [DOI] [PubMed] [Google Scholar]
- 8.Anastas PT, Williamson TC. Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes. Oxford Science Publications; New York: 1998. [Google Scholar]
- 9.Sheldon RA. Chem Ind. 1992:903–906. [Google Scholar]
- 10.Jacobsen EN, Finney NS. Chem Biol. 1994;1:85–90. doi: 10.1016/1074-5521(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 11.Cave GWV, Raston CL, Scott JL. Chem Commun. 2001:2159–2169. [PubMed] [Google Scholar]
- 12.Metzger JO. Angew Chem, Int Ed. 1998;37:2975–2978. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2975::AID-ANIE2975>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka K, Toda F. Chem Rev. 2000;100:1025–1074. doi: 10.1021/cr940089p. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka K. Solvent-Free Organic Synthesis. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- 15.Mojtahedi MM, Abaee MS, Abbasi H. Can J Chem. 2006;84:429–432. [Google Scholar]
- 16.Jeon SJ, Li H, Walsh PJ. J Am Chem Soc. 2005;127:16416–16425. doi: 10.1021/ja052200m. [DOI] [PubMed] [Google Scholar]
- 17.Yuan Y, Zhang X, Ding K. Angew Chem, Int Ed. 2003;42:5478–5480. doi: 10.1002/anie.200352535. [DOI] [PubMed] [Google Scholar]
- 18.Shibahara F, Nozaki K, Hiyama T. J Am Chem Soc. 2003;125:8555–8560. doi: 10.1021/ja034447u. [DOI] [PubMed] [Google Scholar]
- 19.Sato I, Kodaka R, Soai K. J Chem Soc, Perkin Trans 1. 2001;22:2912–2914. [Google Scholar]
- 20.Chesney A. Green Chemistry. 1999;1:209–219. [Google Scholar]
- 21.Martinez LE, Leighton JL, Carsten DH, Jacobsen EN. J Am Chem Soc. 1995;117:5897–5898. [Google Scholar]
- 22.Jacobsen EN, Pfaltz A, Yamamoto H. Comprehensive Asymmetric Catalysis. Springer; Berlin: 1999. [Google Scholar]
- 23.Tokunaga M, Larrow JF, Kakuichi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]
- 24.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 25.Ready JM, Jacobsen EN. J Am Chem Soc. 2001;123:2687–2688. doi: 10.1021/ja005867b. [DOI] [PubMed] [Google Scholar]
- 26.White DE, Jacobsen EN. Tetrahedron: Asymmetry. 2003;14:3633–3638. [Google Scholar]
- 27.Dossetter AG, Jamison TF, Jacobsen EN. Angew Chem, Int Ed. 1999;38:2398–2400. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 28.Long J, Hu J, Shen X, Ji B, Ding K. J Am Chem Soc. 2002;124:10–11. doi: 10.1021/ja0172518. [DOI] [PubMed] [Google Scholar]
- 29.Dolman SJ, Sattely ES, Hoveyda AH, Schrock RR. J Am Chem Soc. 2002;124:6991–6997. doi: 10.1021/ja012534l. [DOI] [PubMed] [Google Scholar]
- 30.Kim JG, García IF, Kwiatokowski D, Walsh PJ. J Am Chem Soc. 2004;126:12580–12585. doi: 10.1021/ja047758t. [DOI] [PubMed] [Google Scholar]
- 31.Yanagisawa A. Comprehensive Asymmetric Catalysis. Vol. 2 Springer; Berlin: 1999. [Google Scholar]
- 32.Denmark SE, Fu J. Chem Rev. 2003;103:2763–2794. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]
- 33.Hanawa H, Kii S, Maruoka K. Adv Synth Catal. 2001;1:57–60. [Google Scholar]
- 34.Casolari S, D’Addario D, Tagliavini E. Org Lett. 1999;1:1061–1063. [Google Scholar]
- 35.Yasuda M, Kitahara N, Fujibayashi T, Baba A. Chem Lett. 1998;8:743–744. [Google Scholar]
- 36.Wadamoto M, Yamamoto H. J Am Chem Soc. 2005;127:14556–14557. doi: 10.1021/ja0553351. [DOI] [PubMed] [Google Scholar]
- 37.Wada R, Kounosuke O, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:8910–8911. doi: 10.1021/ja047200l. [DOI] [PubMed] [Google Scholar]
- 38.Teo YC, Goh JD, Loh TP. Org Lett. 2005;7:2743–2745. doi: 10.1021/ol051018n. [DOI] [PubMed] [Google Scholar]
- 39.Lu J, Hong ML, Ji SJ, Teo YC, Loh TP. Chem Commun. 2005:4217–4218. doi: 10.1039/b507768k. [DOI] [PubMed] [Google Scholar]
- 40.Prieto O, Woodward S. J Organomet Chem. 2006;691:1515–1519. [Google Scholar]
- 41.Costa AL, Piazza MG, Tagliavini C, Umani-Ronchi A. J Am Chem Soc. 1993;115:7001–7002. [Google Scholar]
- 42.Keck GE, Tarbet K, Geraci LS. J Am Chem Soc. 1993;115:8467–8468. [Google Scholar]
- 43.Keck GE, Geraci LS. Tetrahedron Lett. 1993;34:7827–7828. [Google Scholar]
- 44.Ho T-L. Tandem Organic Reactions. Wiley; New York: 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Procedures and full characterization of new compounds (PDF) is available free of charge via the internet at http://pubs.acs.org.