

Abstract
It has been shown by us in a recent communication that homopolymers, in which each repeat unit contains a hydrophilic and a hydrophobic head group, are capable of forming environment dependent micellar or inverse micellar assemblies. A systematic structure-property relationship study is carried out here to test the scope of the design. We show here that the molecular design is indeed broadly applicable and that there is a significant gain in the critical aggregation concentrations of these polymers, compared to the small molecule counterparts. We also show that the design can be tuned to achieve vesicle-type assemblies, which further expands the repertoire of amphiphilic homopolymers in a variety of areas. Characterizations of these assemblies have been carried out using transmission electron microscopy, dynamic light scattering, static light scattering, and dye incorporation experiments.
Introduction
Molecules based on amphiphilic building blocks are capable of providing diverse self-assembled structures, because of their differential interaction energy with the solvent surface.1 Such amphiphilic assemblies could find use in a variety of applications ranging from biology to materials.2 There is significant interest in polymer-based supramolecular assemblies, because of the enhanced stabilities and lower critical aggregation concentrations that these macromolecules are capable of providing.2d Block copolymers have been a popular choice for these types of assemblies, where one of the blocks is relatively more incompatible with the solvent milieu.3 We have recently developed a molecular design based on homopolymers in which both the hydrophilic and the hydrophobic moieties are incorporated within the monomer unit.4 In that preliminary communication, we had demonstrated that such homopolymers are capable of providing both micelle-like and inverse micelle-like assemblies depending on the solvent environment, which is an amplified consequence of the molecular level conformational changes in each monomer unit. Since then, we have shown that such polymer assemblies are indeed unique and therefore could find complementary applications in a broad range of areas.5–7 For example, we have reported that the micellar interiors of these polymers could be used as nanocontainers to carry out organic photochemical reactions and that the selectivities in these reactions are much higher than those obtained with micellar containers based on block copolymers or small molecule surfactants.5 We have also demonstrated that these environment-dependent polymer assemblies are unique in that these are kinetically trapped in the solvent used for initial assembly.6 We have demonstrated that such a feature could be used as nanocontainers for separation of organic molecules.
Considering the possible implications in a variety of areas, it is important that we fully test the scope of our molecular design. Here, we describe the details of a systematic structure-property relationship study. We address the following in this paper: (i) could one apply the structural guidelines developed using our styrene-based polymers for the formation of responsive micelles to other polymer backbones? In other words, are the design guidelines applicable broadly? (ii) How does the nature of the micellar interior vary with the backbone of the polymer and the hydrophobic functionality? (iii) What are the critical micelle concentrations (cmc) of these polymers? How does this vary with the structure of the building blocks of the polymer? How different are these from the small molecule surfactants? (iv) Could one use a similar design principle to achieve vesicle-like assemblies? What are the critical structural requirements for such assemblies? (v) Are the vesicle-like assemblies responsive to the solvent environment? We describe here the design and syntheses of the monomers and the polymers, followed by the characterization of the assemblies formed by these structures using transmission electron microscopy (TEM), dynamic light scattering (DLS), static light scattering studies (SLS), dye-based studies to estimate the polarity of the micellar interior and vesicular membranes, and estimation of critical aggregation constants for micelles8a and vesicles8b.
Results and Discussion
Design and Synthesis of Monomers and Polymers
In our original molecular design, we had stipulated that the hydrophilic and the hydrophobic functionalities should be on either sides of a rigid scaffold such that the monomer is facially amphiphilic. As a consequence of this presumed criterion, we had substituted the hydrophilic and the hydrophobic moieties at the 3-, and 5- positions of a benzene ring relative to the polymerizable double bond, as illustrated by structures PS6M, PS8M and PS10M (Chart 1).9 Such a pre-condition in the molecular design narrows down the scope of these amphiphilic homopolymers. Therefore, we have been interested in identifying whether a simple placement of a hydrophilic and hydrophobic moiety with a flexible linker within a monomer unit is sufficient to achieve the observed environmentally sensitive assemblies. Demonstration of such a feature will obviously expand the scope of the homopolymers in amphiphilic assemblies.
Chart 1.

Structure of the amphiphilic homopolymers studied.
To test this, we have designed and synthesized polymers PA6M, PA8M and PA10M, shown in Chart 1. In these structures, glycine has been derivatized with alkyl chains of different lengths. The secondary amine is then attached to an acryl moiety. The resultant acrylamide polymer has simple design features, i.e. a flexible side chain functionality of the polymer in which each repeat unit contains both a hydrophilic and a hydrophobic moiety. If our molecular design can indeed be applied to a wide variety of structures, then polymers PA6M should form micelles or inverted micelles depending on the solvent environment, just as with polymers PS6M, PS8M, and PS10M.
Also, in order to test whether we could use similar design guidelines to achieve vesicle-type assemblies, polymers PA5V, PA7V, PA10V and PA15V containing bolamphiphilic10 moieties were synthesized. The key structural feature of these polymers is the presence of two hydrophilic carboxylic functionalities connected by alkyl chains of varying lengths. Our hypothesis is that such a presentation of the functionalities in a homopolymer side chain could provide a more direct pathway to achieve vesicular assemblies. For example, the only difference between structures PA10M and PA10V is that the polymer PA10V contains an additional -CO2H functionality at the end of the long alkyl chain. Would PA10M form a micelle-like assembly, while PA10V forms a vesicle-type assembly?
Monomers for the synthesis of acrylamide polymers were synthesized from the ethyl ester of glycine, haloalkanes (or methyl ester of haloalkanoic acid), and acryloyl chloride, as shown in Scheme 1. Synthesis of 11-17 was achieved using the reversible addition-fragmentation-termination (RAFT) polymerization method.11 This controlled radical polymerization technique afforded the polymers 11-17 with a reasonable control in polydispersity, as illustrated in Scheme 1. Saponification of the ester using potassium hydroxide followed by acidification afforded the polymers. This hydrolysis step proceeded with >95% yield, as discerned from the complete disappearance of the alkanol part of the ester peaks in the 1H NMR of the polymers. Details of the synthesis and characterization of these polymers are outlined in the Supporting Information.
Scheme 1.
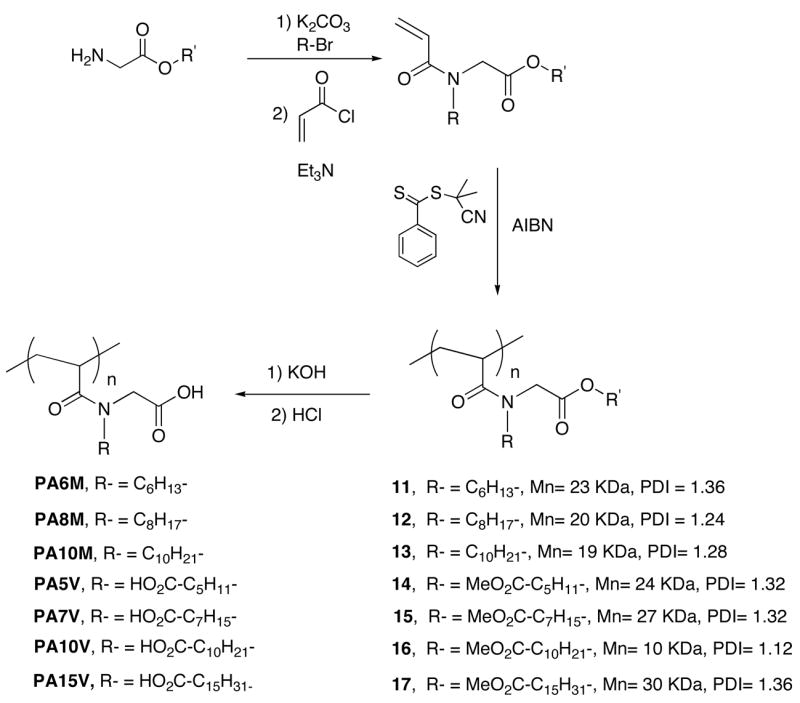
General synthetic approach to polymers PA6M, PA8M, PA10M, PA5V, PA7V, PA10V and PA15V.
Characterization of the micelle-type assemblies
First, we were interested in identifying whether the polymers PA6M, PA8M, and PA10M would be capable of forming solvent-dependent micelle and inverse micelle-type assemblies. This will test whether the design of homopolymers to form micelles or inverse micelles is more broadly applicable. Transmission electron microscopy (TEM) has been a popular and effective tool for characterizing amphiphilic supramolecular assemblies.12 To prepare a micellar solution of the polymer, it had to be dissolved in water. For this purpose, polymers were converted to the corresponding metal carboxylates using lithium hydroxide as the base. The TEM obtained from an aqueous solution of the polymer PA6M is shown in Figure 1a, which suggests that this polymer does afford a micelle-type assembly in water. Analysis of these pictures suggests that the average particle size of the micellar assembly in the aqueous phase is about 70 nm. Similarly, the polymer PA6M was dissolved in toluene and the TEM picture obtained from this solution suggests that an inverse micelle-type assembly is formed (Figure 1b). The average size of these assemblies was determined to be about 120 nm. Similar environment-dependent nanoassemblies were also obtained with polymers PA8M and PA10M.
Figure 1.

TEM pictures from a) solution of PA6M in water b) solution of PA6M in toluene, c) Absorption spectra of pyrene in water and in aqueous solution of PA6M. d) Emission spectra of absorption-matched rhodamine 6G in water and in toluene solution of PA6M.
One could be understandably skeptical of the particle size measurements obtained from TEM, since the solution-based sample is dried in the process. Laser light scattering is a non-invasive method of analyzing the aggregates in solution. In order to investigate the correlation between the assemblies in an aqueous solution compared to a dried sample in TEM, we measured the size of the particles obtained from polymer PA6M using dynamic light scattering (DLS). The hydrodynamic radius (RH), obtained using DLS, for the aqueous solution of PA6M was about 53 nm (comparable to RTEM of 35 nm). We also determined the radius of gyration (RG) of these nanoassemblies using static light scattering (SLS). The RG of PA6M in the aqueous phase was found to be about 41 nm.13 Ratio of the radius of gyration vs. the hydrodynamic radius (RG/RH), known as ρ, is sensitive to the nature of the assembly in solution.14 In the case of polymer PA6M, this ratio is about 0.77, which indicates the formation of a micelle-type assembly.
It is to be noted that the sizes we obtain from TEM and DLS seem rather large to be called micelles. The micelles obtained from these polymers are expected to be closer to those obtained from small molecule surfactants, such as SDS. The rather large sizes obtained here might lead one to suggest that these assemblies might be akin to vesicles. Note that the size alone does not qualify these assemblies as vesicles; they are indeed micellar assemblies, because: (i) the TEM images obtained from micelles are darker in the interior, while the water-filled interiors of vesicles are lighter than the periphery. The former was indeed the case with these polymers and therefore resembles micelles.13 (ii) Micelles are capable of sequestering hydrophobic guest molecules, but not hydrophilic ones, in water. Vesicles are capable of sequestering hydrophilic guest molecules in their interior, and hydrophobic molecules within their membranes. These polymers do not exhibit the ability to sequester hydrophilic guest molecules and do sequester hydrophobic guests (vide infra), consistent with micelle formation. (iii) Micellar interiors are typically more hydrophobic compared to the membranes in typical vesicles. This was indeed the case with the polymers under study (vide infra). (iv) There is sufficient literature precedence for micellar assemblies affording sizes bigger than the expected, both with small molecule surfactants and with amphiphilic block copolymers.15 Al though various investigators call these with different names, the underlying theme in all these observation is that these are aggregation of micelles. It is possible that our polymers also fall under this category.
If the polymers do indeed form micelle-type and inverse micelle-type assemblies in water and apolar solvents respectively, these polymers should be capable of acting as nanocontainers for apolar guest molecules in water and vice versa. To investigate this possibility, we first used pyrene as the guest molecule in water. Pyrene also exhibits an emission spectrum, the fine structure of which is sensitive to the microenvironment in which it is present.16 Therefore, this guest molecule also provides the opportunity to investigate the polarity of the micellar interior formed by these polymers. Pyrene is not very soluble in water by itself, as could be discerned from the absorption spectrum of pyrene obtained in water (Figure 1c). However, in a 10−4 M solution of the polymer PA6M in water, an absorbance of 0.4 was obtained at 337 nm. This result suggests that the micelle-type assembly is capable of sequestering apolar guest molecules in water. If this apolar guest molecule is indeed present in the hydrophobic interior of a micelle-type assembly, the ratio of the intensity of the fluorescence peaks (I1/I3) should be much lesser than 1.8. Indeed, the value obtained for the polymer PA6M is about 1.2. This value compares well with the micellar interior formed from small molecule surfactants such as SDS, which exhibits a value of 1.2.17
Similarly, to investigate the formation of inverse micelles, rhodamine-6G (R6G) that is not soluble in toluene is used as the probe.18 In the presence of the polymer PA6M, it is clear that R6G can be dissolved in the solution from its color and the linear absorption spectra. Since this dye molecule exhibits self-quenching properties, it is also possible to investigate whether it is indeed sequestered inside a nanoscale inverse micelle-type particle. If a certain amount of R6G is uniformly distributed in a solvent that it is soluble, such as water, it could fluoresce without any self-quenching effects (Figure 1d). However, when the same amount of the dye molecule is sequestered inside a 120 nm particle, the local concentration of R6G within the inverse micelle increases dramatically, although the overall solution concentration of R6G is the same as the aqueous solution. This increase in local concentration would result in self-quenching. This is indeed the observed result with this dye molecule and the inverse micelle formed by polymer PA6M, as shown in Figure 1d. Similar behavior was also observed with polymers PA8M and PA10M.
The sensitivity of pyrene emission to its microenvironment has been utilized to determine the CMC of the micelle.19 Since these polymers can be considered to be polymerized surfactants, it is useful to compare the CMC of these polymers with the corresponding building block units. For this purpose, we have synthesized the corresponding building blocks 18-20 by reducing the double bond of the acrylamide monomers, as shown in Scheme 2. When comparing the small molecules with the corresponding polymers, comparison of the CMCs using concentration can be misleading; because, the molecular weights of the polymers are high and these macromolecules contain several copies of the surfactants. Therefore, comparison of the CMCs based on weight is more appropriate. For example, comparison of PA6M and 18 based on concentration suggests an increase in the CMC for the polymer by four orders of magnitude (Table 1). However, the comparison using weight suggests that this increase corresponds to only about two orders of magnitude. It is gratifying to note a significant enhancement in the CMC for the polymer PA6M. The CMC understandably decreases in the small molecule as well as in the polymer, when the length of the hydrophobic chain is increased. But, the magnitude of the gain in CMCs upon going from the monomer to a polymer seems to vary with the nature of the alkyl group; the CMC decreases about 39 times for the octyl and 226 times for the decyl chain. This difference in stage at which a significant difference in CMC is observed for the polymer and the small molecule is attributed to the relative packing of the hydrophobic chains in the small molecule vs. the polymer. In the polymer, the relative distance between an amphiphilic functionality and its neighbor is dictated by the covalent bonds in the polymer backbone. On the other hand, this packing is more adjustable in the small molecule.
Scheme 2.

Synthesis of the building block surfactant.
Table 1.
CAC and I1/I3 values for polymers and building blocks
| Compound | CAC | I1/I3above the CAC | |
|---|---|---|---|
| mg/L | M | ||
| PS6M | 7 | 5.8×10−7 | 1.10 |
| PS8M | 6 | 3.9×10−7 | 1.10 |
| PS10M | 5 | 3.3×10−7 | 0.98 |
| PA6M | 5 | 2.5×10−7 | 1.20 |
| PA8M | 5 | 2.4×10−7 | 1.16 |
| PA10M | 0.6 | 3.3×10−8 | 1.14 |
| PA5V | NA | NA | 1.85 |
| PA7V | NA | NA | 1.75 |
| PA10V | 50 | 5.8×10−6 | 1.52 |
| PA15V | 2 | 7.4×10−8 | 1.22 |
| 18 | 450 | 2.1×10−3 | 1.00 |
| 19 | 193 | 7.9×10−4 | 0.85 |
| 20 | 136 | 5×10−4 | 1.10 |
| 21 | 180 | 5.7×10−4 | 0.90 |
| 22 | 140 | 3.6×10−4 | 0.78 |
It is also interesting to note that the I1/I3 values for the small molecules above CMC is lower than those for the corresponding polymers; i.e. small molecules afford a better hydrophobic environment compared to the polymers. This can be explained by the same argument above. That is, the packing mode in the polymer is dictated by the repeat units, whereas small molecules can better accommodate themselves relative to their neighbors to optimize the packing. This could afford a more hydrophobic environment in the latter case. While this is a reasonable explanation, there is also an alternate possibility. Since the CMCs of the small molecules are inherently higher, the I1/I3 values for these are calculated at a higher concentration. This also means that the number of micellar containers (or more appropriately the relative volume of the micellar containers) is larger in the small molecules. The I1/I3 values are really a measure of the distribution coefficient of pyrene between the bulk solvent and the micellar container. When there is a greater volume of the hydrophobic containers available, the relative I1/I3 is likely to be lower. To distinguish these two possibilities, we measured the I1/I3 for pyrene in the presence of polymers PA6M and PA10M at concentrations similar to those above the CMC of 18 and 20. We found that the I1/I3 value did not change with this huge increase in concentration of the polymer. This suggests that the former mechanism (differential packing for the polymer vs. the small molecule) is operational.
Previously, we had reported on the TEM behavior and the environment-dependent container properties of the amphiphilic polystyrenes.4 However, the CMC of these polymers have not been reported. It is interesting to compare the CMCs obtained from these polymers compared to the corresponding acrylamide polymers PA6M, PA8M and PA10M. The CMCs of the polystyrenes are similar to those of polyacrylamides, when the hydrophobic functionalities are based on hexyl and the octyl groups. However, this difference increases to about an order of magnitude in the case of decyl moiety. This is attributed to the relatively flexible nature of the acrylamide backbone to accommodate the formation of a micellar assembly, relative to the polystyrene backbone. It is also interesting to note that the hydrophobicity of the interior in the case of the polystyrenes is higher than the corresponding polyacrylamides. This is understandable, since the polymer backbone itself is more hydrophobic in the case of polystyrenes relative to the polyacrylamides.
The observed CMCs for the polymers PS6M, PS8M and PS10M could seem contradictory to our preliminary communication, at first glance.4 In our previous report, we had reported that we have observed the micellar assemblies using TEM even at nM concentrations. However, here we report that the CMC of these types of polymer are in the range of 10−7 M. This apparent inconsistency can be easily understood, when the underlying theme in both these measurements are understood. The TEM measurement represents the picture of a dried sample of an amphiphilic polymer assembly at various concentrations. On the other hand, the above-mentioned CMC measurements are based on the distribution ratio of a guest molecule between the micellar interior formed by the polymer and the bulk solvent. This distribution ratio will obviously change with the concentration of the polymer. Therefore, it is very possible that at lower concentrations the polymer still forms a spherical assembly. However, this assembly does not effectively compete for the guest molecule with the bulk solvent at low concentrations. Note that the inherent solubility of pyrene in water is aboutl 10−7 M.19a
Characterization of the vesicle-type assemblies
It is interesting to ask whether the structures of the building blocks within the homopolymers could be tuned to achieve vesicular assemblies. We hypothesized that incorporating two hydrophilic head groups on either side of an alkyl chain should be able to afford vesicle structures. Polymer vesicles based on amphiphilic polymers have been previously achieved using block copolymers or polymers containing two hydrophobic tails for every hydrophilic head group.20 In our case, we ask whether polymerized bolamphiphiles are capable of providing vesicle-type assemblies.
First, we studied the lithium carboxylate of polymer, PA10V, in water. It was gratifying to note that the TEM picture of this solution exhibited a vesicular morphology, as shown in Figure 2a. Our hypothesis is that a vesicle-like assembly is obtained from the polymer PA10V, because one of the carboxylate moieties is presented in the exterior while the other carboxylate is presented in the water-filled interior of the vesicle. It is reasonable to expect the length of the alkyl chain between the two carboxylate moieties to impact the nature of the amphiphilic assembly formed from the polymers. Accordingly, we studied polymers PA5V, PA7V, and PA15V, where the nitrogen of the acrylamide functionality and the non-glycine carboxylate functionality are separated by five, seven, and fifteen methylene units respectively. TEM images obtained from an aqueous solution of PA15V clearly exhibit a vesicle-type assembly, while those from PA5V do not exhibit anything that resembles a vesicle, as exemplified in Figure 2b.
Figure 2.

TEM pictures from a) PA10V in water, b) PA5V in water c) PA10V in toluene.
We were intrigued by the fact that these polymers are also soluble in apolar organic solvents. In order to investigate the nature of the assembly in these solvents, TEM images were obtained from the toluene solution of polymer PA10V (Figure 2c). These images indicate the formation of large inverted micelle-like assemblies. We hypothesize that the long alkyl chains could bend to form a hydrophobic corona and a hydrophilic core. However, we note that the size of the assembly is much larger than even the vesicles from aqueous solutions. These assemblies are thus reminiscent of the so-called compound-micelles.21 Compound micelles are considered to be larger aggregates of smaller inverted-micellar assemblies. The presence of the inverse micelle-type assembly was further confirmed by a dye encapsulation study (vide infra). We propose a simple model that in the case of polymers PA10V and PA15V, the chain could fold and thereby presenting a hydrophobic surface while tucking in the hydrophilic carboxylic acid moieties. The proposed model for these polymers is schematically represented in Figure 3.
Figure 3.
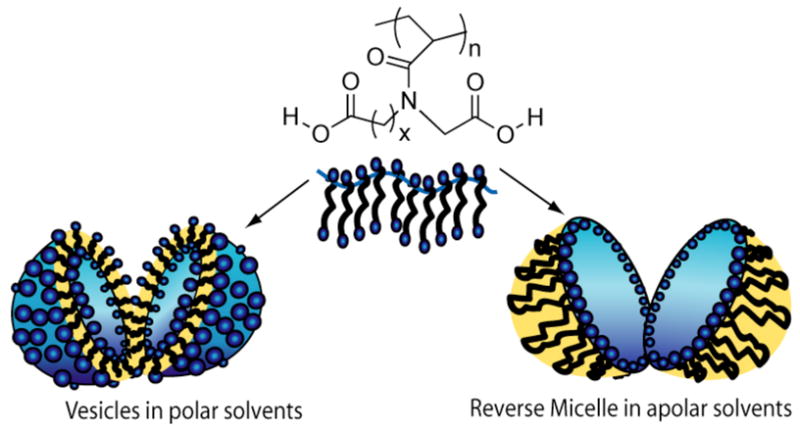
Schematic representation of vesicle and reverse micelle formation.
We measured the size of the particles obtained from polymer PA10V using dynamic light scattering. The hydrodynamic radius (RH) of the particle was found to be about 28 nm. Note that the size of the particles obtained from TEM is the diameter of the particle and therefore the particle sizes obtained from the two measurements are very similar. To obtain additional evidence for the vesicle formation in this solution, we also carried out static light scattering measurements that afforded a radius of gyration (RG) of about 29 nm. As mentioned earlier, the RG/RH (ρ) is sensitive to a particular morphology. The fact that this ratio is 1.04 further supports the formation of vesicle-like assemblies in solution. The TEM picture of polymers PA10V and PA15V also suggest that these assemblies are unilamellar in nature.
The formation of these assemblies can be further ascertained by studying the container properties. Interestingly, the vesicle assemblies should be able to sequester both hydrophilic dye molecules as well as hydrophobic dye molecules. The hydrophilic dye molecules can be sequestered within the polar, water-containing interiors of the vesicles; the apolar guest molecules can be incorporated within the apolar membranes of the vesicles. We utilized the self-quenching nature of rhodamine-6G to probe the nature of the vesicle assemblies. R6G was incorporated into the polymer assembly from PA10V in water and dialyzed the solution to remove any unbound dye molecules. The fluorescence from this solution was compared with an absorbance-matched solution of R6G in water without polymer PA10V. The enhanced quenching in the presence of the polymer supports the presence of vesicle-type structures, as shown in Figure 4a. Since the polymer is negatively charged and R6G is positively charged, the observed quenching could be attributed to a simple surface binding of R6G and thus increase in local concentration and self-quenching. In order to address this issue, we carried out these experiments with a negatively charged polar dye (calcein) that also exhibits self-quenching at higher concentrations. Similar results were observed in this system, as shown in Figure 4b, further supporting the presence of vesicle structures. The amount of R6G inside the vesicular interiors is greater than calcein, as discerned from the absorbance of the solutions and extent of quenching in these two dyes. This is understandable, considering the electrostatic complementarity of R6G with the carboxylates of the polymer.
Figure 4.

Absorbance matched emission spectra of a) rhodamine 6G and b) calcein in water and in PA10V.
Similarly, the self-quenching behavior of R6G was also used to investigate the formation of inverse micelles in toluene with polymers PA10V and PA15V. R6G is insoluble in toluene by itself. The fact that significant amount of the dye is solubilized in toluene in the presence of the polymer PA10V combined with the self-quenching behavior suggests the formation of inverse micelle-type assemblies in toluene.
As mentioned above, hydrophobic guest molecules can also be incorporated within the vesicle membranes in water. When pyrene was incorporated within the aqueous polymer assemblies of PA5V, PA7V, PA10V and PA15V in water, I1/I3 ratios were found to be 1.85, 1.75, 1.52 and 1.22 respectively.13 It is immediately obvious that polymers PA5V and PA7V do not provide environments that are sufficiently different from bulk water for pyrene. This supports the absence of vesicle or micelle type structures for the polymers, as discerned from our TEM studies above. Both polymers PA10V and PA15V provide sufficiently different environments compared to water. It is more so in the case of polymer PA15V, which is understandable due to the longer alkyl chain length and therefore an opportunity for better isolation of pyrene from the bulk solvent (water). It is remarkable that among the micelle-forming polymers including PA6M, which contains only a hexyl hydrophobic chain, exhibits a much less polar environment compared to polymer PA15V containing a pentadecyl alkyl chain linker. We attribute this to the fact that micellar interiors are more capable of excluding water from its interior compared to the vesicular membranes. In the latter case, the guest molecule is in an environment that is surrounded by water on both sides, while the interior does not contain water in the former scenario.8
We utilized changes in the excitation spectra of pyrene to evaluate the critical aggregation concentration of polymers PA5V, PA7V, PA10V and PA15V. No change in the excitation spectra was observed with increasing concentration of polymers PA5V and PA7V. This is understandable, since we have already noted from our TEM studies that these polymers do not form well-defined amphiphilic assemblies in water. The CAC of the polymers PA10V and PA15V is 50 and 2 mg/L respectively. We have also synthesized the corresponding small molecule building block units 21 and 22 for comparison. The CACs for these molecules were found to be 180 and 140 mg/L respectively (Table 1). Note that the difference between the decyl and hexadecyl linkers in the case of small molecules is much smaller than in the case of polymer (Figure 5). We attribute this to the possibility that pre-organization of a large number of amphiphiles in a polymer chain lends itself to making the critical jump in the CAC upon increasing the alkyl chain lengths earlier (even between decyl and hexadecyl), compared to the small molecules. Here too, the hydrophobicity of the aggregates formed by the small molecules is much higher than the corresponding polymers, which can be explained by the differential packing of the amphiphiles in these two classes of molecules, as explained above.
Figure 5.
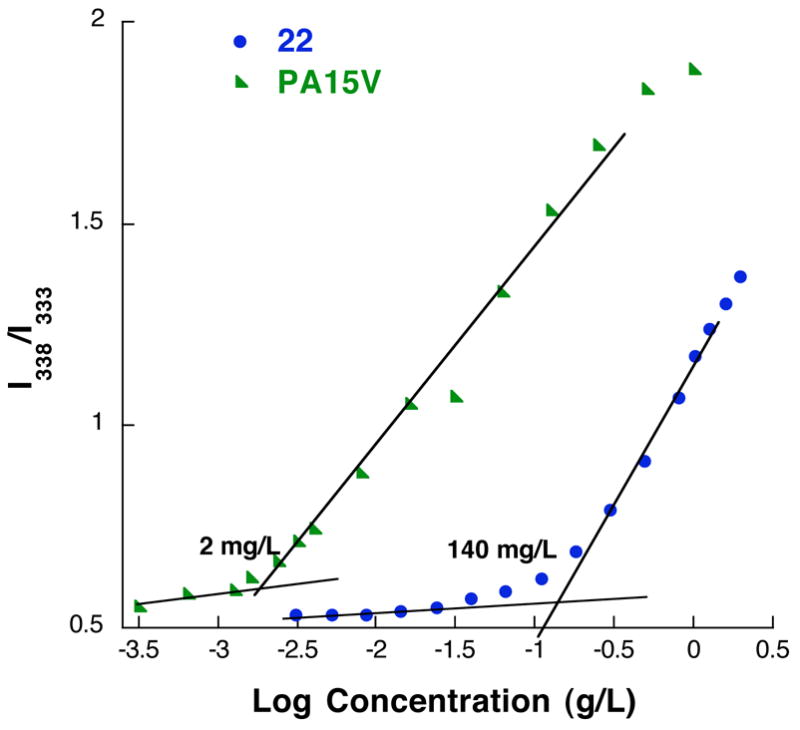
Plot of I338/I333 in the excitation spectra vs concentrations of PA15V and 22.
Summary
We have synthesized a series of amphiphilic homopolymers and the corresponding building block units as model compounds to develop a systematic structure-property relationship. We show here that: (i) the amphiphilic homopolymer design is indeed applicable broadly. We have shown that polymers containing hydrophilic and hydrophobic groups within a flexible building block do indeed provide environment-dependent amphiphilic assemblies. (ii) When two hydrophilic head groups linked by a long, hydrophobic alkyl chain are incorporated on to the repeat units of the homopolymer, vesicular assemblies are obtained. A critical chain length is necessary to obtain such assemblies. (iii) The vesicle-type assemblies are also environment-dependent; i.e. these polymers also form inverted micelle type assemblies in apolar solvents such as toluene, (iv) Compared to the small molecule building blocks, the critical aggregation concentrations for these polymers are much lower. The polarity of the interior of the polymer-based assemblies is however less hydrophobic compared to the building blocks, which has been attributed to the relative packing abilities in the assemblies, (v) The micellar, inverse micellar, and the vesicular assemblies are all capable of acting as nanocontainers for the corresponding small organic molecules. However, we do not yet understand the exact reason for the rather large sizes of the micellar assemblies formed from the amphiphilic homopolymers. (vi) Finally, it is important to note that all these assemblies are obtained by merely dissolving the polymer in the solvents mentioned. The fact that we do not need extensive processing to achieve these amphiphilic supramolecular assemblies is likely to render them broadly applicable in a variety of applications.
Supplementary Material
Experimental details including synthesis and characterization of monomers and polymers. This material is available free of charge at http://pubs.acs.org.
Acknowledgments
We thank the NIGMS of the National Institutes of Health for support. We also acknowledge the NSF for a CAREER award and a MRSEC facility at the University of Massachusetts for the infrastructure used in these characterizations.
References
- 1.(a) Israelachvili JN. Intermolecular and Surface Forces. 2. Academic Press; New York: 1991. [Google Scholar]; (b) Muthukumar M, Ober CK, Thomas EL. Science. 1997;277:1225–1232. [Google Scholar]; (c) Bucknall DG, Anderson HL. Science. 2003;302:904–905. doi: 10.1126/science.1091064. [DOI] [PubMed] [Google Scholar]; (c) Mckee MG, Layman JM, Cashion MP, Long TE. Science. 2006;311:353–355. doi: 10.1126/science.1119790. [DOI] [PubMed] [Google Scholar]; (d) Chen WH, Shao XB, Regen SL. J Am Chem Soc. 2005;127:12727–12735. doi: 10.1021/ja053527q. [DOI] [PubMed] [Google Scholar]; (e) Loewik DWPM, van Hest JCM. Chem Soc Rev. 2004;33:234–245. doi: 10.1039/b212638a. [DOI] [PubMed] [Google Scholar]; (d) Menger FM, Keiper JS. Angew Chem Int Ed. 2000;39:1906–1920. doi: 10.1002/1521-3773(20000602)39:11<1906::aid-anie1906>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]; (e) Ryu EH, Zhao Y. Org Lett. 2004;6:3187–3189. doi: 10.1021/ol048679p. [DOI] [PubMed] [Google Scholar]; (e) Cornelissen JJLM, Fischer M, Sommerdijk NAJM, Nolle RJM. Science. 1998;280:1427–1430. doi: 10.1126/science.280.5368.1427. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]; (b) Gaucher G, Dufresne M-H, Sant VP, Kang N, Maysinger D, Leroux J-C. J Controlled Release. 2005;109:169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]; (c) Riess JG, Krafft MP. Biomaterials. 1998;19:1529–1539. doi: 10.1016/s0142-9612(98)00071-4. [DOI] [PubMed] [Google Scholar]; (d) Torchilin VP. J Controlled Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]; (e) Shuai X, Ai H, Nasongkla N, Kim S, Gao J. J Controlled Release. 2004;98:415–426. doi: 10.1016/j.jconrel.2004.06.003. [DOI] [PubMed] [Google Scholar]; (f) Trubetskoy VS, Torchilin VP. Adv DrugDeliv Rev. 1995;16:311–320. [Google Scholar]; (g) Gao Z, Lukyanov AN, Singhal A, Torchilin VP. Nano Lett. 2002;2:979–982. [Google Scholar]; (h) Wang YL, Banziger J, Dubin PL, Filippelli G, Nuraje N. Environ Sci Technol. 2001;35:2608–2601. doi: 10.1021/es001662r. [DOI] [PubMed] [Google Scholar]
- 3.For examples, see: Moffitt M, Khougaz K, Eisenberg A. Acc Chem Res. 1996;29:95–102.Pochan DJ, Chen Z, Cui H, Hales K, Qi H, Wooley KL. Science. 2004;306:94–97. doi: 10.1126/science.1102866.Jain S, Bates FS. Science. 2003;300:460–464. doi: 10.1126/science.1082193.Li Y, Lokitz BS, McCormick CL. Angew Chem Int Ed. 2006;45:5792 –5795. doi: 10.1002/anie.200602168.
- 4.Basu S, Vutukuri DR, Shyamroy S, Sandanaraj BS, Thayumanavan S. J Am Chem Soc. 2004;126:9890–9891. doi: 10.1021/ja047816a. [DOI] [PubMed] [Google Scholar]
- 5.Arumugam S, Vutukuri DR, Thayumanavan S, Ramamurthy V. J Am Chem Soc. 2005;127:13200–13206. doi: 10.1021/ja051107v. [DOI] [PubMed] [Google Scholar]
- 6.Basu S, Vutukuri DR, Thayumanavan SJ. Am Chem Soc. 2005;127:16794–16795. doi: 10.1021/ja056042a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandanaraj BS, Simard J, Vutukuri D, Rotello VM, Thayumanavan S. J Am Chem Soc. 2005;127:10693–10698. doi: 10.1021/ja051947+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Micelles are defined here as containers that have hydrophilic exterior and hydrophobic interior. These assemblies are capable of encapsulating hydrophobic guest molecules, but not hydrophilic ones, (b) Vesicles are containers with hydrophilic environment in both exterior and interior. These assemblies are capable of encapsulating hydrophilic guests in the interior and hydrophobic guests within their membranes.
- 9.The polymers are here named as PS for polystyrene (PA for polyacrylamide), the number following this indicates the length of the alkyl chain, and M (or V) indicates whether an assembly is expected to provide micelles or vesicles, based on our molecular design.
- 10.For a few small molecule examples of bolaamphiphiles, see: Fuhrhop JH, Wang T. Chem Rev. 2004;104:2901–2937. doi: 10.1021/cr030602b.Escamilla GH, Newkome GR. Angew Chem Int Ed Engl. 1994;33:1937–1940.Fuhrhop JH, Fritsch D. Ace Chem Res. 1986;19:130–137.
- 11.(a) Barner-Kowollik C, Davis TP, Heuts JPA, Stenzel MH, Vana P, Whittaker M. J Polym Sci Part A: Polym Chem. 2003;41:365–375. [Google Scholar]; (b) Moad G, Rizzardo E, Thang SH. Austr J Chem. 2005;58:379–410. [Google Scholar]; (c) Chiefari J, Chong YK, Ercole F, Kristina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Macromolecules. 1998;31:5559–5562. [Google Scholar]
- 12.(a) Disher DE, Eisenberg A. Science. 2002;297:967–973. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]; (b) Discher BM, Won YY, Ege DS, Lee JCM, Bates FS, Discher DE, Hammer HA. Science. 1999;284:1143–1146. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]; (c) Xu J, Zubarev ER. Angew Chem Int Ed. 2004;43:5491–5496. doi: 10.1002/anie.200460906. [DOI] [PubMed] [Google Scholar]; (d) Kim BS, Hong DJ, Bae J, Lee M. J Am Chem Soc. 2005;727:16333–16337. doi: 10.1021/ja055999a. [DOI] [PubMed] [Google Scholar]
- 13.See supporting information for details.
- 14.(a) Burchard W. Adv Polym Sci. 1983;48:1–167. [Google Scholar]; (b) Rodriguez-Hernandez J, Lecommandoux S. J Am Chem Soc. 2005;127:2026–2027. doi: 10.1021/ja043920g. [DOI] [PubMed] [Google Scholar]; (c) Zhou S, Burger C, Chu B, Sawamura M, Nagahama N, Toganoh M, Hackler UE, Isobe H, Nakamura E. Science. 2001;291:1944–1947. doi: 10.1126/science.291.5510.1944. [DOI] [PubMed] [Google Scholar]; (d) Giacomelli C, Men LL, Borsali R, Lai-Kee-Him J, Brisson A, Armes SP, Lewis AL. Biomacromolecules. 2006;7:817–828. doi: 10.1021/bm0508921. [DOI] [PubMed] [Google Scholar]
- 15.(a) Chakraborty H, Sarkar M. Langmuir. 2004;20:3551–3558. doi: 10.1021/la0361417. [DOI] [PubMed] [Google Scholar]; (b) Erhsrdt R, Zhang M, Boker A, Zettl H, Abetz C, Frederic P, Kraush G, Abetz V, Miiller AHE. J Am Chem Soc. 2003;125:3260–3267. doi: 10.1021/ja028982q. [DOI] [PubMed] [Google Scholar]; (c) Wang C, Ravi P, Tarn KC. Langmuir. 2006;22:2927–2930. doi: 10.1021/la052191v. [DOI] [PubMed] [Google Scholar]; (d) Kujawa P, Tanaka R, Winnik FM. Macromolecules. 2006;39:3048–3055. [Google Scholar]; (e) Petrov P, Bozukov M, Burkhardt M, Muthukrishnan S, Miiller AHE, Tsvetanov CB. J Mater Chem. 2006;16:2192–2193. [Google Scholar]
- 16.Kalyanasundaram K, Thomas JK. J Am Chem Soc. 1977;99:2039–2044. [Google Scholar]
- 17.(a) Goddard ED, Turro NJ, Kuo PL. Langmuir. 1985;1:352–355. doi: 10.1021/la00063a015. [DOI] [PubMed] [Google Scholar]; (b) Lee J, Moroi Y. J Colloid Interface Sci. 2004;273:645–650. doi: 10.1016/j.jcis.2004.01.079. [DOI] [PubMed] [Google Scholar]
- 18.Jung HM, Price KE, McQuade DT. J Am Chem Soc. 2003;125:5351–5355. doi: 10.1021/ja0271983. [DOI] [PubMed] [Google Scholar]
- 19.(a) Wilhelm M, Zhao CL, Wang Y, Xu R, Winnik MA, Mura JL, Riess G, Croucher MD. Macromolecules. 1991;24:1033–1040. [Google Scholar]; (b) Astafieva L, Zhong XF, Eisenberg A. Macromolecules. 1993;26:7339–73352. [Google Scholar]; (c) Kwon GS, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Langmuir. 1993;9:945–949. [Google Scholar]
- 20.(a) Schillen K, Bryskhe K, Melnikova S. Macromolecules. 1999;32:6885–6888. [Google Scholar]; (b) Liu F, Eisenberg A. J Am Chem Soc. 2003;125:15059–15064. doi: 10.1021/ja038142r. [DOI] [PubMed] [Google Scholar]; (c) Nardin C, Thoeni S, Widmer S, Winterhalter M, Meier W. Chem Commun. 2000:1433–1434. [Google Scholar]; (d) Du J, Armes SP. J Am Chem Soc. 2005;127:12800–12801. doi: 10.1021/ja054755n. [DOI] [PubMed] [Google Scholar]; (e) Ringsdorf H, Schlarb B, Venzmer J. Angew Chem. 1988;100:117–162. [Google Scholar]; (f) Tsuchida E, Nishide H, Yuasa M, Babe T, Fukuzumi M. Macromolecules. 1989;22:66–72. [Google Scholar]
- 21.Zhang L, Eisenberg A. J Am Chem Soc. 1996;118:3168–3181. [Google Scholar]; b) Yu Y, Zhang L, Eisenberg A. Langmuir. 1997;9:2578–2581. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details including synthesis and characterization of monomers and polymers. This material is available free of charge at http://pubs.acs.org.