

Abstract
Vinylcarbenes are versatile synthetic intermediates, capable of asymmetric cyclopropanation and insertion into unactivated CH bonds. The vinyldiazolactone precursor to the metal vinylcarbene possesses superior stability in comparison to previously known vinyldiazoacetates and, for the first time, allows the use of Z-vinylcarbenes in asymmetric C-H insertion and cyclopropanation reactions. Dirhodium azetidinone ligated compounds are optimal catalysts. Cyclopropanation coupled with the Cope rearrangement provides access to trans-3,4-disubstituted hydroazulenes.
Rhodium(II)-stabilized vinylcarbenes are valuable synthetic intermediates capable of undergoing a spectrum of transformations in a highly selective fashion.1 Their versatility is illustrated in applications directed to the construction of complex molecular architectures.2 However, limitations of this catalytic methodology have become apparent. Vinyldiazoacetates undergo a spontaneous [1,5]-cyclization to yield pyrazoles,3 and this transformation has limited their applications with catalysts that are less reactive towards diazo decomposition than dirhodium tetracarboxylates. In addition, although E-styrylcarbenes readily undergo intermolecular addition and insertion reactions, the Z-vinylcarbene isomer produces the intramolecular aromatic substitution product exclusively,4 and asymmetric intermolecular reactions of Z-vinylcarbenes have been observed only when the Z-substituent is alkyl.5 In the course of our research with vinyldiazoacetates,2c,d we became intrigued by the possibility of utilizing vinyldiazolactones as carbene precursors. Restriction of the vinyldiazo moiety within a ring was expected to prevent the undesirable intramolecular processes that occur with acyclic vinyldiazoacetates and vinylcarbene intermediates. We report here the first synthesis of a vinyldiazolactone and it’s unexpected suitability for asymmetric induction in intermolecular insertion and addition reactions with a chiral dirhodium carboxamidate.
The synthesis of vinyldiazolactone 1 was accomplished in one step from commercially available 5,6-dihydro-2H-pyran-2-one using direct diazo transfer conditions that are reported in the preparation of select vinyldiazoacetates.6 However, unlike vinyldiazoacetates, lactone 1 exhibited a long shelf-life. Throughout this study, 1 was stored for several weeks at 4° C and used without further purification.
Perhaps the most remarkable property of rhodium(II)-stabilized vinylcarbenes is their applicability toward intermolecular C-H insertion reactions.7 Carbenes generated from vinyldiazoacetates and aryldiazoacetates in the asymmetric environment of Rh2(DOSP)4 (Figure 1) and related structures are unique in their ability to undergo insertion into unactivated C-H bonds with synthetically viable yields and enantioselectivities. To investigate the utility of 1 in intermolecular C-H insertion reactions, asymmetric dirhodium(II) carboxylate and carboxamidate catalysts were screened in the reaction of 1 with 1,4-cyclohexadiene (Table 1).
Figure 1.
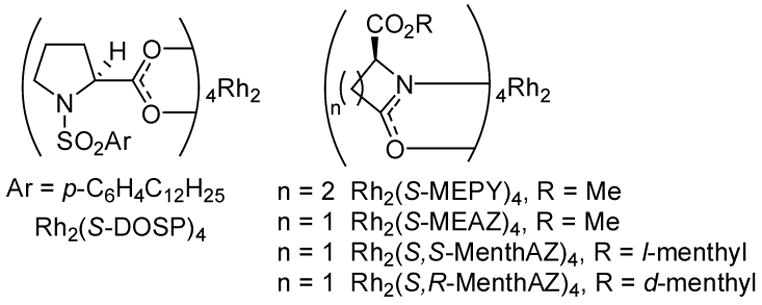
Asymmetric dirhodium catalysts.
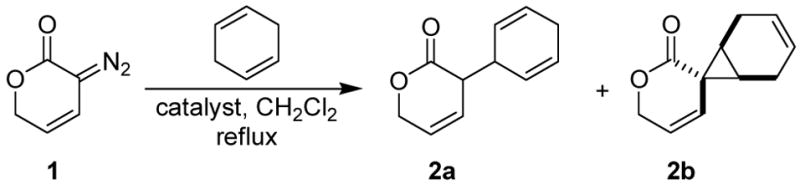
Table 1.
C-H insertion of 1 into 1,4-cyclohexadiene.a
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | 2a:bb | Yield (%) | ee (%)c |
| 1 | Rh2(OAc)4 | 2:3 | 53 | – |
| 2d | Rh2(S-DOSP)4 | 1:9 | 56 | 18, R |
| 3 | Rh2(S-MEPY)4 | 4:1 | 47 | 8, R |
| 4 | Rh2(S-MEAZ)4 | 9:1 | 43 | 60, R |
| 5 | Rh2(S,S-MenthAZ)4 | 9:1 | 42 | 79, R |
| 6 | Rh2(S,R-MenthAZ)4 | 9:1 | 50 | 80, R |
Unless otherwise noted, to a solution of 5 equivalents diene and 1.0 mol% of catalyst in refluxing CH2Cl2 (20 mL) under N2 was added a solution of 1 (1.3 mmol) in CH2Cl2 (10 mL) over 8h.
Determined by 1H NMR spectroscopy prior to chromatography.
%ee of 2a determined by GC.
Reaction run at room temperature.
Surprisingly, Rh2(S-DOSP)4,8 ordinarily the optimal catalyst for enantioselective C-H insertion reactions of vinyldiazoacetates,7 was not suitable in this application. Not only was the enantiomeric excess of C-H insertion product 2a negligible, but the propensity of this catalyst for C-H insertion relative to cyclopropanation was the lowest of all catalysts evaluated (Table 1, entry 2). The carboxamidate catalyst Rh2(S-MEPY)4,9 which has seen some use in C-H insertion reactions but was found to be unsuitable for reactions with vinyldiazoacetates,3 provided a greater chemoselectivity for 2a than Rh2(S-DOSP)4, but enantiocontrol with this catalyst was low (Table 1, entry 3). With the azetidinone ligated catalyst Rh2(S-MEAZ)4 (Table 1, entry 4),10 however, promising selectivities were obtained. Optimization of the azetidinone catalyst structure led to the use of diastereomeric catalysts Rh2(S,S-MenthAZ) and Rh2(S,R-MenthAZ)4,11 the selectivities from which were high and comparable (Table 1, entries 5 and 6). Absolute stereochemistry of 2a was determined by oxidative aromatization of the diene of 2a, followed by reduction of the olefin to provide the known compound α-phenyl-γ-valerolactone. Comparison of the optical rotation to that reported for (R)-α-phenyl-γ-valerolactone12 allowed the assignment of absolute stereochemistry of 2a as shown (Table 1). All of the dirhodium catalysts evaluated provided comparable isolated yields of 2a,b.
Encouraged by the results of this C-H insertion study, we turned our attention to asymmetric cyclopropanation of 1 (Table 2). Reactions occurred cleanly and in high yield with notable diastereoselectivities. As with the previously described C-H insertion, azetidinone ligated dirhodium catalysts are the optimal catalysts for the cyclopropanation of styrene [Rh2(S-DOSP)4, 3a = 60% yield, 14% ee, dr > 20:1); Rh2(S-MEPY)4, 3a = 69% yield, 10% ee, dr > 20:1; Rh2(S,R-MenthAZ)4, 3a = 74% yield, 84% ee, dr > 20:1]. Other cyclopropanation reactions catalyzed by Rh2(S,R-MenthAZ)4 provided enantioselectivities ranging from 73–86% ee, with the E-cyclopropane as the dominant or exclusive diastereomer.
Table 2.
Asymmetric cyclopropanation of selected alkenes with 1.a
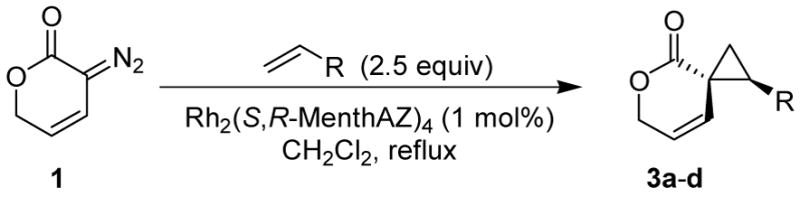
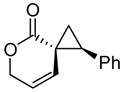
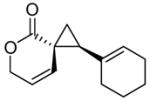
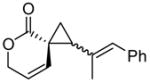
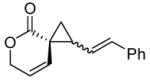
To olefin (2.5 equiv) and catalyst (1.0 mol %) in refluxing CH2Cl2 (20 mL) under N2 was added a solution of 2 (1.3 mmol) in CH2Cl2 (10 mL) over 8h. E:Z ratio determined by 1H NMR prior to chromatography. Reported yields obtained upon isolation via silica gel chromatography.
E-cyclopropane.
Extensive studies of vinyldiazoacetates by Davies have demonstrated that upon formation of cis-divinylcyclopropanes a Cope rearrangement occurs to provide hydroazulenes.13 Construction of seven-membered carbocycles is important in natural product synthesis, and the development of stereoselective strategies toward these targets remains an ongoing challenge.14 However, unlike the transient cis-divinylcyclopropanes formed from vinyldiazoacetates, conformational restrictions on spirocyclic cyclopropanes 3b-c prevent subsequent Cope rearrangement and allow isolation of the cyclopropane products without rearrangement. Cleavage of the lactone overcomes this restriction and allows the dienes to access the necessary conformation for rearrangement.
Cope rearrangement product 4 was obtained upon reduction of lactone E-3d with LiAlH4 in refluxing THF (eq 1). Recrystallization provided a facile means of separating 4 from the cyclopropane-diol arising from Z-3d and, concomitantly, the enantiomeric excess of 4 was enriched to 92%.15 Thus, in two steps the hydroazulene 4 is conveniently accessed in high enantiomeric excess from the diastereomeric mixture of 3d. The relative stereochemistry of 4 was confirmed by X-ray diffraction.
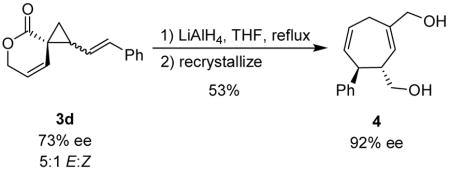 |
(1) |
Vinyldiazolactone 1 is an effective and stable vinylcarbene precursor. Over the course of this study comparisons were made of the reactivity of 1, which had been stored for several weeks, with that of freshly purified 1; no significant diminution in yield or selectivity was observed, emphasizing the operational convenience of 1 as a vinylcarbene precursor. The striking differential in selectivity between dirhodium(II) carboxylates and carboxamidates was unexpected but is probably a result of conformational influences in the reacting carbenes.8,16 The spirocyclic structure of 3b-d allows the isolation of unrearranged divinylcyclopropanes, and reduction of divinylcyclopropane lactones (i.e., 3d) provides a convenient route to hydroazulenes such as 4. The stereochemistry for substitution at the 3,4-position of 4 is opposite that which would be obtained from reactions of E-substituted vinyldiazoacetates. Future research will be focused on evaluating the scope of cyclic vinyldiazo compounds as carbene precursors and providing a better understanding of the unique selectivities obtained with azetidinone ligated dirhodium catalysts.
Supplementary Material
Experimental details, characterization data, synthesis of 1, absolute stereochemical assignment of 2a, and X-ray crystal structure of 4. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful to Prof. H. M. L. Davies for providing a sample of Rh2(S-DOSP)4. Peter Y. Zavalij of the University of Maryland X-ray Crystallographic Center obtained the X-ray crystal structure of 4. Funding for this work was provided by the National Institutes of Health (GM 46503) and the National Science Foundation. DB would like to thank NSERC (Natural Sciences and Engineering Research Council of Canada) for a postgraduate scholarship.
References
- 1.(a) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. 1998. p. 652. [Google Scholar]; (b) Davies HML, Walji AM. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Wiley-VCH; 2005. p. 301. [Google Scholar]
- 2.(a) Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]; (b) Nyong AM, Rainier JD. J Org Chem. 2005;70:746. doi: 10.1021/jo0482413. [DOI] [PubMed] [Google Scholar]; (c) Yan M, Jacobsen N, Hu W, Gronenberg Luisa S, Doyle MP, Colyer John T, Bykowski D. Angew Chem, Int Ed Engl. 2004;43:6713. doi: 10.1002/anie.200461722. [DOI] [PubMed] [Google Scholar]; (d) Doyle MP, Yan M, Hu W, Gronenberg LS. J Am Chem Soc. 2003;125:4692. doi: 10.1021/ja029745q. [DOI] [PubMed] [Google Scholar]; (e) Davies HML, Stafford DG, Hansen T. Org Lett. 1999;1:233. doi: 10.1021/ol9905699. [DOI] [PubMed] [Google Scholar]; (f) Davies HML, Doan BD. J Org Chem. 1998;63:657. doi: 10.1021/jo971577a. [DOI] [PubMed] [Google Scholar]
- 3.Davies HML, Huby NJS, Cantrell WR, Jr, Olive JL. J Am Chem Soc. 1993;115:9468. [Google Scholar]
- 4.Davies HML, Hodges LM, Matasi JJ, Hansen T, Stafford DG. Tetrahedron Lett. 1998;39:4417. [Google Scholar]
- 5.(a) Bulugahapitiya P, Landais Y, Parra-Rapado L, Planchenault D, Weber V. J Org Chem. 1997;62:1630. [Google Scholar]; (b) Landais Y, Planchenault D, Weber V. Tetrahedron Lett. 1994;35:9549. [Google Scholar]
- 6.Baum JS, Shook DA, Davies HML, Smith HD. Synthetic Communications. 1987;17:1709. [Google Scholar]
- 7.(a) Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (b) Davies HML, Antoulinakis EG. J Organomet Chem. 2001;617–618:47. [Google Scholar]
- 8.Davies HML. Eur J Org Chem. 1999:2459. [Google Scholar]
- 9.Doyle MP, Van Oeveren A, Westrum LJ, Protopopova MN, Clayton TW., Jr J Am Chem Soc. 1991;113:8982. [Google Scholar]
- 10.Doyle MP, Davies SB, Hu W. Org Lett. 2000;2:1145. doi: 10.1021/ol005730q. [DOI] [PubMed] [Google Scholar]
- 11.Hu W, Timmons DJ, Doyle Michael P. Org Lett. 2002;4:901. doi: 10.1021/ol017276b. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura Y, Takeuchi S, Ohgo Y, Yamaoka M, Yoshida A, Mikami K. Tetrahedron. 1999;55:4959. [Google Scholar]
- 13.Davies HML, Stafford DG, Doan BD, Houser JH. J Am Chem Soc. 1998;120:3326. [Google Scholar]
- 14.For recent synthetic advances Wender PA, Haustedt LO, Lim J, Love JA, Williams TJ, Yoon JY. J Am Chem Soc. 2006;128:6302. doi: 10.1021/ja058590u.Sperry JB, Wright DL. J Am Chem Soc. 2005;127:8034. doi: 10.1021/ja051826+. For a recent review Battiste MA, Pelphrey PM, Wright DL. Chem Eur J. 2006;12:3438. doi: 10.1002/chem.200501083.
- 15.Prior to recrystallization, measured enantiomeric excess of 5 matched that of 4d.
- 16.Doyle MP, Winchester WR, Hoorn JAA, Lynch V, Simonsen SH, Ghosh R. J Am Chem Soc. 1993;115:9968. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details, characterization data, synthesis of 1, absolute stereochemical assignment of 2a, and X-ray crystal structure of 4. This material is available free of charge via the Internet at http://pubs.acs.org.