

Abstract
Human transglutaminase 2 (TG2) is believed to play an important role in the pathogenesis of various human disorders including celiac sprue, certain neurological diseases, as well as some types of cancer. Selective inhibition of TG2 should therefore enable further investigation of its role in physiology and disease, and may lead to effective clinical treatment. Recently we showed that certain 3-halo-4-,5-dihydroisoxazole containing compounds are selective inhibitors of human TG2 with promising pharmacological activities. Here we present definitive evidence that this class of compounds targets the active site of human TG2. Structure-activity relationship studies have provided insights into the structural prerequisites for selectivity, and have led to the discovery of an inhibitor with ca. fifty-fold higher activity than a prototypical dihydroisoxazole inhibitor with good in vivo activity. A method for preparing enantiomerically enriched analogues was also developed. Our studies show that the 5-(S-) dihydroisoxazole is a markedly better inhibitor of human TG2 than its 5-(R-) stereoisomer.
Introduction
The transglutaminase family of enzymes plays important roles in many biological functions by cross-linking selected γ-glutaminyl and ε-lysine residues in proteins. (1–3) Expressed in many tissues, transglutaminase 2 (TG2) is known to have extracellular functions in fibronectin cross-linking, cell migration, adhesion, and proliferation.(1–3) When bound to GTP inside the cell, TG2 has no known catalytic activity, but rather acts as a G protein in the phospholipase C signal transduction cascade.(3) Its many biological and biochemical roles implicate it in a number of neurological diseases including Huntington’s, Alzheimer’s and Parkinson’s Diseases.(4–5) Importantly, TG2 knockout mice suffer no apparent physiological, developmental or reproductive defects. (6) That the critical functions of TG2 may be absorbed by redundant enzymes in its absence, and that its roles in the pathogenesis of disease may be curtailed by selective inhibition, make TG2 an attractive therapeutic target. Several recent reports have described small molecule inhibitors against TG2 (most often using guinea pig TG2 as a surrogate target).(7–11)
Our interest in the inhibition of human TG2 began as a consequence of our interest in celiac sprue, a common disorder for which there is no current non-dietary therapy.(12–14) Celiac sprue is an autoimmune-like disease in which genetically susceptible individuals experience gut damage in response to gluten, a group of ubiquitous dietary proteins. Gluten proteins contain proline- and glutamine-rich sequences, some of which are selectively deamidated by TG2. The resultant peptides are presented to immune system cells, leading to a combined T-cell and B-cell mediated inflammatory response in the small intestinal mucosa. (15,16) Thus, the development of specific TG2 inhibitors may lead to compounds capable of limiting deamidation of gluten peptides in the gut, thereby curtailing the immune response to gluten in celiac patients.
We have previously investigated mechanism-based TG2 inhibitors (10,17) based on a 3-halo-, 4-,5-dihydroisoxazole pharmacophore, whose structure derives from the naturally occurring glutamine isostere, acivicin. Decarboxylated acivicin derivatives had increased activity as TG2 inhibitors. (18) The inhibitors studied were mostly comprised of α-amino acids functionalized with N-carbamate moieties and coupled to the dihydroisoxazole electrophile. Several trends in the structure-activity relationships of this class of TG2 inhibitors emerged. (17) Increases in affinity (as determined by KI) correlated with increases in the bulk of carbamate moiety, e.g. OCH2-2-naphthyl > OCH2CH2Ph > OCH2Ph (Table 1). Unfortunately, the ten-fold increase in affinity gained by changing to a naphthyl-2-yl-methyl carbamate from a benzyl-carbamate was outweighed by a twelve-fold decrease in reactivity (as determined by kinh), leading to a virtually unchanged specificity (as determined by kinh/KI). Increases in affinity were also observed with the existence of heteroatom hydrogen bond acceptors in the carbamate groups, e.g. OCH2-3-picolyl > OCH2-N-phthalimidyl > OCH2-3-dioxobenzothiophene. However, the ten-fold increase in affinity observed with the change from a benzyl-carbamate moiety to a picolin-3-yl-methyl carbamate moiety was still met with a four-fold decrease in reactivity (Table 1). Finally, our previous studies varying the amino acid side chain showed an increase in affinity with the presence of heteroatoms, e.g. compounds derived from 5-OH-tryptophan > tryptophan > tyrosine > phenylalanine. Here too, the increase in affinity (seven-fold) with the change from 5-OH-tryptophan to phenylalanine corresponded to a decrease (four-fold) in reactivity.
Table 1.
Summary of key results from our previous study.(17)
| Cmpd | R= | KI(mM) | kinh(min−1) | kinh/KI (M−1min−1) | |
|---|---|---|---|---|---|
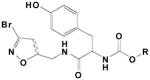
|
1 | -benzyl | 0.42 | 0.86 | 1000 |
| 2 | -3-picolyl | 0.078 | 0.21 | 2700 | |
| 3 | -CH2CH2Ph | 0.061 | 0.093 | 1500 | |
| 4 | -CH2Napthyl | 0.043 | 0.070 | 1600 | |
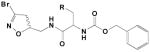
|
5 |
|
0.087 | 0.38 | 4300 |
| 6 | -phenyl | 0.74 | 1.3 | 1900 | |
| 8 | -3-indole | 0.31 | 0.78 | 2500 | |
| 9 | -5-OH-indole | 0.11 | 0.31 | 2800 | |
|
R = -5-OH-indole | ||||
| R′= | |||||
| 10 |
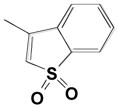
|
0.079 | 0.54 | 6800 |
The best inhibitor found in our previous studies was the 5-OH-tryptophan derivative 10 containing a dioxobenzothiophene in the N-carbamate moiety. This compound combined a heteroatom-containing amino acid with a bulky, H-bond acceptor-containing carbamate moiety. However, its metabolic instability made it an unsuitable candidate for in vivo investigations. Therefore, compound 10 served as a starting point for our current study.
RESULTS AND DISCUSSION
In this study we have demonstrated directly via mass spectrometry that 3-halo-4,5-dihydroisoxazole inhibitors bind to the active site cysteine of human TG2. Furthermore, we have systematically investigated: 1) the importance of the identity and position of heteroatoms on the amino acid side chain, 2) the significance of alternative hydrogen bond acceptors in a bulky, aromatic carbamate moiety, 3) the effect of stereochemistry in the amino acid side chain, and 4) the stereochemistry of the dihydroisoxazole.
To verify that dihydroisoxazole inhibitors such as the compounds shown in Table 1 do indeed covalently bind to the active site cysteine of TG2, recombinant human TG2 was incubated in the presence or absence of the Cbz-tyrosine derivative 1, digested with trypsin, and analyzed by LC-MS/MS. While the samples incubated with (Figure 1B) and without (Figure 1A) derivative 1 both contained peaks at 1787 (+1) m/z and 894 (+2) m/z corresponding to the predicted proteolytic fragment containing the active site cysteine, only the sample incubated with the inhibitor showed a significant 1092 (+2) m/z peak corresponding to the mass of the active site cysteine containing fragment (1787) plus the mass of the dihydroisoxazole inhibitor without bromine (395). To confirm that these mass peaks did in fact correspond to the TG2 proteolytic fragment containing the active site cysteine, MS/MS analysis (Figure 1C) was performed and 18–22 of the 32 possible b and y peaks were successfully assigned. Similar analysis was repeated for four different dihydroisoxazole inhibitors, including compounds 1, 4, 14 and a dansyl-carbamate protected tyrosine derivative (data not shown). All compounds were found to bind the active site cysteine of human TG2. Inhibitor binding to other proteolytic fragments was not observed.
Figure 1.

Dihydroisoxazole inhibitors covalently bind the active site cysteine of transglutaminase 2. Human TG2 was incubated with or without inhibitor 1 before being digested with trypsin and analyzed by mass spectrometry. LC-MS chromatogram of uninhibited TG2 (A) and inhibited TG2 (B) filtered to only display peptide peaks corresponding to the predicted weight of the peptide fragment (YGQCWVFAAVACTVLR) containing the active site cysteine (1787 a.m.u. (+1 peak) + 894 amu (+2 peak)) or inhibited active site cysteine (1092 amu (+2 peak)). The fragment containing the active site cysteine is the smaller peak eluting at 23.9 minutes. Note that there is a log difference in the scale of the 1092 plots. MS/MS spectrum (C) of the 1092 peak for the inhibited TG2 sample with 15 “y” and “b” peaks identified, proving the identity of the peptide fragment.
Because the tryptophan derivatives were among the best inhibitors in our previous study, we prepared and tested additional tryptophan derivatives. Commercially available 4-fluoro-, 5-fluoro-, 6-fluoro-, and 7-aza indole derivatives of (D-/L-) tryptophan were functionalized with N-benzyl-carbamates and coupled to the racemic dihydroisoxazole pharmacophore (compounds 13-16). The 5-fluoro-indole derivative 14 was the best inhibitor of TG2 with a KI of 0.019mM and a kinh of 0.071/min. However, it is interesting to note that the 6-fluoro- and 7-aza- derivatives 15 and 16 had comparable affinity (KI = 0.019 and 0.022mM respectively), but their reactivity was only about half that of the 5-fluoro-compound (kinh = 0.046 and 0.033min−1, respectively). It is also noteworthy that the 4-fluoro-tryptophan derivative 13 is both the least specific and most reactive of these compounds. Even though there is a ca. ten-fold increase in TG2 affinity of these compounds relative to the Cbz-tryptophan derivative 8, that increase is met with a nearly equal decrease in reactivity.
We changed the identity of the carbamate moiety from benzyl- to quinoline-3-yl-methyl for several reasons. We had observed the benefit to TG2 affinity of aromatic groups more distal to the amino acid N-terminus: the naphthyl-2-yl-methyl-carbamate 4 and the phenethyl- carbamate 3 gave greater affinity than the benzyl- carbamate 1. In tests with compounds containing H-bond acceptors in the carbamate moiety, other compounds with second aromatic rings had also shown promise: the OCH2-3-dioxobenzothiophene carbamate had only half the affinity of the naphthyl-2-yl-methyl compound, but 5-fold greater reactivity. From this we surmised that benefits to affinity accruing from the existence of a second aromatic ring could be augmented by H-bond acceptors in the carbamate moiety. This drew into focus the 3-picolyl carbamate compound 2. Although 2 had only one ring in its carbamate moiety, its affinity was slightly better than the other H-bond acceptor containing compounds, and its reactivity was 3-fold better than that of naphthyl-2-yl-methyl compound 4. Our hypothesis was that the enzyme formed stronger or geometrically more favorable hydrogen-bonds to the picolyl- protecting group than to the dioxobenzothiophene. Thus, if the binding modes of the picolyl-, naphthylmethyl- and dioxobenzothiophene derivatives were similar, a quinoline-3-yl-methyl carbamate moiety might retain the reactivity of the picolyl- compound without losing the affinity of the naphthyl- compound. To test this hypothesis, quinoline-containing derivatives of tyrosine, 18, and 5-fluoro-tryptophan, 21, were prepared. Also, to further explore the effects of changes in functional groups on the tryptophan side chain, derivatives of 5-hydroxy-tryptophan 19 and 5-methoxytryptophan 20 were prepared with the quinoline-containing carbamate.
Changing from the picolyl- to the quinoline-3-yl-methyl carbamate increased the TG2 affinity of the tyrosine-derived compounds two fold (KIpicolyl- = 0.08mM, KIquinolyl- =0.04mM) with no change in reactivity (kinhpicolyl- =0.21min−1, kinhquinolyl- = 0.21min−1). No effect was seen on the affinity of the 5-fluoro-tryptophan derived compounds in the change from the benzyl- to quinolyl- carbamate (KI/benzyl- =0.019mM, KIquinolyl-=0.018mM), but a nearly three-fold increase in reactivity was observed (Kinhbenzyl-=0.071min−1, Kinhquinolyl-= 0.186min−1). The 5-hydroxy- and 5-methoxy-substituents on the indole moiety in derivatives 19 and 20 gave lower TG2 affinity than the 5-fluoro-substituent in 21, although the reactivity of the MeO-tryptophan compound 20 was ~10% higher. Encouraged by these results, and because only racemic mixtures of 5-fluoro-tryptophan had been used thus far, we prepared a quinoline-containing, 5-fluorotryptophan derivative from (S-) 5-fluorotryptophan. Testing this compound, 22, showed the importance of (S-) side chain stereochemistry on the activity of the inhibitor. The (S-) tryptophan derivative had a KI four-fold better than the (R-/S-) mixture, and an equivalent kinh (Table 2).
Table 2.
Investigation of amino acid side chains and carbamate protecting groups.
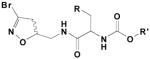
| Cmpd | R= | KI (mM) | kinh (min−1) | kinh/KI (min−1M−1) | |
|---|---|---|---|---|---|
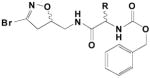
|
13 | (S-/R-)-CH2-3-(4-F-indolyl) | .099 | .09 | 925 |
| 14 | (S-R-)CH2-3-(5-F-indolyl) | .019 | .071 | 3600 | |
| 15 | (S-R-)CH2-3-(6-F-indolyl) | .018 | .046 | 2500 | |
| 16 | (S-/R-)-CH2-3-(7-aza-indolyl) | .022 | .033 | 1500 | |
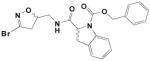
|
17 | (S-/R-)-2,3-dihydro-indole-1-carbamic acid benzyl ester | .138 | .171 | 1250 |
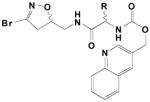
|
18 | (S-)-CH2-p-OH-phenyl | .041 | .209 | 5100 |
| 19 | (S-/R-)-CH2-3-(5-OH-indolyl) | .034 | .136 | 4000 | |
| 20 | (S-/R-)-CH2-3-(5-OMe-indolyl) | .051 | .211 | 4100 | |
| 21 | (S-/R-)-CH2-3-(5-F-indolyl) | .018 | .186 | 10,000 | |
| 22 | (S-)CH2-3-(5-F-indolyl) | 0.004 | .189 | 45,000 | |
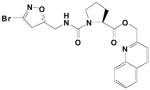
|
23 | (S-)-proline | 0.062 | .377 | 6100 |
In the context of celiac sprue, TG2 has high specificity for the second glutamine in a gluten-derived sequence PQPQLPY.(7) Because the dihydroisoxazole pharmacophore is derived from acivicin, a known mimic of glutamine, we prepared the quinolyl-carbamate functionalized (S-) proline compound 23 to test whether a compound with more structural similarity to the natural substrate might improve inhibition. Although the affinity of 23 was considerably poorer than the (S-)5-fluoro-tryptophan derivative 22 (KIprolyl- =0.062mM, KI5-F-tryptophan =0.004mM), its reactivity was modestly higher (kinhprolyl-=0.377min−1, kinh5-F-tryptophan = 0.189min−1).
To determine which stereoisomer of the dihydroisoxazole pharmacophore gave better inhibition, enantiomerically enriched versions of the dihydroisoxazole pharmacophore were prepared by enzymatic chiral resolution, as described below, and coupled to several N-protected amino-acids. The (S-) isomer of the dihydroisoxazole retained its potency as a pharmacophore (Table 3), whereas inhibitors containing the (R-) dihydroisoxazole isomer showed little or no inhibition. The quinoline-carbamate functionalized proline with the (R-) dihydroisoxazole showed <5% of the inhibitory activity of the (S-) isomer at 0.048mM. The quinoline-carbamate functionalized tyrosine was indistinguishable from negative control at 0.048mM. as was the Cbz-tyrosine compound. Compound 25A, with a kinh/KI of 57,000 M− min− , is 50-fold more potent than 1, which has shown promising activity in rodent models of glioblastoma. (11, 19) The (R-) isomer of quinoline-carbamate functionalized 5-fluoro-tryptophan showed ~5% of the inhibitory activity of its (S-) diastereomer. Because the enzymatic resolution of the two isomers of the dihydroisoxazole was imperfect, this inhibition may be an artifact arising from the presence of small amounts of (S-) isomer in the sample.
Table 3.
TG2 inhibitory activity of enantiomerically enriched dihydroisoxazoles.
| Cmpd | KI (mM) | kinh (min−1) | kinh/KI(M−1min−1) | |
|---|---|---|---|---|
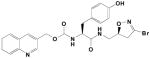
|
24A | 0.030 | 0.139 | 4,600 |
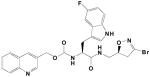
|
25A | 0.0013 | 0.072 | 57,000 |
|
|
26A | 0.011 | 0.110 | 10,300 |
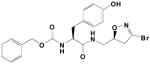
|
27A | 0.068 | 0.155 | 2,300 |
CONCLUSIONS
Heteroatom-containing amino acid side chains, hydrogen bond acceptors in the N- carbamate moieties of those amino acids, and (S-) stereochemistry at the 5- position of the dihydroisoxazole pharmacophore all confer increased activity on TG2 inhibitors of this class. The improvement in inhibition correlating to the change from our previously reported lead compound, Cbz-tyrosyl-halo-dihydroisoxazole 1 (kinh/KI = 1000 min−1M−1), to the quinoline-carbinolic-5-fluorotryptophan-halo-(S-) dihydroisoxazole 25A (kinh/KI = 57,000 min−1M−1) is significant.
The emerging structure-activity relationships also suggest further opportunities for improvement. The order of specificity among our most promising lead compounds predominantly tracks inversely with reactivity. In the context of the four compounds that have been synthesized with the (S-) dihydroisoxazole, the most reactive compound 27A, is also the least specific, and the most specific compound, 25A, is the least reactive. The other two compounds, 24A and 26A, also follow this trend. A likely explanation for this phenomenon is that moieties in these compounds which enhance non-covalent interactions with the inhibitor binding site do so in a way that curtails access by the active site nucleophile to the dihydroisoxazole electrophile. Unless the inhibitor shifts out of that binding mode in a specific manner, the irreversible nucleophilic attack cannot occur. The orthogonality between specificity and reactivity modes might be because the binding mode holds the pharmacophore electrophile too far away from the active site nucleophile, or because the binding mode orients the electrophile such that an unfavorable rotation barrier must be overcome before it is in position to undergo nucleophilic attack. Future compounds may be designed to test this hypothesis by allowing better access to the active site from the binding mode. For example, it may be feasible to combine the specificity-engendering indole of the quinolyl-5-fluoro-tryptophan compound 25A with the reactivity-engendering conformational bias of the quinolyl-proline compound 26A. Development of increasingly effective and selective inhibitors of transglutaminase 2 should facilitate illumination of the role of TG2 in a variety of human disorders and may ultimately lead to effective clinical treatments for those disorders.
EXPERIMENTAL METHODS
Mass Spectrometry
Recombinant human transglutaminase 2 (50 μl of a 20 μM stock) was diluted 1:1 with Tris buffered saline containing 10 mM calcium chloride, and 1 μl of inhibitor dissolved in DMSO was added to give a final concentration of 100 μM. The sample was incubated 1.5 h at 37 C to allow for irreversible inhibition. Next, 1 μl of a 15 mg/ml stock of trypsin (Sigma) was added and incubated 1 h at 37 C to completely digest the protein. The reaction was quenched by diluting the sample 1:1 with formic acid and stored at −20 deg C until submitted for LC-MS/MS analysis. Fifteen of 20 expected proteolytic fragments from trypsin digestion were found, of those, only the active site cysteine-containing fragment was labeled with inhibitor.
General Techniques
All reactions were carried out under anhydrous conditions unless otherwise stated. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. Reagents were purchased at the highest commercial quality and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60F-254) using UV light as a visualizing agent and cerium molybdate stain, ninhydrin stain, permanganate stain and heat as developing agents. E. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. Preparative thin-layer chromatography (PTLC) separations were carried out on 0.25 mm E. Merck silica gel plates (60F-254). NMR spectra were recorded on Varian Inova 500 or Varian 200 instruments and calibrated using residual undeuterated solvent as an internal reference. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, b = broad, bs = broad singlet. Mass spectra were recorded on a Finnigan LCQ quadrupole ion trap mass spectrometer using electrospray ionization.
Dihydroisoxazole synthesis
The synthesis of compounds 1–10 was reported earlier.(17) Synthesis of compounds 13–17 was accomplished similarly by reaction of commercially available Cbz-Cl with individual amino acids, followed by coupling of the resulting products to the dihydroisoxazole pharmacophore using EDCI/HOBT in DMF. The dihydroisoxazole was synthesized by [3+2] cycloaddition of dibromoformaldoxime with allylamine.(20)
Compounds 18–23 were synthesized similarly except the amino acids were conjugated to the appropriate quinoline-containing carbamate (Scheme 1). This quinoline-containing carbamate was synthesized by reducing commercially available 3-quinolylcarbaldehyde with lithium borohydride in THF, followed by an aqueous workup and flash chromatography yielding 3-quinolylcarbinol. Para-nitro-phenylchloroformate was reacted with 3-quinolylcarbinol in methylene chloride and N-methyl morpholine to give a carbonate of 3-quinoline methanol and para-nitro-phenol. After purification by flash chromatography, this carbonate was reacted with the methyl ester of the amino acid in DMF and N-methyl-morpholine to give the N-quinolylmethyl carbamate-protected amino acid methyl ester. Base hydrolysis of the methyl ester followed by EDCI coupling of the resultant acid to the halo-dihydroisoxazole gave the indicated compounds as a mixture of diastereomers. Compound 17 was synthesized by coupling of the dihydroisoxazole pharmacophore to commercially available 2-,3-dihydro-indole-1-carbamic acid benzyl ester with EDCI.
Scheme 1.

General synthesis of inhibitors in this study. a) LiBH4, THF b) p-nitro-phenylchloroformate, N-methylmorpholine, CH2Cl2 c) 1. amino acid methyl ester, 11, DMF 2. MeOH, THF, H2O, LiOH d) EDCI, HOBT, DMF, 12.
The preparation of chiral dihydroisoxazoles (Scheme 2) was achieved by [3+2] cycloaddition of dibromoformaldoxime with allyl acetate. The racemic product was resolved into enantiomers by treatment with Amano Lipase PS in phosphate buffer, pH7, containing 15% acetone. These reaction conditions selectively hydrolyze the ester borne on the (R−) dihydroisoxazole, leaving the (S−) enantiomer unchanged. The (R−) dihydroisoxazole alcohol is readily separated from the (S−) dihydroisoxazole ester by flash chromatography. Hydrolysis of the (S−) dihydroisoxazole ester in methanol/water/potassium carbonate afforded the second enantiomer of the dihydroisoxazole.(21) The alcohols yielded from the chiral resolution had optical rotations of −135.5° (S-isomer) and +111.4° (R-isomer), consistent with published literature.(21) Both compounds were converted to the corresponding amines by means of the Mitsunobu reaction using tetra-chloro-phthalimide as nucleophile, followed by aminolysis with ethylenediamine in ethanol/acetonitrile/THF. The enantiomerically enriched dihydroisoxazole 32 and its enantiomer were coupled to N-functionalized amino acids under the same conditions as before.
Scheme 2.
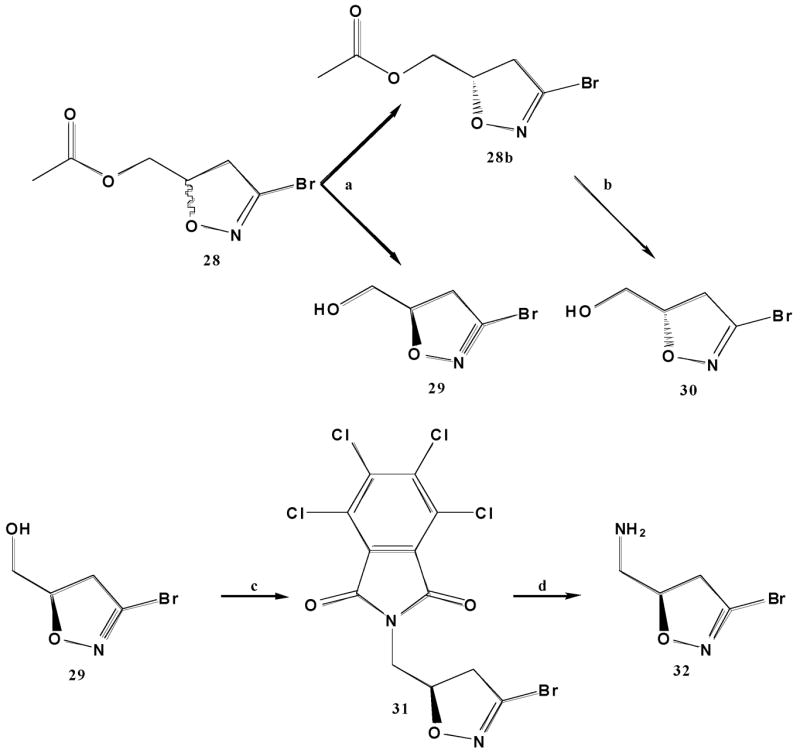
Chiral resolution of dihydroisoxazoles. a) 15% v/v acetone/0.1M pH7 phosphate buffer, Amano Lipase PS b) MeOH, 20% aq. K2CO3 c) DIAD, PPh3, tetrachlorophthalimide, THF d) ethylenediamine, ACN, THF, EtOH.
[1-[(3-Bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamoyl]-2-(4-fluoro-1H-indol-3-yl)-ethyl]-carbamic acid benzyl ester (13)
10mg (0.045mmol) commercially available racemic 4-fluorotryptophan was dissolved in 450ul of 1:1 dioxane water (0.1M), and cooled in a water/ice bath. Excess potassium carbonate was added followed by 2 equiv. of Cbz-Cl (15.35mg) and stirred vigorously overnight. The reaction mixture was diluted to 10ml with EtOAc, acidified with HCl to pH 1, washed with brine (10 × 1ml), dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by preparative TLC in 90% methylene chloride, 9% methanol and 1% HOAc. (11mg, 0.03mmol, 69%). Without further characterization the compound was dissolved in 300ul neat DMF (0.1M), and placed in a water/ice bath. 6mg of 7-amino-3-bromo,4-,5-dihydroisoxazole (0.045mmol, 1.5 equiv) were added to the reaction mixture followed by 9.8mg (0.06mmol, 2 equiv) of hydroxybenzotriazole hydrate, and 14mg of EDCI (0.06mmol, 2 equiv). The reaction was allowed to run for 12 hrs, was followed by an aqueous workup with 1M phosphate buffer at pH 3, extraction with EtOAc (5×5ml), drying over sodium sulfate, concentration under reduced pressure and purification by prep. TLC in 4:1 dichloromethane: acetone. (13mg, 0.025mmol, 83%). 1H NMR (500MHz, CDCl3): δ 3.14–3.196 (dd, 1H, J=10.5, 17.5), 3.23–3.30 (m, 1H), 3.368–3.409 (dd, 1H, J=6.0, 15.0), 3.452–3.524 (m, 2H), 4.483–4.527 (m, 1H), 4.65–4.73 (bm, 1H), 5.046–5.057 (d, 2H, J=5.5), 5.531–5.545 (d, 1H, J=7.0), 6.544–6.574 (bm, 1H), 6.753–6.796 (m, 1H), 6.998–7.001 (d, 1H, J=1.5), 7.076–7.118 (m, 1H), 7.140–7.156 (d, 1H, J=8.0), 7.315–7.344 (m, 5H), 8.048 (s, 1H), 8.412 (bs, 1H).
[1-[(3-Bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamoyl]-2-(5-fluoro-1H-indol-3-yl)-ethyl]-carbamic acid benzyl ester (14)
Compound 14 (25mg, 0.049mmol, 53%) was prepared by the method used for compound 13. 1H NMR (500MHz, CDCl3): δ 3.8–3.9 (m, 1H), 3.1–3.2 (m, 1H), 3.3–3.4 (m, 1H), 3.4–3.5 (m, 1H), 4.43–4.505 (m, 1H), 4.46–4.47 (m, 1H), 5.1–5.2 (m, 2H), 5.3–5.5 (m, 1H), 6.1–6.25 (m, 1H), 6.95–7.0 (m, 1H), 7.09–7.15 (d, 1H), 7.17–7.21 (m, 1H), 7.27–7.4 (m, 8H), 8.1–8.2 (s, 1H); MS (ESI) m/z 518.9 [M+H]+.
[1-[(3-Bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamoyl]-2-(6-fluoro-1H-indol-3-yl)-ethyl]-carbamic acid benzyl ester (15)
Compound 15 (70.0mg, 0.136mmol, 68%) was prepared by the method used for compound 13. 1H NMR (500MHz, CDCl3): δ 2.783–2.849 (m, 1H), 3.094–3.182 (m, 2H), 3.283–3.440 (m, 2H), 4.45–4.59 (m, 1H), 4.639 (m, 1H), 5.069–5.147 (m, 2H), 5.501–5.549 (dd, 1H, 1=7.0, 17.5), 6.348–6.380 (d, 1H, J=16.0), 6.860–6.892 (dd, 1H, J=8.5), 7.020–7.056 (m, 2H), 7.3–7.4 (m, 5H), 7.557 (bs, 1H), 8.075 (s, 1H), 8.328 (bs, 1H).
[1-[(3-Bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamoyl]-2-(1H-pyrrolo[2,3-b]pyridin-3-yl)-ethyl]-carbamic acid benzyl ester (16)
Compound 16 (14.5mg, 0.029mmol, 47%) was prepared by the method used for compound 13. 1H NMR (500MHz, CDCl3): δ 3.1–3.3 (m, 4H), 4.577(m, 1H), 4.695 (m, 1H), 5.087–5.118 (m, 2H), 5.548–5.605 (m, 2H), 5.694 (bs, 1H), 7.265–7.279 (m, 2H), 7.3–7.46 (m, 5H), 7.596–7.637 (m, 2H), 8.221–8.276 (m, 1H), 8.514–8.561 (dd, 1H, J=5.0, 18.5).
2-[(3-Bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamoyl]-2,3-dihydro-indole-1-carboxylic acid benzyl ester (17)
Commercially available 1-[(benzyloxy)carbonyl]-2-indoline-carboxylic acid (100mg, 0.336mmol) was dissolved in neat DMF (3.36ml, 0.1M). With rapid stirring, 7-amino-3-bromo,4-,5-dihydroisoxazole (90.2mg, 0.504mmol, 1.5 equiv) was added followed by HOBT-hydrate (68.1mg, 0.504mmol, 1.5equiv.), and EDCI (96.6mg, 0.504mmol, 1.5 equiv.). The reaction was stirred 12hrs, was followed by an aqueous workup with 1M phosphate buffer at pH 3, and extraction with EtOAc (5×25ml). The crude product was dried over sodium sulfate, concentrated under reduced pressure and purified by prep. TLC in 7:1 dichloromethane: acetone. (319mg, 0.235mmol, 70%). 1H NMR (500MHz, CDCl3): δ 2.7–3.6 (m, 6H), 4.5–4.7 (m, 1H), 4.8–4.92 (m, 1H), 5.15–5.35 (m, 2H), 6.4 (bs, 1H), 6.9–7.0 (m, 1H), 7.03–7.2 (m, 2H), 7.25–7.4 (m, 5H), 7.61 (bs, 1H).
4-nitrophenyl quinolin-3-ylmethyl carbonate (11)
Commercially available 2-quinolinecarbaldehyde (651mg, 4.14mmol, 1equiv.) was dissolved in ethanol (11.7ml, 0.1M) and cooled in a dry ice/acetone bath. Lithium borohydride 1M in THF was added drop wise slowly and followed by TLC. When the reaction was complete, it was quenched with 1N HCl (1ml), and stirred 1hr while warming to room temp. The reaction mixture was concentrated under reduced pressure, diluted to 50ml with EtOAc, and washed with 1M pH 7 buffer (3×10ml), and with brine (3×5ml), dried over sodium sulfate, concentrated under reduced pressure and used without further purification. A 250mg aliquot of the resulting quinoline-carbinol was added drop wise to a solution 0.16M in p-nitrophenylchloroformate (1.5 equiv) with rapid stirring in a bath of ice/brine. After 2 minutes of stirring 4-methylmorpholine (1.6 equiv) was added in one aliquot. The mixture was stirred 10min. and then removed from the bath and allowed to warm to room temp. The reaction was followed by TLC and complete in about 7 hrs. The reaction was diluted with 50ml EtOAc, and washed with 1M HCl (2×10ml) to remove the morpholines, then with sat. aq. bicarb. (2×10ml) to begin removal of p-nitrophenol. The organic phase was dried over sodium sulfate and then pushed through a plug of silica topped by a plug of anhydrous potassium carbonate to remove most remaining p-nitrophenol. The resultant liquor was concentrated under reduced pressure, and purified by flash chromatography in 10–70% EtOAc/hexanes. (219mg, 43%). 1H NMR (200MHz, CDCl3): δ 5.450 (s, 2H), 7.308–7.354 (d, 2H, J=23), 7.525–7.605 (dddd, 1H, J=1, 7), 7.698–7.782 (dddd, 1H, J=1, 7), 7.806–7.852 (dd, 1H, J=1.2, 8.0), 8.097–8.141 (d, 1H, J=8.8), 8.203–8.250 (d, 2H, J=23), 8.950–8.961 (d, 1H, J=2.2).
(S)-quinolin-3-ylmethyl 1-((3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(5-fluoro-1H-indol-3-yl)-1-oxopropan-2-ylcarbamate (22)
Commercially available (S-)5-F-tryptophan methyl ester (75mg, 0.315mmol, 1 equiv.) was dissolved in 1ml DMF (0.315M). 11 (219mg, 0.615mmol, 2.14 equiv.) was added in one aliquot. The reaction was stirred overnight, then diluted with 50ml CH2Cl2 and 75ml EtOAc, and washed with sat. aq. bicarb (3×25ml). The organic phase was dried over sodium sulfate, then pushed through a pad of silica topped with a pad of anhydrous potassium carbonate. The resultant liquor was concentrated under reduced pressure and purified by flash chromatography in equal parts EtOAc/hexanes - EtOAc/hexanes+5%EtOH. (79mgs, 0.187mmol, 60%). The resultant methyl ester was dissolved in 1.5ml THF. 1 equivalent of LiOH was added as 1M soln in water (187ul). Methanol was added dropwise till the reaction mixture became homogeneous, and stirred 3hrs. The reaction mixture was acidified and extracted with EtOAc (4×10ml), dried over sodium sulfate and concentrated under reduced pressure. (70mg, 0.172mmol, 94%). The free acid was dissolved in DMF (1.7ml, 0.1M). HOBT (23.24mg, 0.172mmol, 1 equiv.) was added followed by 12 (30mg, 0.172mmol, 1 equiv.) and EDCI (33mg, 0.172mmol, 1 equiv.). The reaction was stirred overnight, diluted with 25ml EtOAc and washed with brine (3×10ml), washed with 1M phosphate buffer pH 7 (3×10ml), then dried over sodium sulfate, concentrated under reduced pressure and purified by column chromatography in 10–50% acetone in dichloromethane -50% acetone/dichloromethane 0–3%EtOH. (73mg, 0.128mmol, 75%). 1H NMR (200MHz, DMSO-d6): δ 2.9–3.55 (m, 4H), 4.387–4.497 (dd, 1H, J=7.6, 14.8), 4.6–4.76 (m, 1H), 5.199 (s, 2H), 6.541–6.575 (d, 1H, J=6.8), 6.770–6.873 (ddd, 1H, J=2.2, 9.0), 7.24–7.38 (m, 4H), 7.515–7.590 (m, 2H), 7.658–7.742 (ddd, 1H, J=1.6, 7.0), 7.849–7.888 (d, 1H, J=8.0), 7.973–8.015 (d, 1H, J=8.4) 8.8 (s, 1H), 10.18 (bs, 1H).
(S)-quinolin-3-ylmethyl 1- ((3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3- (4-hydroxy-phenyl)-1-oxopropan-2-ylcarbamate (18)
The compound 18 (6.8mg, 0.0129mmol, 3.2% for three steps) was prepared by the method used to prepare compound 22. 1H NMR (200MHz, CDCl3): δ 2.6–3.0 (m, 4H), 4.0–4.6 (m, 1H), 4.49–4.7 (m, 1H), 5.094–5.114 (d, 2H, J=4.0), 6.543–6.584 (d, 2H, J=8.2), 6.966–7.008 (d, 2H, J=8.4), 7.47–7.596 (m, 2H), 7.655–7.738 (ddd, 1H, J=1.2, 7.0), 7.82–7.978 (m, 2H), 8.1–8.35 (m, 2H), 8.779–8.789 (d, 1H, J=2.0), 9.124 (s, 1H).
(S)-quinolin-3-ylmethyl 1-((3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(5-hydroxy-lH-indol-3-yl)-1-oxopropan-2-ylcarbamate (19)
The compound 19 (5.6mg, 0.0099mmol, 3.6% for three steps) was prepared by the method used to prepare compound 22. 1H NMR (200MHz, CDCl3): δ 2.8–3.4 (m, 4H), 4.1–4.2 (m, 1H), 4.45–4.66 (m, 1H), 5.101–5.132 (d, 2H, J=6.2), 6.492–6.545 (dd, 1H, J= 2.0, 8.8), 6.84–6.9 (m, 1H), 6.96–7.07 (m, 2H), 7.25–7.589 (m, 3H), 7.659–7.729 (dd, 1H, J=1.2, 7.0), 7.852–7.971 (dd, 2H, J= 8.4, 16.6), 8.15–8.3 (m, 2H), 8.516 (s, 1H), 8.794–8.804 (d, 1H, J=2.0), 10.440 (bs, 1H).
(S)-quinolin-3-ylmethyl 1-((3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(5-methoxy-1H-indol-3-yl)-l-oxopropan-2-ylcarbamate (20)
The compound 20 (9.0mg, 0.0155mmol, 6.2% for three steps) was prepared by the method used to prepare compound 22. 1H NMR (200MHz, CDCl3): δ 2.8–3.4 (m, 4H), 3.680 (s, 3H), 4.1–4.3 (m, 1H), 4.5–4.7 (m, 1H), 5.087–5.121 (d, 2H, J=6.8), 6.604–6.659 (dd, 1H, J=2.3, 8.7), 7.116–7.159 (d, 1H, J=8.6), 7.462–7.588 (m, 2H), 7.661–7.729 (dd, 1H, J=1, 6.8), 7.838–7.970 (dd, 2H, J=8.4, 17.8), 8.167 (s, 1H), 8.22–8.38 (m, 1H), 8.781–8.791 (d, 1H, J=2.0), 10.590 (s, 1H).
quinolin-3-ylmethyl 1-((3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(5-fluoro-1H-indol-3-yl)-1-oxopropan-2-ylcarbamate (21)
The compound 21 (4.7mg, 0.0083mmol, 2.4% for three steps) was prepared by the method used to prepare compound 22. 1H NMR (200MHz, DMSO-d6): δ 2.9–3.55 (m, 4H), 4.387–4.497 (dd, 1H, J=7.6, 14.8), 4.6–4.76 (m, 1H), 5.199 (s, 2H), 6.541–6.575 (d, 1H, J=6.8), 6.770–6.873 (ddd, 1H, J=2.2, 9.0), 7.24–7.38 (m, 4H), 7.515–7.590 (m, 2H), 7.658–7.742 (ddd, 1H, J=1.6, 7.0), 7.849–7.888 (d, 1H, J=8.0), 7.973–8.015 (d, 1H, J=8.4) 8.8 (s, 1H), 10.18 (bs, 1H).
(S)-quinolin-3-ylmethyl 2-(((3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (23)
The compound 23 (5.2mg, 0.0113mmol, 5.1% for three steps) was prepared by the method used to prepare compound 22. 1H NMR (200MHz, CDCl3): δ 1.6–1.8 (m, 4H), 2.8–3.4 (m, 6H), 4.05–4.25 (m, 1H), 4.45–4.7 (m, 1H), 5.1–5.25 (m, 2H), 7.522–7.738 (m, 2H), 7.866–7.977 (m, 2H), 8.15–8.3 (m, 2H), 8.765–8.859 (d, 1H, J=18.4).
quinolin-3-ylmethyl (S)-1-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(4-hydroxyphenyl)-1-oxopropan-2-ylcarbamate (24a) and quinolin-3-ylmethyl (S)-1-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(4-hydroxyphenyl)-1-oxopropan-2-ylcarbamate (24b)
The compounds 24a and 24b (24a: 40.6mg, 0.077mmol, 77% in the coupling reaction; 24b: 26.0mg, 0.049mmol, 58%) were prepared by the method used to prepare compound 22, with the exception that the enantiomerically enriched dihydroisoxazoles, 32 and 32b, were used. 24a:1H NMR (500MHz, DMSO-d6): δ 2.799–2.836 (dd, 1H, J=4.5, 13.5), 2.935–2.985 (dd, 1H, J=7.5, 17.5), 3.179–3.228 (ddd, 1H, J=5.0, 5.0, 14.0), 3.293–3.350 (dd, 1H, J=11.0, 18.0), 3.378–3.430 (m, 1H), 4.146–4.193 (m, 1H), 4.653–4.709 (m, 1H), 5.144–5.229 (dd, 2H, J=13.0, 29.5), 6.644–6.661 (d, 2H, J=8.5), 7.062–7.079 (d, 2H, J=8.5), 7.952–7.609 (d, 1H, J=8.5), 7.625–7.655 (ddd, 1.0, 7.0, 7.0), 7.761–7.794 (ddd, 1H, J=1.0, 8.5, 8.5), 7.952–7.968 (d, 1H, J=8.0), 8.029–8.046 (d, 1H, J=8.5), 8.245–8.247 (d, 1H, J=1.0), 8.331–8.355 (dd, 1H, J=6.0, 6.0), 8.865–8.869 (d, 1H, J=1.5), 9.213 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 173.2, 156.5, 156.4, 151.3, 147.8, 138.9, 135.2, 130.8, 130.3, 129.5, 128.7, 127.9, 127.7, 115.6, 80.8, 64.0, 57.4, 44.2, 41.9, 37.6, 28.58, 23.4; [α]22D(c=0.5, DMSO-d6) = +40.0 HRMS (TOF MS ES+) m/z calc’d for C24H23N4O5Br, 527.0930, found, 527.0915 (M + H)+. 24b: 1H NMR (500MHz, CD3OD): 2.790–2.835 (dd, 1H, J=8.5, 13.5), 2.965–3.012 (m, 2H), 3.187–3.243 (dd, 1H, J=10.5, 17.5), 3.328–3.454 (ddd, 2H, J=10.2, 14.0, 44.0), 4.279–4.308 (dd, 1H, J=6.5, 8.0), 4.652–4.709 (m, 1H), 5.294 (s, 2H), 6.688–6.705 (dd, 2H, J=2.0, 6.5), 7.048–7.066 (d, 2H, 9.0), 7.641–7.671 (dd, 1H, J=7.0, 7.0), 7.953–7.97 (d, 1H, J=8.5), 8.041–8.058 (d, 1H, 8.5), 8.304 (s, 1H), 8.859–8.862 (d, 1H, J=1.5); 13C NMR (125 MHz, CD3OD) δ 173.7, 156.7, 156.2, 150.2, 147.0, 138.0, 136.0, 130.5, 130.2, 128.2, 128.1, 127.8, 127.3, 115.1, 80.5, 57.2, 43.7, 41.5, 37.1; [α]22D (c=0.5, CD3OD) = -38.8. HRMS (TOF MS ES+) m/z calc’d for C24H23N4O5Br, 527.0930, found, 527.0942 (M + H)+.
quinolin-3-ylmethyl (S)-1-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(5-fluoro-1H-indol-3-yl)-1-oxopropan-2-ylcarbamate (25a) and quinolin-3-ylmethyl (S)-1-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(5-fluoro-1H-indol-3-yl)- 1-oxopropan-2-ylcarbamate (25b)
The compounds 25a and 25b (25a: 12.2mg, 0.0214mmol, 67% in the coupling reaction; 25b: 14.6mg, 0.026mmol, 81%) were prepared by the method used to prepare compound 22, with the exception that the enantiomerically enriched dihydroisoxazoles, 32 and 32b, were used. 25a:1H NMR (500MHz, DMSO-d6): δ 2.87–3.1 (m, 3H), 3.186–3.325 (ddd, 1H, J=5.0, 5.0, 14.5), 3.257–3.313 (dd, 1H, J=10.5, 17.5), 3.98–3.54 (m, 1H), 4.247–4.293 (m, 1H) 4.644–4.700 (m, 1H) 5.111–5.226 (dd, 2H, J=8.0, 44.5) 6.833–6.925 (ddd, 1H, J=3.0, 9.5, 9.5) 7.261–7.265 (d, 1H, J=2.0) 7.311–7.338 (dd, 1H, J=4.5, 4.5) 7.473–7.498 (dd, 1H, J=2.5, 10.0) 7.604–7.636 (dd, 1H, J=8.0, 8.0) 7.633–7.652 (dd, 1H, J=1.5, 7.0) 7.757–7.790 (ddd, 1H, J=1.5, 7.0, 8.0) 7.927–7.945 (dd, 1H, 1=1.0,8.0) 8.023–8.040 (d, 1H, J=8.5) 8.236–8.239 (d, 1H, J=1.5) 8.441–8.464 (dd, 1H, J=6.0, 6.0) 8.856–8.860 (d, 1H, J=2.0) 10.944–10.948 (d, 1H, J=2.0); 13C NMR (125 MHz, DMSO-d6) δ 173.4, 158.3, 156.5, 151.3, 147.8, 138.8, 135.2, 133.4, 130.7, 130.3, 129.5, 128.7, 128.1, 127.9, 127.7, 127.0, 112.9, 111.1, 109.8, 109.6, 104.2, 80.8, 64.9, 56.2, 44.1, 42.0, 28.7) [α]22D (c=0.5, DMSO-d6) = +18.7 HRMS (TOF MS ES+) m/z calc’d for C26H23N5O4FBr, 568.0996, found, 568.0094 (M + H)+. 25b: 1H NMR (500MHz, DMSO-d6): 52.867–2.916 (dd, 1H, J=10.0, 14.5), 2.995–3.052 (m, 1H), 3.257–3.337 (m, 1H), 4.236–4.287 (m, 1H), 4.664–4.721 (m,1H), 5.112–5.232 (dd, 2H, J=13.0, 47.0) 6.880–6.921 (ddd, 1H, J=2.0, 8.0, 8.0), 7.274 (s, 1H), 7.308–7.335 (dd, 1H, J=5.0, 9.0), 7.471–7.496 (dd, 1H, 1=2.0,20.0), 7.578–7.594 (d, 1H, J=8.0), 7.621–7.651 (dd, 1H, 1=7.0,7.0), 7.757–7.790 (ddd, 1H, J=1.0, 8.0, 8.0), 7.929–7.945 (d, 1H, J=8.0), 8.023–8.040 (d, 1H, J=8.5), 8.427–8.451 (dd, 1H, 1=6.0,6.0), 8.858–8.861 (d, 1H, J=1.5), 10.947 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 173.2, 158.3, 156.5, 151.3, 147.8, 139.0, 135.2, 133.4, 130.8, 130.3, 129.5, 128.7, 128.0, 127.7, 127.0, 112.9, 111.1, 109.7, 109.5, 104.0, 80.8, 69.7, 56.1, 44.0, 41.7, 28.5); [α]22D (c=0.5, DMSO-d6) = -30.8. HRMS (TOF MS ES+) m/z calc’d for C26H23N5O4FBr, 568.0996, found, 568.0980 (M + H+)
(S)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5- yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (26a) and (S)-quinolin-3-ylmethyl 2-((((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (26b)
Compounds 26a and 26b (26a: 22.2mg, 0.048mmol, 48.2% in the coupling reaction; 26b: 26.0mg, 0.056mmol, 66%) were prepared by the method used to prepare compound 22, with the exception that the enantiomerically enriched dihydroisoxazoles, 32 and 32b, were used. 26a: 1H NMR (500MHz, CD3CN) δ 1.867–1.972 (bm, 3H), 2.909–3.059 (ddd, 1H, J=7.5, 17.5, 50.0), 3.153–3.209 (dd, 1H, J=10.0, 17.5), 3.273–3.396 (m, 2H), 3.462–3.538 (m, 2H), 3.573–3.616 (m, 1H), 4.205–4.279 (ddd, 1H, J=3.0, 8.5, 25.0), 4.587–4.644 (m, 1H), 4.755–4.811 (m, 1H), 5.233–5.371 (dd, 1H, J=13.0, 57.0), 5.358 (s, 1H), 6.904–6.971 (bd, 1H, J=33.5), 7.637–7.664 (dd, 1H, J=7.0, 7.0), 7.781–7.810 (dd, 1H, J=8.5, 8.5), 7.946–7.991 (dd, 1H, J=8.0, 14.5), 8.231–8307 (d, 1H, J=38.0), 8.892–8.969 (d, 1H, J=38.5); 13C NMR (125 MHz, CD3CN) δ 173.5, 173.1, 155.2, 154.6, 151.0, 148.0, 137.9, 135.0, 134.8, 130.4, 129.8, 129.3, 128.3, 127.9, 127.2,80.8, 80.6, 64.7, 61.3, 60.7, 47.5, 47.1, 43.9, 43.7, 41.6, 41.2, 31.6, 300.2, 24.4, 23.6. [α]22D (c=0.5, CD3CN) = +17.2. HRMS (TOF MS ES+) m/z calc’d for C20H21N4O4Br, 461.0824, found, 461.086 (M + H)+. 26b: 1H NMR (500MHz, CD3OD): δ 1.900–2.035 (m, 3H), 2.201–2.370 (m, 1H), 2.892–2.943 (dd, 1H, J=8.0, 17.5), 3.266–3.324 (m, 1H), 3.358–3.434 (dd, 1H, J=4.5, 33.5), 3.387–3.406 (dd, 1H, J=4.5, 4.5), 3.522–3.649 (m, 2H), 4.272–4.341 (ddd, 1H, J=3.5, 8.5, 22.0), 4.618–4.815 (m, 1H), 5.292–5.442 (dd, 1H, J=13.0, 62.5), 5.388 (s, 1H), 7.651–7.683 (ddd, 1H, J=1.0, 8.0, 8.0), 7.800–7.833 (ddd, 1H, J=1.0, 8.0, 8.0), 7.982–8.011 (dd, 1H, J=7.0, 7.0), 8.052–8.069 (d, 1H, J=9.0), 8.356–8.416 (d, 1H, J=30.0), 8.862–8.936 (d, 1H, J=42.0). 13C NMR (125 MHz, CD3OD) δ 174.8, 150.4, 150.3, 147.1, 137.6, 136.4, 136.2, 130.2, 128.2, 127.8, 127.4, 80.7, 80.3, 60.9, 43.7, 41.8, 41.3, 31.6, 30.3, 24.3, 23.4. [α]22D (c=0.54, CD3OD) = −68.0. HRMS (TOF MS ES+) m/z calc’d for C20H21N4O4BrNa, 483.0664, found, 483.0646 (M + H)+.
benzyl (S)-1-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(4-hydroxyphenyl)-1-oxopropan-2-ylcarbamate (27a) and benzyl (S)-1-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-3-(4-hydroxyphenyl)-1-oxopropan-2-ylcarbamate (27b)
Compounds 27a and 27b (27a: 72.3mg, 0.152mmol, 76% in the coupling reaction; 27b: 80.8mg, 0.170mmol, 84.8%) were prepared by the method used to prepare compound 22, with the exception that the enantiomerically enriched dihydroisoxazoles, 32 and 32b, were used. 27a: 1H NMR (500MHz, CD3OD) δ 2.609–2.657 (dd, 1H, J=10.5, 13.5), 2.795–2.832 (dd, 1H, J=4.5, 14.0), 2.939–39.989 (dd, 1H, J=7.0, 17.5), 3.181–3.230 (ddd, 1H, J=5.0, 5.0,14.0), 3.299–3.356 (dd, 1H, J=11.0, 17.5), 4.134–4.181 (m,1H), 4.659–4.716 (m, 1H), 4.946–4.953 (d, 2H, J=3.5), 6.648–6.665 (d, 2H, J=8.5), 7.055–7.071 (d, 2H, J=8.5), 7.240–7.254 (d, 1H, J=7.0), 7.282–7.359 (m, 3H), 7.462–7.479 (d, 1H, J=8.5), 8.294–8.318 (dd, 1H, J=6.0, 6.0), 9.238 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 173.1, 156.5,138.9, 137.7,130.8, 129.0,128.7, 128.4, 128.2, 115.6, 80.8, 65.9, 57.3, 44.2, 41.9, 37.6. [α]22D (c=0.5, DMSO-d6) = +45.5. HRMS (TOF MS ES+) m/z calc’d for C21H22N3O4Br-Na, 498.06, found, 498.0641 (M + Na)+.27b: 1H NMR (500MHz, CD3OD) δ 2.607–2.655 (dd, 1H, J=7.0, 13.5), 2.806–2.843 (dd, 1H, J=4.5, 13.5), 3.002–3.053 (dd, 1H, J=7.5, 17.5), 3.303–3.365 (m, 4H), 4.122–4.168 (m, 1H), 4.671–4.720 (m, 1H), 4.965 (s, 2H), 6.646–6.663 (d, 2H, J=8.5), 7.059–7.076 (d, 2H, J=8.5), 7.247–7.361 (m, 5H), 7.457–7.474 (d, 1H, J=8.5), 8.273–8.296 (dd, 1H, J=6.0, 6.0), 9.207 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 173.1, 156.6, 156.5, 139.0, 137.8, 130.8, 129.0, 128.8, 128.4, 128.2, 115.6, 80.8, 65.9, 57.3, 44.1, 41.8, 37.5. [α 22D (c=0.5, CD3CN) = -68.2. HRMS (TOF MS ES+) m/z calc’d for C21H22N3O4Br, 498.06, found, 498.0641 (M + Na)+.
(3-bromo-4,5-dihydroisoxazol-5-yl)methyl acetate. (28)
To 100.1 mg (1 mmol) of allylacetate in 10 ml of 1:1 H2O/EtOAc were added 202.8 mg (1 mmol) of dibromoformaldoxime with vigorous stirring at room temperature. 10 ml of 1M pH7 phos. buffer were added slowly and reaction progress followed by TLC. Upon completion, aqueous NaOH was added drop wise to pH9, and the organic phase decanted. The aqueous phase was extracted with 50ml of EtOAc, and the combined organic phases dried with sodium sulfate and the solvent removed under reduced pressure. The resultant oil was purified by flash chromatography in 10–40% EtOAc in hexanes. (158mg, 0.72mmol, 71%) 1H NMR (200MHz, CDCl3): δ 1.990 (s, 3H), 2.911–3.348 (ddd, 2H, J=7.6, 17.4, 59.2), 4.052–4.240 (m, 2H), 4.774–4.885 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 170.1, 136.3, 78.2, 63.6, 42.8, 20.1.
(R)-(3-bromo-4,5-dihydroisoxazol-5-yl)methanol (29) and ((S)3-bromo-4,5-dihydroisoxazol-5-yl)methyl acetate. (28b)
To 8.5g (38.5mmol) of the racemic dihydroisoxazole acetate 27 were added 1.15L of 15% v/v acetone in 0.1M pH7 phos. buffer, followed by 38mg (1 mg/mmol substrate) Amano Lipase PS. The reaction progress as monitored by TLC stalled after 2hrs. NaCl (~1 teacup) was added, and the reaction mixture was extracted with EtOAc 10×125ml. The combined organic layers were dried with sodium sulfate and evaporated under red. press. The chiral alcohol and ester were purified and separated by flash chromatography in 40%EtOAc in hexanes. The reaction yielded about 1g of alcohol and the remaining acetate was re-treated with the lipase under identical conditions. After three rounds of resolution, the total yield of alcohol was 3.26g, (18.23mmol, 47%), and of 4.02g of acetate (18.18mmol, 47%). 29: 1H NMR (200MHz, CDCl3): δ 1.818–2.1 (bs, 1H), 2.544–2.772 (m, 2H), 4.305–4.475 (m, 2H), 4.79–4.88 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 138.2, 82.4, 63.0, 42.8. [α]22D (c=0.5, CDCl3) = −135.7. 28b: 1H NMR (200MHz, CDCl3): δ 1.990 (s, 3H), 2.911–3.348 (ddd, 2H, J=7.6, 17.4, 59.2), 4.052–4.240 (m, 2H), 4.774–4.885 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 170.1, 136.3, 78.1, 63.6, 42.8, 20.1. [α]22D (c=0.5, CDCl3) = +101.0.
(S)-(3-bromo-4,5-dihydroisoxazol-5-yl)methanol (30)
Acetate 27b, 1.21g, 5.5mmol, was hydrolyzed in 31.5ml MeOH and 16ml 20% aq. potassium carbonate. After 15min, the reaction was complete by TLC, and the reaction mixture was diluted with 200ml EtOAc, and washed 10 × 10ml with brine. The organic phase was dried with sodium sulfate, evaporated under red. press, and purified by flash chromatography in 40%EtOAc/hexanes. (808mg, 4.51mmol, 82%). Note: The (S−) enantiomer of this compound was spectroscopically indistinct from the (R−) enantiomer with the exception of its optical rotation: [α]22D (c=0.5, CDCl3)= +111.2.
(S)-2-((3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-4,5,6,7-tetrachloroisoindoline-1,3-dione. (31)
To 1g (4.97mmol) of DIAD in 45ml THF were added at room temperature 1.3g (4.97mmol) of triphenylphosphine. After 15 min. of vigorous stirring, the reaction was cooled in an ice/water bath and 808mg (4.51mmol) of alcohol 29 were added. The reaction was stirred for 45 minutes whereupon 1.42g (4.97mmol) of tetra-chloro-phthalimide was added. The reaction was allowed to warm to room temperature and stir overnight. The reaction was diluted with 200ml EtOAc and washed with brine 10 × 30ml. The organic phase was dried with sodium sulfate and evaporated under reduced pressure. The resulting solid was purified by flash chromatography in neat methylene chloride. (694mg, 1.55mmol, 34%). 1H NMR (500MHz, CDCl3): δ 3.113–3.430 (ddd, 2H, J=7.0, 17.5, 130.5), 3.825–4.059 (ddd, 2H, J=5.5, 14.50, 96.0), 4.876–5.025 (m, 1H); 13C NMR (125MHz, CDCl3): δ 163.6, 140.8, 137.5, 130.6, 130.3, 127.6, 78.0, 45.4, 41.4, 31.2. [α]22D (c=0.5, CDCl3)= +108.6. Note: The (R−) enantiomer of this compound was spectroscopically indistinct from the (S−) enantiomer with the exception of its optical rotation: [α]22D (c=0.5, CDCl3)= −98.2.
(S)-(3-bromo-4,5-dihydroisoxazol-5-yl)methanamine (32)
2.03g (4.54mmol) of 30 were dissolved in 46ml of 2:1:1 acetonitrile, THF, ethanol. Ethylenediamine was added in 1/10th equivalent aliquots hourly until 2 equivalents had been added. The reaction was stirred overnight, diluted with 250ml EtOAc, and washed with brine 3×25ml. The organic phase was dried with sodium sulfate, evaporated under reduced pressure, and the resultant oil purified by flash chromatography in 0–60% acetone in methylene chloride. (335mg, 1.87mmol, 41%). 1H NMR (500MHz, CDCl3): δ 2.855–2.894 (dd, 1H, J=6.0, 13.5), 3.013–3.048 (dd, 1H, J=4.0, 13.5), 3.079–3.130 (dd, 1H, J=7.0, 17.5), 3.244–3.300 (dd, 1H, J= 10.5, 17.0), 4.73–4.79 (m, 1H). 13C NMR (125MHz, CDCl3): δ 137.65, 83.22, 45.12, 43.99. The (R−) enantiomer of this compound (32b) was spectroscopically indistinct by NMR.
TG2 Inhibition Assay
Recombinant human TG2 was expressed in E. coli and purified to >90% homogeneity as before.(17) Compounds were assayed in a reaction mixture containing 200 mM MOPS (pH = 7.2), 5 mM CaCl2, 1 mM EDTA, 10 mM α-ketoglutarate, 18 U/ml glutamate dehydrogenase, 0.4 mM NADH, 3.3% (v/v) DMSO, 0.5 piM TG2, 11.2 mM Cbz-Gln-Gly (KM = 6.28 mM, kcat = 37 min−1). Concentrations of inhibitors were varied from 0.001mM to 0.3mM. The enzyme reaction was started by addition of TG2 and the consumption of NADH was monitored by UV spectroscopy (340 nm, Σ= 6220 cm−1M−1). Kinetic parameters were obtained by plotting the reaction progress curves against theoretical equations for irreversible enzyme inhibition.(22)
Acknowledgments
We thank Tracy Holmes and the Vincent Coates Mass Spectrometry facility for technical assistance. This research was supported by a grant from the NIH (R01 DK 063158) to C.K. M.S. is a recipient of a predoctoral fellowship from the Stanford-NIH Biotechnology Training Program.
References
- 1.Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002;27:534–539. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- 2.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 4.Kim SY, Jeitner TM, Steinert PM. Transglutaminases in disease. Neurochem Int. 2002;40:85–103. doi: 10.1016/s0197-0186(01)00064-x. [DOI] [PubMed] [Google Scholar]
- 5.Hoffner G, Djian P. Transglutaminase and Diseases of the Central Nervous System. Frontiers in Bioscience. 2005;10:3078–3092. doi: 10.2741/1764. [DOI] [PubMed] [Google Scholar]
- 6.Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino G, Moreno S, Pavone F, Oliverio S, Fesus L, et al. Tissue transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- 7.de Macédo P, Marrano C, Keillor JW. Synthesis of dipeptide-bound epoxides and alpha,beta-unsaturated amides as potential irreversible transglutaminase inhibitors. Bioorg Med Chem. 2002;2:355–60. doi: 10.1016/s0968-0896(01)00292-9. [DOI] [PubMed] [Google Scholar]
- 8.Marrano C, de Macédo P, Gagnon P, Lapierre D, Gravel C, Keillor JW. Synthesis and evaluation of novel dipeptide-bound 1-,2-,4-thiadiazoles as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg Med Chem. 2001;12:3231–41. doi: 10.1016/s0968-0896(01)00228-0. [DOI] [PubMed] [Google Scholar]
- 9.Marrano C, de Macédo P, Keillor JW. Evaluation of novel dipeptide-bound alpha,beta-unsaturated amides and epoxides as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg Med Chem. 2001;7:1923–8. doi: 10.1016/s0968-0896(01)00101-8. [DOI] [PubMed] [Google Scholar]
- 10.Hausch F, Halttunen T, Mäki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol. 2003;10:225–31. doi: 10.1016/s1074-5521(03)00045-0. [DOI] [PubMed] [Google Scholar]
- 11.Choi K, Siegel M, Piper J, Yuan L, Cho E, Strnad P, Omary B, Rich K, Khosla C. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;4:469–75. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002;346:180–188. doi: 10.1056/NEJMra010852. [DOI] [PubMed] [Google Scholar]
- 13.Green PH, Jabri B. Coeliac disease. Lancet. 2003;362:383–391. doi: 10.1016/S0140-6736(03)14027-5. [DOI] [PubMed] [Google Scholar]
- 14.Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2:647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 15.Piper JL, Gray GM, Khosla C. High selectivity of human tissue transglutaminase for immunoactive gliadin peptides: implications for celiac sprue. Biochemistry. 2002;41:386–393. doi: 10.1021/bi011715x. [DOI] [PubMed] [Google Scholar]
- 16.Molberg O, McAdam S, Lundin KE, Kristiansen C, Arentz-Hansen H, Kett K, Sollid LM. T cells from celiac disease lesions recognize gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur J Immunol. 2001;31:1317–1323. doi: 10.1002/1521-4141(200105)31:5<1317::AID-IMMU1317>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 17.Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469–75. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Castelhano AL, Billedeau R, Pliura DH, Bonaventura BJ, Krantz A. Synthesis, chemistry, and absolute-configuration of novel transglutaminase inhibitors containing a 3-halo-4,5-dihydroisoxazole. Bioorg Chem. 1988;16:335–340. [Google Scholar]
- 19.Yuan L, Siegel M, Choi K, Khosla C, Miller CR, Jackson EN, Piwnica-Worms D, Rich KM. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2006 doi: 10.1038/sj.onc.1210048. in press. [DOI] [PubMed] [Google Scholar]
- 20.Rohloff JC, Robinson J, Gardner JO. Bromonitrile oxide [3+2] cycloadditions in water. Tetrahedron Lett. 1992;33:3113–3116. [Google Scholar]
- 21.De Amici M, Magri P, De Micheli C, Cateni F, Bovara R, Carrea G, Riva S, Casalone G. Nitrile Oxides in Medicinal Chemistry. 4. Chemoenzymatic Synthesis of Chiral Heterocyclic Derivatives. J Org Chem. 1992;57:825–2829. [Google Scholar]
- 22.Gray PJ, Duggleby RG. Analysis of kinetic data for irreversible enzyme inhibition. Biochem J. 1989;257:419–424. doi: 10.1042/bj2570419. [DOI] [PMC free article] [PubMed] [Google Scholar]