

Abstract
A functionally distinct subset of CD103+ dendritic cells (DCs) has recently been identified in murine mesenteric lymph nodes (MLN) that induces enhanced FoxP3+ T cell differentiation, retinoic acid receptor signaling, and gut-homing receptor (CCR9 and α4β7) expression in responding T cells. We show that this function is specific to small intestinal lamina propria (SI-LP) and MLN CD103+ DCs. CD103+ SI-LP DCs appeared to derive from circulating DC precursors that continually seed the SI-LP. BrdU pulse-chase experiments suggested that most CD103+ DCs do not derive from a CD103− SI-LP DC intermediate. The majority of CD103+ MLN DCs appear to represent a tissue-derived migratory population that plays a central role in presenting orally derived soluble antigen to CD8+ and CD4+ T cells. In contrast, most CD103− MLN DCs appear to derive from blood precursors, and these cells could proliferate within the MLN and present systemic soluble antigen. Critically, CD103+ DCs with similar phenotype and functional properties were present in human MLN, and their selective ability to induce CCR9 was maintained by CD103+ MLN DCs isolated from SB Crohn's patients. Thus, small intestinal CD103+ DCs represent a potential novel target for regulating human intestinal inflammatory responses.
DCs are well recognized for their role in the priming and differentiation of naive T cells (1, 2). Murine conventional DCs have been extensively studied and have traditionally been divided into five distinct subsets based on their expression of CD11b, CD4, CD8α, and Langerin. The spleen contains three of these subsets (CD11b+CD4+, CD11b+CD4− CD8α−, and CD11b− CD8α+), whereas LNs contain two additional populations (CD4− Langerin+ and CD4− CD8α− CD11blo) that display a more mature phenotype and appear to represent tissue-derived migratory DCs (3, 4). Although the developmental relationships between these populations remains unclear, several of these subsets have been described to have distinct functional properties (5, 6). Nevertheless, surprisingly few studies have attempted to characterize DC subsets in human LN (4, 7–10) and, thus, the possibility of targeting distinct conventional DC subsets for regulating human immune responses remains unclear. Indeed, to our knowledge functionally distinct conventional DC subsets in human LN have yet to be described.
Recent evidence suggests that the tissue environment in which DCs reside has a major impact on their phenotypic and functional properties. This is particularly apparent in the intestinal mucosa. Intestinal DCs are located throughout the villus lamina propria (LP) and in intestinal lymphoid tissue (Peyers Patches [PP], solitary isolated lymphoid tissue [SILT], and mesenteric LN [MLN]), where they play a central role in sampling and processing luminal as well as peripheral self antigen for presentation to T cells (11). Several functional properties have recently been ascribed to murine intestinal DCs. These include an enhanced ability to promote FoxP3+ regulatory T cell and IgA-secreting B cell differentiation and induction of gut-homing receptors (CCR9 and α4β7) on responding T and B cells (8, 12–17). Remarkably, these diverse functions appear, at least in part, to be driven through intestinal DC–mediated retinoic acid receptors (RAR) signaling. Thus, the vitamin A metabolite retinoic acid (RA) induces expression of gut-homing receptors and enhances FoxP3+ T cell and IgA B cell differentiation, whereas RAR antagonists block the ability of intestinal DCs to generate these populations (8, 18–20).
Only a subset of MLN DCs, which express the integrin α chain CD103, are efficient at inducing gut-homing receptors on responding T cells and FoxP3+ T cell differentiation in vitro (15–17). CD103+ MLN DCs express higher levels of aldh1a2, the gene encoding RALDH2 (15) which is a key enzyme involved in metabolizing retinal to RA, compared with their CD103− MLN counterparts. Consistent with this, CD103+ MLN DCs induce enhanced RAR dependent signaling in responding T cells, compared with splenic or CD103− MLN DCs (21). Despite these findings, the relationship between CD103+ and CD103− MLN DC subsets and the functional properties of these subsets in other tissues, including the small intestinal and colonic LP, remains unknown. Critically, it also remains to be determined whether these cells represent a functionally distinct population in human MLN and, thus, the potential of targeting CD103+ DCs for regulating intestinal immunity in humans is unclear.
RESULTS
Murine small intestinal LP and MLN CD103+ DCs are unique in their capacity to efficiently induce CCR9 and RAR signaling in responding T cells
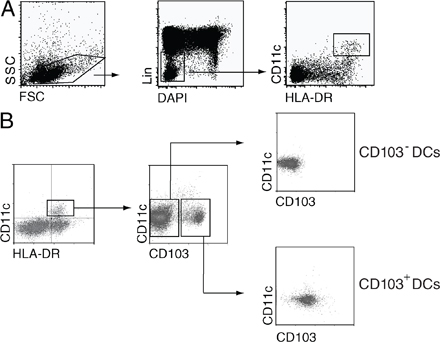
CD103+ DCs are present in a wide range of mucosal murine tissues and secondary lymphoid organs (Fig. 1 A; 15, 17, 22–25). Although CD103+ and CD103− DCs isolated from mucosal tissues (small intestine LP [SI-LP], colonic LP, and lung) were CCR7− CD40lo, the majority of CD103+ DCs in LN (MLN, lung draining mediastinal LN, and peripheral [axillary and brachial] LN) expressed CCR7 and high levels of CD40 and, thus, displayed a more mature phenotype than their peripheral counterparts and CD103− LN DCs (Fig. S1, A and B, available at http://www.jem.org/cgi/content/full/jem.20080414/DC1; and not depicted). To determine whether the ability to efficiently induce CCR9 on T cells is a feature of all CD103+ DCs, CFSE-labeled CD8+OT-I cells were primed with OVA peptide (pOVA; 2000 pM)–pulsed CD103+ and CD103− DCs from MLN, SI-LP, Colonic-LP, PP, and lung-draining LNs, and expression of tissue-homing receptors on responding T cells was assessed 6 d later. As expected, CD103+, but not CD103−, MLN DCs induced CCR9 on responding OT-I cells (Fig. 1 C). CD103+ SI-LP DCs also efficiently induced CCR9 on responding T cells, whereas CD103− SI-LP, CD103+ and CD103− colonic LP, PP, and lung-draining LN DCs failed to do so (Fig. 1, B and C). In contrast, all DC populations induced some α4β7 on responding OT-I cells (Fig. S2). CD103+ DCs and, in particular, intestinal CD103+ DCs induced higher levels of α4β7 compared with their CD103− counterparts. Although CD103− DCs induced less α4β7 expression, they were more efficient at inducing E-selectin ligand (which is required for effector cell migration to extraintestinal sites of inflammation) on responding T cells than CD103+ DCs, irrespective of their tissue origin (Fig. S2). We have recently demonstrated that OT-I cells express higher levels of CCR9 when primed with CD103+ and CD103− MLN DCs that have been pulsed with a low-dose peptide (21). Because CCR9 expression by OT-I cells, in addition to DCs, is also regulated by antigen dose, we next determined the ability of the different DC preparations to induce expression of CCR9 at a lower and more permissive peptide dose. When we pulsed DCs with a 10-fold lower peptide dose (200 pM), CD103+ PP and, to a lesser extent, CD103− PP DCs induced CCR9 on responding T cells, although the percentage of cells expressing CCR9 remained significantly lower than that observed after priming with CD103+ MLN DCs (Fig. 1 C). CD103+ colonic LP and lung-draining LN DCs also induced limited CCR9 expression under these conditions, whereas CD103− colonic LP, lung-draining LN, and splenic DC subsets failed to efficiently induce CCR9 (Fig. 1 C and not depicted). Thus, although the mechanism underlying the antigen dose-dependent regulation of CCR9 expression by OT-I cells and whether CCR9 expression is influenced by antigen dose in other TCR transgenic systems are currently not known, these results demonstrate that at a lower peptide dose, there is also an inherent difference between CD103+ and CD103− DCs, as well as between CD103+ DCs derived from different tissues, in their ability to efficiently confer CCR9 expression to OT-I cells.
Figure 1.

Small intestinal LP and MLN CD103+ DCs are unique in their capacity to efficiently induce CCR9 on responding T cells. (A) Percentage of DCs (MHC class II+CD11c+) expressing CD103 in indicated organs. Results are the mean and SD from three to eight independent experiments (two to five mice per experiment). (B and C) CD103+ and CD103− DCs were sorted from the indicated organs by FACS, pulsed with 2 nM (B and C) or 200 pM (C) pOVA257–264, and incubated with OT-I cells. CCR9 expression on responding OT-I cells was assessed after 6 d by flow cytometry. Representative FACS plots (B) and mean and SD (C) of the indicated number of separate experiments are shown. (D and E) CD103+ and CD103− DCs were sorted from the indicated organs by FACS, pulsed with 200 pM pOVA257–264, and incubated with DR5.OT-I cells. Luciferase activity was determined after 22 h of culture. Data are normalized to the luciferase expression observed in CD103+ MLN DC:DR5.OT-I cell cultures within each experiment. −, DR5.OT-I cells alone. Results are the mean and SD of the indicated number of separate experiments. (E) Luciferase activity was blocked when 1 μM of the pan-RAR antagonist LE540 was added to MLN DC-T cell cultures. Results are the mean and SD of three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with CD103+ MLN DCs). LDLN, Lung draining LN; Co-LP, colonic LP.
CD103+ MLN DCs express greater aldh1a2 levels compared with CD103− MLN DCs (15), and their efficiency in inducing CCR9 and, presumably, Foxp3+ T reg cell differentiation lies, at least in part, in an enhanced ability to induce RAR signaling in T cells (21). To determine whether CCR9 induction correlated with enhanced DC-mediated RAR signaling, OT-I cells from OT-I.DR5 transgenic mice (which express luciferase under control of three RA response element (DR5) repeats [reference 21]) were primed with pOVA (200 pM)-pulsed CD103+ or CD103− DCs, and luciferase activity in responding cells was assessed after 22 h (Fig. 1 D) at a time when OT-I cells have yet to divide (not depicted). We have previously demonstrated that DCs pulsed with 2,000 or 200 pM pOVA induce similar levels of RAR signaling in responding T cells (21). CD103+ MLN DCs induced significantly greater luciferase activity in OT-I.DR5 cells than their CD103+ PP and colonic counterparts (Fig. 1 D). In contrast, CD103+ SI-LP DCs induced similar levels of RAR signaling as CD103+ MLN DCs (Fig. 1 D). DC-induced luciferase activity was dependent on RAR signaling, as it was inhibited by addition of the pan-RAR antagonist LE540 to the cell cultures (Fig. 1 E). Thus, the ability of CD103+ MLN and SI-LP+ DCs to efficiently induce CCR9 correlates with an enhanced ability to induce RAR signaling in responding T cells. Although CD103+ DCs in general appeared to induce slightly enhanced luciferase activity compared with CD103− DCs (consistent with their enhanced ability to induce α4β7), these results demonstrate that the ability to induce high levels of RAR signaling and efficient CCR9 induction is not a general property of CD103+ DCs or intestinal DCs but is selective for CD103+ DCs that reside in the SI-LP and MLN.
CD103 is not required for intestinal DCs' ability to generate gut tropic T cells
The results in the previous section clearly demonstrate that CD103 signaling is not sufficient to imprint DCs with the ability to efficiently induce CCR9 and enhanced RAR signaling in responding T cells. However, it remained possible that CD103-mediated adhesion to E-cadherin on epithelial cells (26) or putative ligands in the small intestinal LP (27, 28) was required to expose CD103+ DCs sufficiently to imprinting signals. To assess this possibility, antigen-pulsed MLN DCs from CD103−/− and WT mice were assessed for their ability to induce CCR9 and α4β7 on responding DO11.10 cells (Fig. 2). From these experiments, it is clear that CD103−/− MLN DCs are as efficient as WT MLN DCs at inducing CCR9. Thus, CD103 is simply a marker of DCs in the SI-LP and MLN that have an enhanced ability to induce RAR signaling in responding T cells.
Figure 2.

CD103 is not required for imprinting MLN DCs with the ability to induce CCR9 on responding T cells. 105 MLN DCs from WT and CD103−/− BALB/c mice were cultured together with 2 × 105 CFSE-labeled DO11.10 SCID CD4+ cells, and OVA and expression of CCR9 and α4β7 was assessed 6 (experiment 1 [Expt. 1]) or 7 (experiment 2) d later. (A) Representative FACS plots. (B) Mean and SD of three mice per group for both experiments.
CD103+ SI-LP DCs derive from DC precursors that continually seed the intestinal LP
CD103 is induced on activated T cells subsequent to their entry into the intestinal mucosa (29–31). Given the differential ability of CD103+ and CD103− SI-LP DCs to induce an RAR signal and CCR9 on responding T cells, we next determined whether CD103+ SI-LP DCs derive directly from a DC precursor or from CD103− SI-LP DCs (MHC class II+ CD11c+ cells) that up-regulate CD103 in the SI-LP. Mice received a single injection of BrdU i.p., and the percentage of BrdU+ CD103+ and CD103− DCs in the SI-LP was determined over time (Fig. 3 A). 3 h after BrdU injection, 8% (± 5.1%) of CD103+ SI-LP DCs were BrdU+, and this number increased to 16% (± 6.5%) by 9 h before leveling off. In contrast, 7% (± 2.0%) of CD103− SI-LP DCs were BrdU+ at 3 h, and this percentage increased only slightly over the subsequent 24-h period (Fig. 3 A). BrdU+ CD103+ SI-LP DCs were Ki67− and, thus, not actively dividing (Fig. 3 B). Thus, there is a rapid turnover of both CD103+ and CD103− DCs in the SI-LP. Further, because CD103+ SI-LP DCs incorporated BrdU with at least as fast kinetics as CD103− DCs (even at 90 min after injection; not depicted), it appears that the majority of CD103+ SI-LP DCs do not derive from a CD103− SI-LP DC intermediate but from an actively proliferating DC precursor population. To examine whether the villus SI-LP DC compartment is replenished by blood-derived or tissue-resident DC precursors, SI from C57BL/6.CD45.2 mice was grafted into C57BL/6.CD45.1 mice (32). The proportions of host- and graft-derived MHC class IIhiCD11c+ DCs were then determined in the small intestinal mucosa of the graft and host after 6 and 45 d by immunohistochemistry (Fig. 3 C). At both time points, almost all DCs in the small intestinal villi of the grafted intestine were of host origin and at similar levels to those found in the host's own SI (Fig. 3 C). Together, these results suggest that the SI-villus DC compartment is not maintained by tissue resident precursors but from DC precursors that continually seed the SI-LP from the circulation.
Figure 3.

Turnover of SI-LP and MLN CD103+ and CD103− DC. (A) Mice were injected i.p. with 2 mg BrdU, and the percentage of BrdU+ CD103+ and CD103− DCs (MHC class II+CD11c+) in the SI-LP and MLN was determined by flow cytometry at the times indicated. Results are the mean and SD of three to seven independent experiments with two mice in each time point except the 72-h time point, which was performed once. (B) BrdU and Ki67 staining on SI-LP and MLN CD103+ and CD103− DCs (MHC class II+ CD11c+) was assessed by flow cytometry 3 and 24 h after BrdU injection. Plots are from one representative experiment of three performed. (C) SI from CD45.2+ mice (graft) was transplanted into CD45.1+ recipients (host) as previously described (32). At days 6 and 45, host and graft intestine were sectioned and stained with antibodies to CD45.2 (red), CD11c (blue), and MHC class II (green). Immunohistochemistry of the 45-d graft is shown. Arrows point to host-derived CD45.2−MHC class IIhiCD11c+ DCs. Bar, 25 μm. The graph is percentage of host-derived (CD45.2 negative) CD11c+MHC class IIhi cells in graft and host small intestinal villus. Results are mean and SD (n = 3 mice per group).
CD103+ and CD103− SI-LP DCs are found primarily in villus LP and solitary isolated lymphoid tissue, respectively
Suspensions of LP cells generated by enzymatic digestion of intestine likely contain DCs from both villus and small-sized lymphoid follicles, which are also referred to as solitary intestinal lymphoid tissue (SILT). Indeed, more than 1,000 SILT are interspersed throughout the small intestinal mucosa, and DCs constitute a major population in these structures. We therefore examined whether the CD103+ and CD103− subsets of DCs in our intestinal preparations could be differentially derived from these sites. Consistent with our previous results (17), CD103+ DCs (MHC class IIhiCD11c+ cells) were detected in the villus LP. In addition, CD103+ DCs were observed in the subepithelial dome of SILT (Fig. 4 A). Quantitative analysis demonstrated that almost all MHC class IIhiCD11c+ cells in the villus expressed CD103, whereas the majority of MHC class IIhiCD11c+ cells in the dome region of SILT were CD103− (Fig. 4 B). Of note, numerous CD11c+ cells that failed to stain or stained very weakly with anti–MHC class II antibody (CD11c+ MHC classsIIlo/− cells) were present within the villus LP (Fig. 4 B), which is consistent with previous results (17); however, these cells did not express CD103 and their function and ontogeny remains to be determined.
Figure 4.

Localization of CD103+ and CD103− MHC class II+CD11c+ cells within the small intestinal mucosa. (A and B) Expression of CD103 by DCs in small intestinal villus and SILT. (A) Images show overlays of CD103 (red) in combination with CD11c (blue) and MHC class II (green), with insets showing DAPI staining. Arrows point toward MHC class IIhiCD11c+ DCs coexpressing CD103. Bars, 50 μm. (B) The graph is percentage of MHC class IIhiCD11c+ cells in SI villus or dome region of SILT that express CD103. Results are mean and SD (n = 5 mice per group). ***, P < 0.001.
The majority of CD103+ and CD103− MLN DCs appear to represent tissue-derived migratory and lymphoid-resident populations, respectively
Because most CD103+ SI-LP DCs appear not to derive from CD103− SI-LP DCs and because CD103+ MLN DCs induced greater RAR signaling in T cells compared with CD103− MLN DCs, we determined whether CD103+ and CD103− were of similar origin in MLN. Mice received a single i.p. injection of BrdU, and the appearance of BrdU+ CD103+ and CD103− DCs in the MLN was assessed over time. BrdU+ CD103+ DCs appeared in the MLN with much slower kinetics than in the SI-LP, with their numbers peaking at 48 h (Fig. 3 A). This is consistent with the idea that the majority of CD103+ DCs in the MLN represent a tissue-derived migratory population that enter the MLN via the afferent lymph (17, 33). In marked contrast, the bulk population of BrdU+ CD103− DCs accumulated within the MLN with similar, if not more rapid, kinetics than in the SI-LP, with their numbers peaking 9 h after BrdU injection (Fig. 3 A). Remarkably, most BrdU+ CD103− MLN DCs were also Ki67+ at 3 h and were thus actively proliferating (Fig. 3 B). The percentage of CD103− MLN DCs that were Ki67+ remained constant (∼6%) throughout the period of the experiment, whereas the proportion of Ki67− BrdU+ cells increased between 3 and 24 h as BrdU-labeled cells exited the cell cycle. Together, these results demonstrate that the majority of CD103+ and CD103− MLN DCs are of different origin. Although most CD103+ MLN DCs appear to represent a tissue-derived migratory population, most CD103− MLN DCs appear to be maintained through continual and local homeostatic proliferation and, potentially, through recruitment of blood-derived precursors.
CD103+ MLN DCs play a key role in presenting orally administered soluble antigen to CD8+ and CD4+ T cells
Although antigen-pulsed CD103+ and CD103− MLN DCs can both prime CD4+ and CD8+ T cells in vitro (15, 17, 22), their distinct origins suggested that each subset might have distinct antigen-presenting function in vivo. To address this question, mice were immunized with OVA orally, killed 17 h later, and the ability of sorted CD103+ and CD103− MLN DCs to prime OT-I or OT-II T cell was assessed in vitro. After oral OVA administration, only CD103+ MLN DCs induced OT-I and OT-II cell proliferation (Fig. 5 A). To determine whether this was caused by a general inability of CD103− MLN DCs to process and present antigen to T cells, similar experiments were performed after i.p. OVA immunization. Under these conditions, both CD103+ and CD103− MLN DCs were capable of inducing OT-I and OT-II proliferation (Fig. 5 A). Notably, CD103− DCs consistently induced more OT-II cell proliferation than CD103+ DCs (Fig. 5 A and not depicted). Thus, although both DC populations are capable of presenting and cross-presenting antigen to CD8+ and CD4+ T cells, CD103+ MLN DCs are of primary importance in driving T cell responses to orally administered soluble antigen. Because the majority of CD103+ MLN DCs appear to represent an LP-derived migratory population and OT-I cells are not primed in the MLN of CCR7−/− mice after oral OVA administration (17), these results further suggest that CD103+ DC migration from the LP is required to drive T cell responses to luminal soluble antigen in the MLN.
Figure 5.

CD103+ DCs present orally administered soluble antigen to CD4+ and CD8+ T cells. (A and B) Mice received PBS 5 mg OVA i.p., or 50 mg orally, and MLN were harvested after 17 h. 105 CD103+ and CD103− MLN DCs were isolated by cell sorting and cocultured with 2 × 105 CFSE-labeled OT-I or OT-II cells. (A) CFSE profile of OT-I and OT-II cells 3.5 d after coculture with indicated DCs. Data are representative of three to five separate experiments using MLN DCs isolated from 15–20 pooled mice per group. (B) Expression of CCR9 and α4β7 was analyzed by FACS after 3.5 d in coculture. The graph shows mean and SEM from five independent experiments. *, P < 0.05 (when comparing CD103+ and CD103− DC–primed cultures).
Finally, because both DC subsets isolated from i.p.-immunized mice induced some OT-I cell proliferation, we compared induction of gut-homing receptors on responding cells (Fig. 5 B). OT-I cells primed with CD103+ DCs expressed significantly higher levels of CCR9 and high levels of α4β7 compared with OT-I cells primed with CD103− DCs (Fig. 5 B). The limited induction of CCR9 on CD103− DC-primed OT-I cells likely reflects the level of peptide being presented by these cells, because CD103− MLN DCs pulsed with low peptide dose are capable of inducing some CCR9 on responding OT-I cells (21).
CD103+ DCs are present in human MLN and efficiently induce CCR9 on responding T cells
Finally, to determine whether studies of murine CD103+ MLN DCs were translatable to the human, we determined whether CD103+ DCs are present in human MLN. CD103+ DCs were readily detected in MLN draining the normal SI, although the percentage of CD103+ DCs showed considerable variation (Fig. 6, A and B; for gating strategy see Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20080414/DC1). As in the mouse (17), human CD103+ MLN DCs displayed a more mature phenotype compared with their CD103− counterparts, as judged by expression of CD40 and CD83 (Fig. 6, C and D). CD103+ DCs were also readily detected in MLN draining the inflamed SI of Crohn's patients (Fig. 6 B). To assess the functionality of CD103+ and CD103− MLN DCs, these subsets were purified from pooled MLN draining normal SI (Fig. S3 B) and incubated together with allogeneic CFSE-labeled PBL. The expression of CCR9 and α4β7 on responding CD8+ T cells was assessed after 7 d (Fig. 7, A–C). As in the mouse, human CD103+ MLN DCs induced significantly higher levels of CCR9 on responding CD8+ T cells than their CD103− counterparts (Fig. 7, A and B). In contrast, both DC populations induced similar expression of α4β7 (Fig. 7, A and C). To determine whether the selective ability of CD103+ MLN DCs to induce CCR9 was maintained in chronic small intestinal inflammatory disease, similar studies were performed with sorted MLN DCs from Crohn's patients. Because these LN were considerably enlarged, pooling of LN was not required. As with DCs from normal MLN, only CD103+ DCs efficiently induced CCR9 on responding T cells (Fig. 7 B), whereas both populations efficiently induced α4β7 (Fig. 7 C). Finally, induction of CCR9 and α4β7 by human CD103+ MLN DCs was dependent on RAR signaling as gut-homing receptor expression was inhibited by addition of LE540 to the cultures (Fig. 7 D). Together, these results demonstrate a remarkable cross-species preservation of the phenotypic and functional properties of CD103+ MLN and demonstrate that these functional properties are maintained in Crohn's disease.
Figure 6.

Phenotypic characterization of human CD103+ and CD103− MLN DCs. (A) Human MLN section stained with anti–DC-LAMP (FITC), CD103 (Cy3), and IgG (Cy5) antibody to identify B cell follicles for orientation in the LN. CD103+ DCs (yellow). Bar, 100 μm. (B) Percent of DCs expressing CD103 in normal and Crohn's MLN, as determined by flow cytometry using the DC gatings described in Fig. S2 A (available at http://www.jem.org/cgi/content/full/jem.20080414/DC1). Filled squares, normal MLN (bladder reconstruction patients; n = 9); open squares, colon cancer patients (n = 4); triangles, SB Crohn's patients (n = 6). (C and D) CD40 and CD83 expression on CD103+ and CD103− normal MLN DCs using the gatings described in Fig. S2 A (available at http://www.jem.org/cgi/content/full/jem.20080414/DC1). Representative FACS plots (C) and mean and SD (D) of three (CD83) or four (CD40) independent experiments. **, P < 0.01. Results are normalized within each experiment to the percent expression on CD103+ DCs.
Figure 7.

Human MLN CD103+ DCs efficiently induce CCR9 on responding T cells. (A–C) CCR9 and α4β7 expression on CD8+ T cells primed with CD103+ or CD103− DCs from normal (A–C) or SB Crohn's (B and C) MLN. DCs were sorted as described in Materials and methods and Fig. S2 B (available at http://www.jem.org/cgi/content/full/jem.20080414/DC1). (A) Representative flow cytometry plots. (B) CCR9 expression on T cells stimulated with CD103+ (empty symbols) or CD103− MLN DCs (filled symbols). Each symbol represents a separate experiment and horizontal line represents the mean value. Boxes in A show the gates that were used to assess CCR9 expression. (C) α4β7 expression on T cells stimulated with CD103+ or CD103− MLN DCs. Normal, mean and SD of four experiments; Crohn's, mean of two experiments. Because α4β7 is induced in a cell cycle–dependent manner (12, 13, 54), α4β7 expression was assessed on cells that had divided six to nine times (arrows in A). (D) Gut-homing receptor induction by CD103+ DCs from normal human MLN was inhibited by addition of 1 μM of the pan-RAR antagonist LE540 to the cell cultures. Results are mean and SD of four separate experiments. ***, P < 0.001.
DISCUSSION
Recent studies have highlighted an important role for intestinal DCs (12, 14, 18) and, in particular, intestinal CD103+ DC (8, 15–17, 21) in inducing RAR signaling and gut-homing receptor expression on responding T cells and FoxP3+ Treg differentiation. In this paper, we show that SI-LP and MLN CD103+ DCs induce greater RAR signals and more efficiently induce CCR9 in responding T cells than other CD103+ or CD103− DC populations. We provide evidence that most CD103+ SI-LP DCs develop directly from a circulating DC precursor, and not from CD103− SI-LP DCs, and that the majority of CD103+ MLN DCs represent a SI-LP–derived migratory population that plays a critical role in presenting orally derived soluble antigen to T cells. Thus, these cells presumably capture orally applied antigens locally in the SI and subsequently migrate into the MLN. In contrast, most CD103− MLN DCs appear to represent a blood-derived LN population that can proliferate in the MLN and present systemic antigen to T cells. Critically, we also demonstrate that CD103+ DCs are present in normal and inflamed human MLN and display similar phenotypic and functional properties to their murine counterparts. Thus, CD103+ MLN DCs represent a potential novel target population for regulating human intestinal immune responses.
Our current results demonstrate that although all DCs appear to be capable of inducing limited RAR signaling in T cells, the ability to induce high RAR signaling and efficient CCR9 expression is not a general property of CD103+ DCs and, notably, not of CD103+ Co-LP DCs but of SI-LP and MLN CD103+ DCs. As the latter population appears to derive from the former (see subsequent Discussion paragraphs), these results argue for a selective imprinting of CD103+ DCs, or their immediate precursors, in the SI-LP. In this regard, vitamin A itself is primarily absorbed through the SI (34), and incubation of monocyte-derived DCs with RA imprinted on these cells the ability to induce CCR9 on responding T cells (35). This activity was blocked with a RAR antagonist, but not a RALDH inhibitor, leading to the suggestion that CCR9 induction may be mediated by RA that has been taken up and stored by DCs. Nevertheless, incubation with high concentration of RA (1,000 nM) was required for this effect, and the in vivo relevance of these findings remains unclear. CD103+ SI-LP and MLN DCs express higher levels of aldh1a2 (unpublished data) (15) than their CD103− counterparts, suggesting that their enhanced ability to induce RAR signaling may lie in an increased capacity to metabolize retinal. Although the signals inducing aldh1a2 expression and the link between aldh1a2 expression and DC-mediated RAR signaling remains unclear, addition of IL-4 to MLN DC-T cell cultures was recently found to enhance CCR9, but not α4β7, expression on responding CD4+ T cells (36). Notably, MLN DCs from IL-4rα−/− mice displayed a reduced capacity to induce CCR9 on CD4+ T cells and had reduced levels of aldh1a2 (36), although the composition of MLN DCs in these mice was not assessed. Moreover, CD8+ T cells in the SI-LP of IL-4rα−/− expressed normal levels of CCR9. In the current study, the inability of CD103− SI-LP DCs to induce enhanced RAR signaling suggests that imprinting signals are not ubiquitously expressed in the SI-LP or that CD103− SI-LP DCs are incapable of responding to such signals. Notably, although CD103 expression on DCs appears to play a critical functional role in T cell–mediated regulation of CD45RBhi T cell–induced colitis (22), our experiments with CD103−/− MLN DCs demonstrate that CD103 is not required for imprinting intestinal DCs with the ability to generate gut tropic T cells.
Irrespective of the imprinting mechanism, the similar initial kinetics of BrdU+ cells within the CD103+ and CD103− LP DC populations suggest that CD103+ SI-LP DCs are not the progeny of CD103− SI-LP (MHC class II+CD11c+) DCs. Moreover, our intestinal transplant experiments suggest that CD103+ SI-LP DCs do not derive from resident proliferating precursors within the LP but are continually replenished by blood-borne precursors seeding the LP. This result, however, should be interpreted with a degree of caution, as we cannot at this point rule out that inflammation associated with the transplant surgery triggers the release of donor-derived DC precursors that are replaced by precursors of host origin. In the rat, small numbers of DCs were found in intestinal lymph that originated from adoptively transferred blood monocytes (37), indicating that at least some intestinal-LP migratory DCs are monocyte derived. Consistent with this possibility, murine CD11b+Gr1+ monocytes generate CD11c+ LP cells when transferred into LP-DC–depleted recipient mice (38), although further phenotypic analysis of the resulting population was not performed. It also remains to be determined whether CD103+ or CD103− SI-LP DCs derive from distinct DC precursors in the steady state or whether the same DC precursor population can generate both subsets depending on the microenvironmental signals they receive. The former possibility is supported by the recent observation that CCR2+Ly6Chi and CX3CR1+Ly6Clo monocyte subsets replenish CD103+ and CD103− lung DCs, respectively (39).
Our current results also provide strong evidence that the majority of CD103+ and CD103− MLN DCs derive from distinct populations of DCs. The delayed appearance of BrdU+ CD103+ DCs in MLN compared with the SI-LP is compatible with the idea that most of these cells represent a tissue-derived migratory population. Consistent with this hypothesis, CD103+ DC numbers are reduced in the MLN of mice lacking CCR7, a chemokine receptor which is required for DC migration from peripheral tissues into the draining LN (17, 24, 40, 41). CD103+ Langerin+ DCs are present in the lung (23) and are reduced in numbers in the LDLN of CCR7−/− mice (24, 25). Furthermore, a radiosensitive CD103+ Langerin+ DC population was recently identified in the skin dermis (42–44) that migrates to the skin, draining LN in the steady state in a CCR7-dependent manner (42–44). Together, these results suggest that most CD103+ DCs in intestinal and extraintestinal LN represent tissue-derived migratory DCs.
In marked contrast to CD103+ DCs, BrdU+ CD103− DCs accumulated with at least as rapid kinetics in the MLN as in the SI-LP, suggesting that the majority of these cells in MLN represent a resident population. In addition, most CD103− LN DCs displayed an immature phenotype under steady state, a proposed characteristic of LN-resident DCs (45, 46). Several mechanisms have been reported to help maintain the splenic and LN-resident DC compartment in the steady state. These include continuous recruitment of DC precursors from the blood and differentiation from tissue resident DC precursors, as well as a limited expansion of DCs themselves (47–50). We found that ∼6% of CD103− DCs in the MLN were Ki67+ at any one time and were thus actively proliferating. Further, in combined Ki67 and BrdU staining, ∼60% of BrdU+ CD103− DCs were Ki67+ 3 h after BrdU administration, suggesting that in the steady state, the CD103− DC population is maintained in part through homeostatic proliferation and in part through the entry of DC precursors from the blood.
Our observations that only CD103+ MLN DCs induce OT-I and OT-II proliferation after oral OVA administration demonstrate a key role for these cells in cross-presenting orally derived soluble antigen and confirm the importance of these cells in presenting oral antigen to CD4+ T cells (15). We further show that this is not caused by a defect in the ability of CD103− MLN DCs to present antigen, as these cells primed OT-I and, especially, OT-II cells after i.p. administration of antigen. These results, together with previous observations that OT-I and OT-II cells fail to respond to orally administered OVA in CCR7−/− mice but respond to i.p. OVA (17, 51), provides further evidence that CD103+ MLN DCs represent the major tissue-derived migratory DC population that presents soluble luminal antigen to T cells.
We have previously demonstrated that CCR9 is more efficiently induced on adoptively transferred OT-I cells in the MLN after oral compared with i.p. OVA administration (12). Thus, efficient induction of CCR9 correlates with enhanced antigen presentation by CD103+ DCs. Further, OT-I cells primed in the MLN of CCR7−/− mice after administration of OVA and adjuvant i.p. fail to express CCR9 (12). Thus, reduced levels of CD103+ DCs in the MLN correlate with reduced CCR9 induction. Together, these correlations strongly indicate that CD103+ MLN DCs are required for efficient CCR9 induction in vivo. Although α4β7 induction is also greatest under conditions of highest CCR9 induction, α4β7 is also induced on OT-I cells after i.p. OVA administration alone in both WT and CCR7−/− mice. Because induction of α4β7 is RAR dependent (21), these results suggest that RAR signaling provided by additional cells in the MLN is sufficient to induce some α4β7 expression on T cells primed at this site. Indeed, α4β7 is induced on proliferating antigen-specific T cells in the MLN after i.p. injection of antigen-loaded BM or peripheral LN DCs (52–54). Determining the precise role of CD103+ MLN DC–mediated RAR signaling in the generation of gut tropic T cells and FoxP3+ T reg cell differentiation will likely require analysis of immune responses in mice selectively depleted of this unique subset of DCs.
To our knowledge, the phenotype and function of distinct DC subsets in human intestinal LN has not previously been examined. Given the unique functional characteristics of murine CD103+ MLN DCs and their potentially important role in regulating intestinal immune responses, it was of central importance to determine the relevance of this population in humans. CD103+ DCs were readily detected in MLN, draining both healthy SI and the SI of patients with Crohns disease. These cells, as in the mouse, displayed a more mature phenotype than their CD103− counterparts. Total human MLN, but not splenic or liver, DCs have previously been shown to efficiently induce gut-homing receptors on responding B cells through an RAR-dependent mechanism (8). Importantly, we demonstrate that the ability to induce CCR9 is not a general property of human MLN DCs but, again, selective for CD103+ MLN DCs. Moreover, the selective ability of CD103+ MLN DCs to induce CCR9 was maintained in Crohn's disease patients. Together, these results highlight the relevance of studying this population in murine models of intestinal homeostasis and inflammation and the future possibility of targeting these cells to regulate human intestinal immune responses in health and disease.
MATERIALS AND METHODS
Mice and human tissues
C57BL/6 (Taconic), DR5 (provided by R. Blomhoff, Institute of Basic Medical Science, University of Oslo, Norway), OT-I, OT-II, and DR5.OT-I mice (21) were bred and maintained at the Biomedical Center animal facility (Lund University, Lund, Sweden). BALB/c, CD103−/− BALB/c, and DO11.10 mice were bred and maintained in animal Facilities at the University of Oxford (Oxford, England, UK). Mice between 6 and 14 wk of age were used for experiments. Animal experiments were performed in accordance with guidelines from Lund/Malmö Animal Ethics Committee, the review board of the Hannover Medical School, or the UK Animals (Scientific Procedures) Act of 1986 where appropriate.
Normal MLN were obtained from patients undergoing bladder reconstruction by cystectomy (n = 9) or surgery for colon cancer (n = 4). Inflamed MLN were obtained from patients with SB Crohn's disease (n = 6). All tissues were obtained with informed patient consent and in accordance with local ethical approval from the Regional Ethics committee in Lund (Sweden) and the South Birmingham Research Ethics committee (England, UK).
Reagents
R10 medium contained the following reagents, all from Invitrogen: RPMI-1640, 10% FCS, 10 mM Hepes, 1 mM sodium pyruvate, 50 μM mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamicin. HBSS was obtained from Invitrogen. Synthetic pOVAs (OT-I, SIINFEKL; and OT-II, ISQAVHAAHAEINEAGR) were obtained from Innovagen. OVA grade VII, LPS (Escherichia coli, serotype 055:B55), DNase I, and collagenase type IV and VIII were obtained from Sigma-Aldrich. OVA was purified using Detoxi-Gel Endotoxin removal gel (Thermo Fisher Scientific). CFSE (Sigma-Aldrich) and LE540 (Wako Chemicals USA, Inc.) were dissolved in DMSO.
Cell staining reagents
The following reagents were used to stain murine cells: unconjugated or directly conjugated anti-CD103 (M290), anti-α4β7 (DATK32), anti-CD62L (MEL-14), anti-MHC class II (2G9), anti-CD11c (N418), and rat IgG2a and IgG2b isotype controls (BD Biosciences); perCP-Cy5.5, Alexa 700–conjugated anti-CD11c (N418), and Pacific blue–conjugated anti–MHC Class II (M5/114.15.2; BioLegend); anti-CCR7 (4B12; eBiosciences); recombinant mouse E-selectin human Fc chimera (R&D Systems); anti-FcRII/III (2.4G2), anti-CD40 (FGK45), anti-CD86 (GL1), and anti-CCR9 antibody (7E7, (56); purified from hybridomas); Cy5-labeled donkey anti–human IgG F(ab′)2 (Jackson ImmunoResearch Laboratories); biotinylated mouse anti–rat IgG2a (RG7/1.30), biotinylated mouse anti–rat IgG2b (G15-337), PE-labeled Ki67, streptavidin-conjugated PE, PECy7, PerCP Cy5.5, and the BrdU FITC flow kit (BD Biosciences); streptavidin–alexa 488 (Invitrogen); and DAPI (Sigma-Aldrich).
The following reagents were used to stain human DCs: APC-conjugated anti-CD40 (5C3), APC-Cy7–conjugated anti–HLA-DR (L243), PE-Cy5–conjugated anti-β7 (FIB504), and APC-Cy7–conjugated anti-CD8 (SK7; BD Biosciences); PE-Cy7–conjugated anti-CD11c (3.9), biotinylated or PE-Cy5–conjugated anti-CD83 (HB15e), PE-conjugated anti-CD103 (BLy7), APC/PE/PE-Cy5– or biotin-conjugated IgG1 isotype controls, and FITC-conjugated anti-CD3 (UCHT1), -CD14 (61D3), -CD16 (CB16), -CD19 (HIB19), and -CD56 (MEM188; eBiosciences); PE-Cy7–conjugated anti-CD3 (UCHT1; Beckman Coulter); APC-conjugated anti-CCR9 (R&D Systems); PE-conjugated anti-CD8 (DK2) and FITC/PE-conjugated anti-CD103 (Ber–ACT8; Dako); FITC/Cy7-conjugated anti-CD11c (BU15) and FITC/Cy5-conjugated anti–HLA-DR (YE2/36HLK; AbD Serotec); and mouse anti–human α4β7 (ACT-1; provided by M. Briskin, Millennium Pharmaceuticals, Inc.).
Murine cell isolation and flow cytometry analysis
Murine T cells and DCs were purified by MACS as previously described (17). T cell purity was 90–95%. DCs were ∼95% CD11c+ and 90% CD11c+MHC class II+. For CD103+ and CD103− DC purification, MACS-enriched CD11c+ DCs were sorted using anti-CD11c, anti-CD103, and anti–MHC class II on a FACSAria (BD Biosciences; 17). DC fractions were routinely 95–99% pure. In Fig. 1 (B–E), DCs were isolated from mice injected with FMS-like tyrosine kinase-3 ligand (Flt3L)-secreting B16 melanoma cells, as previously described (17). Such treatment does not influence the selective ability of CD103+ MLN DCs to induce CCR9 or the phenotype of intestinal DCs (17, 21). CFSE labeling of T cells and flow cytometry analysis was performed as previously described (55). Data acquisition was performed on a FACSCalibur or FACSAria (BD Biosciences) and analyzed using FlowJo software (Tree Star, Inc.).
Human cell isolation and flow cytometry analysis
Lymphocytes were isolated from peripheral blood using LymphoLyte-H (Cedarlane Laboratories.). Human MLN DCs were isolated using one of two protocols: (a) MLN were cut into small pieces and incubated in R10 with 500 μg/ml collagenase IV and 50 U/ml DNase I for 10 min at 37°C on an orbital shaker (Innova 2300; New Brunswick Scientific) at 250 rpm. Remaining tissue was mechanically crushed and, together with the cell suspension, passed through a 70-μm cell strainer. Cells were stained and either analyzed or sorted using a FACSAria. (b) MLN pieces were mechanically disaggregated in HBSS supplemented with 5% FCS and 5 mM EDTA (Sigma-Aldrich) at 230 rpm for 5 min in a circulator (Stomacher 400; Seward). The resulting sample was passed through a fine mesh and washed with HBSS. Supernatant was mixed with OptiPrep (AxisShield) 1:3 and overlayered with 11.5% OptiPrep and HBSS. Samples were centrifuged at 600 g for 20 min and the DC fraction was harvested. Remaining lymphocytes were depleted with anti-CD3 dynabeads (Invitrogen) and DCs isolated by CD11c expression using EasySep (StemCell Technologies Inc.). CD11c+ HLA-DR+ cells were FACS sorted into CD103+ and CD103− DC fractions. Both protocols yielded similar proportions of CD103+ and CD103− MLN DCs. Data acquisition was performed using FACSAria or Cyan flow cytometer (Dako) and data analyzed using FlowJo or Summit software (Dako).
In vitro and ex vivo cell cultures
Murine cell cultures.
DCs were pulsed with 2 nM or 200 pM SIINFEKL peptide for 1 h at 37°C, washed extensively, and incubated (105 cells) with 2 × 105 CFSE-labeled OT-I cells in flat bottom 96-well plates (Thermo Fisher Scientific) in 200 μl of complete R10 medium. The phenotype of responding OT-I cells was assessed by flow cytometry after 3.5 d or after expansion for an additional 2 d as previously described (21). For luciferase measurements, DR-5.OT-I cells and antigen-pulsed DCs were cocultured and luciferase activity was assessed after 22 h as previously described (21). For ex vivo DC cultures, CD103+ and CD103− MLN DCs were FACS sorted from OVA-immunized mice and cultured (105 cells) together with 2 × 105 CFSE-labeled OT-I cells for 3.5 d before analysis.
Human cell cultures.
CFSE-labeled PBL were cultured together with 5 × 105 allogeneic CD103− or CD103+ MLN DCs at a 1:1 ratio in R10 medium supplemented with 400 IU/ml IL-2 (Proleukin). Proliferation and CCR9/α4β7 expression on CD8+ T cells were measured after 7 d by flow cytometry.
Small bowel transplantation
The small bowel transplantation was performed as previously described (32).
Immunohistochemistry
Staining of mouse tissue.
Staining was performed as previously described (17) using CD11c-APC (N418), CD103-PE (M290), MHC class II–biotin (2G9), DAPI, and streptavidin–Alexa 488.
Staining of human tissue.
Acetone-fixed MLN sections were blocked with 10% NGS (Sigma-Aldrich) and avidin-biotin blocking kit (Vector Laboratories). Tissue was stained with anti–DC-LAMP (104.G4; Beckman Coulter) followed by FITC-labeled goat anti–mouse antibody (Jackson ImmunoResearch Laboratories). After washing and blocking with 10% NMS, sections were stained with biotin anti-CD103 (28C12) and visualized using a tyramide signal amplification kit (PerkinElmer Life Science) and Cy3-conjugated streptavidin (Biosite). Sections were costained with Cy5-labeled donkey anti–human IgG F(ab′)2 (Jackson ImmunoResearch Laboratories). Images were acquired with a microscope (Axiovert 200M; Carl Zeiss, Inc.) and Volocity software (Improvision) or AxioVision 4.0 software (Carl Zeiss, Inc.).
Statistical analysis
Statistical analyses were performed using paired or unpaired two-tailed Student's t test with Welch correction or one sample t test where appropriate.
Online supplemental material
Fig. S1 shows the flow cytometry gating strategy used for identifying CD103+ and CD103− DCs and the maturation phenotype of these populations in different tissues. Fig. S2 shows the ability of CD103+ and CD103− DC subsets to induce α4β7 and E-selectin ligand on responding T cells. Fig. S3 displays the flow cytometry gating strategy used to phenotype and sort human CD103+ and CD103− intestinal DCs. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20080414/DC1.
Supplementary Material
Acknowledgments
The authors would like to thank Professor R. Blomhoff (University of Oslo, Norway) and Cgene (Cgene AS, Oslo, Norway) for providing the DR5-luciferase mice. We also wish to thank Professors A. Mowat (University of Glasgow, UK) and D. Adams (Birmingham University, UK) for valuable input, Dr T. Ismail (Birmingham University) and Prof W. Månsson (Lund University Hospital) for intestinal samples, and Ann-Charlotte Selberg for valuable technical assistance.
W.W. Agace is supported by grants from the Swedish Medical Research Council, the Crafoordska, Wenner-Gren, Österlund, LEO Pharma A/S, Åke Wiberg, Nanna Svartz, and Kocks Foundations, the Royal Physiographic Society, the Wellcome Trust, and the Swedish Foundation for Strategic Research INGVAR II program. F. Powrie is funded by the Wellcome Trust and J.L. Coombes by an MRC studentship.
The authors have no conflicting financial interests.
Abbreviations used: LP, lamina propria; MLN, mesenteric LN; pOVA, OVA peptide; PP, Peyer's patch; RA; retinoic acid; RAR, retinoic acid receptor; SI, small intestine; SI-LP, small intestinal LP; SILT, solitary isolated lymphoid tissue.
References
- 1.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 2.Steinman, R.M., and H. Hemmi. 2006. Dendritic cells: translating innate to adaptive immunity. Curr. Top. Microbiol. Immunol. 311:17–58. [DOI] [PubMed] [Google Scholar]
- 3.Henri, S., D. Vremec, A. Kamath, J. Waithman, S. Williams, C. Benoist, K. Burnham, S. Saeland, E. Handman, and K. Shortman. 2001. The dendritic cell populations of mouse lymph nodes. J. Immunol. 167:741–748. [DOI] [PubMed] [Google Scholar]
- 4.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 5.Pulendran, B., J.L. Smith, G. Caspary, K. Brasel, D. Pettit, E. Maraskovsky, and C.R. Maliszewski. 1999. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. USA. 96:1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villadangos, J.A., and P. Schnorrer. 2007. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 7:543–555. [DOI] [PubMed] [Google Scholar]
- 7.Cox, K., M. North, M. Burke, H. Singhal, S. Renton, N. Aqel, S. Islam, and S.C. Knight. 2005. Plasmacytoid dendritic cells (PDC) are the major DC subset innately producing cytokines in human lymph nodes. J. Leukoc. Biol. 78:1142–1152. [DOI] [PubMed] [Google Scholar]
- 8.Mora, J.R., M. Iwata, B. Eksteen, S.Y. Song, T. Junt, B. Senman, K.L. Otipoby, A. Yokota, H. Takeuchi, P. Ricciardi-Castagnoli, et al. 2006. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 314:1157–1160. [DOI] [PubMed] [Google Scholar]
- 9.O'Mahony, L., L. O'Callaghan, J. McCarthy, D. Shilling, P. Scully, S. Sibartie, E. Kavanagh, W.O. Kirwan, H.P. Redmond, J.K. Collins, and F. Shanahan. 2006. Differential cytokine response from dendritic cells to commensal and pathogenic bacteria in different lymphoid compartments in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 290:G839–G845. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi, K., K. Asagoe, J. Zaishun, H. Yanai, T. Yoshino, K. Hayashi, and T. Akagi. 1998. Heterogeneity of dendritic cells in human superficial lymph node: in vitro maturation of immature dendritic cells into mature or activated interdigitating reticulum cells. Am. J. Pathol. 153:745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johansson, C., and B.L. Kelsall. 2005. Phenotype and function of intestinal dendritic cells. Semin. Immunol. 17:284–294. [DOI] [PubMed] [Google Scholar]
- 12.Johansson-Lindbom, B., M. Svensson, M.A. Wurbel, B. Malissen, G. Marquez, and W. Agace. 2003. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J. Exp. Med. 198:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stagg, A.J., M.A. Kamm, and S.C. Knight. 2002. Intestinal dendritic cells increase T cell expression of alpha4beta7 integrin. Eur. J. Immunol. 32:1445–1454. [DOI] [PubMed] [Google Scholar]
- 14.Mora, J.R., M.R. Bono, N. Manjunath, W. Weninger, L.L. Cavanagh, M. Rosemblatt, and U.H. Von Andrian. 2003. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 424:88–93. [DOI] [PubMed] [Google Scholar]
- 15.Coombes, J.L., K.R. Siddiqui, C.V. Arancibia-Carcamo, J. Hall, C.M. Sun, Y. Belkaid, and F. Powrie. 2007. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid–dependent mechanism. J. Exp. Med. 204:1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun, C.M., J.A. Hall, R.B. Blank, N. Bouladoux, M. Oukka, J.R. Mora, and Y. Belkaid. 2007. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204:1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johansson-Lindbom, B., M. Svensson, O. Pabst, C. Palmqvist, G. Marquez, R. Forster, and W.W. Agace. 2005. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 202:1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwata, M., A. Hirakiyama, Y. Eshima, H. Kagechika, C. Kato, and S.Y. Song. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 21:527–538. [DOI] [PubMed] [Google Scholar]
- 19.Kang, S.G., H.W. Lim, O.M. Andrisani, H.E. Broxmeyer, and C.H. Kim. 2007. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J. Immunol. 179:3724–3733. [DOI] [PubMed] [Google Scholar]
- 20.Mora, J.R., G. Cheng, D. Picarella, M. Briskin, N. Buchanan, and U.H. von Andrian. 2005. Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. J. Exp. Med. 201:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Svensson, M., B. Johansson-Lindbom, F. Zapata, E. Jaensson, L. Austenaa, R. Blomhoff, and W.W. Agace. 2008. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunology. 1:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Annacker, O., J.L. Coombes, V. Malmstrom, H.H. Uhlig, T. Bourne, B. Johansson-Lindbom, W.W. Agace, C.M. Parker, and F. Powrie. 2005. Essential role for CD103 in the T cell–mediated regulation of experimental colitis. J. Exp. Med. 202:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sung, S.S., S.M. Fu, C.E. Rose Jr., F. Gaskin, S.T. Ju, and S.R. Beaty. 2006. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176:2161–2172. [DOI] [PubMed] [Google Scholar]
- 24.Hintzen, G., L. Ohl, M.L. del Rio, J.I. Rodriguez-Barbosa, O. Pabst, J.R. Kocks, J. Krege, S. Hardtke, and R. Forster. 2006. Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J. Immunol. 177:7346–7354. [DOI] [PubMed] [Google Scholar]
- 25.del Rio, M.L., J.I. Rodriguez-Barbosa, E. Kremmer, and R. Forster. 2007. CD103- and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J. Immunol. 178:6861–6866. [DOI] [PubMed] [Google Scholar]
- 26.Cepek, K.L., S.K. Shaw, C.M. Parker, G.J. Russell, J.S. Morrow, D.L. Rimm, and M.B. Brenner. 1994. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 372:190–193. [DOI] [PubMed] [Google Scholar]
- 27.Schon, M.P., A. Arya, E.A. Murphy, C.M. Adams, U.G. Strauch, W.W. Agace, J. Marsal, J.P. Donohue, H. Her, D.R. Beier, et al. 1999. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J. Immunol. 162:6641–6649. [PubMed] [Google Scholar]
- 28.Strauch, U.G., R.C. Mueller, X.Y. Li, M. Cernadas, J.M. Higgins, D.G. Binion, and C.M. Parker. 2001. Integrin alpha E(CD103)beta 7 mediates adhesion to intestinal microvascular endothelial cell lines via an E-cadherin-independent interaction. J. Immunol. 166:3506–3514. [DOI] [PubMed] [Google Scholar]
- 29.Ericsson, A., M. Svensson, A. Arya, and W.W. Agace. 2004. CCL25/CCR9 promotes the induction and function of CD103 on intestinal intraepithelial lymphocytes. Eur. J. Immunol. 34:2720–2729. [DOI] [PubMed] [Google Scholar]
- 30.Lefrancois, L., C.M. Parker, S. Olson, W. Muller, N. Wagner, M.P. Schon, and L. Puddington. 1999. The role of β7 integrins in CD8 T cell trafficking during an antiviral immune response. J. Exp. Med. 189:1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Asady, R., R. Yuan, K. Liu, D. Wang, R.E. Gress, P.J. Lucas, C.B. Drachenberg, and G.A. Hadley. 2005. TGF-β–dependent CD103 expression by CD8+ T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J. Exp. Med. 201:1647–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pabst, O., H. Herbrand, T. Worbs, M. Friedrichsen, S. Yan, M.W. Hoffmann, H. Korner, G. Bernhardt, R. Pabst, and R. Forster. 2005. Cryptopatches and isolated lymphoid follicles: dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur. J. Immunol. 35:98–107. [DOI] [PubMed] [Google Scholar]
- 33.Agace, W.W. 2006. Tissue-tropic effector T cells: generation and targeting opportunities. Nat. Rev. Immunol. 6:682–692. [DOI] [PubMed] [Google Scholar]
- 34.Yeum, K.J., and R.M. Russell. 2002. Carotenoid bioavailability and bioconversion. Annu. Rev. Nutr. 22:483–504. [DOI] [PubMed] [Google Scholar]
- 35.Saurer, L., K.C. McCullough, and A. Summerfield. 2007. In vitro induction of mucosa-type dendritic cells by all-trans retinoic acid. J. Immunol. 179:3504–3514. [DOI] [PubMed] [Google Scholar]
- 36.Elgueta, R., F.E. Sepulveda, F. Vilches, L. Vargas, J.R. Mora, M.R. Bono, and M. Rosemblatt. 2008. Imprinting of CCR9 on CD4 T cells requires IL-4 signaling on mesenteric lymph node dendritic cells. J. Immunol. 180:6501–6507. [DOI] [PubMed] [Google Scholar]
- 37.Yrlid, U., C.D. Jenkins, and G.G. MacPherson. 2006. Relationships between distinct blood monocyte subsets and migrating intestinal lymph dendritic cells in vivo under steady-state conditions. J. Immunol. 176:4155–4162. [DOI] [PubMed] [Google Scholar]
- 38.Varol, C., L. Landsman, D.K. Fogg, L. Greenshtein, B. Gildor, R. Margalit, V. Kalchenko, F. Geissmann, and S. Jung. 2007. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 204:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jakubzick, C., F. Tacke, F. Ginhoux, A.J. Wagers, N. van Rooijen, M. Mack, M. Merad, and G.J. Randolph. 2008. Blood monocyte subsets differentially give rise to CD103+ and CD103- pulmonary dendritic cell populations. J. Immunol. 180:3019–3027. [DOI] [PubMed] [Google Scholar]
- 40.Jang, M.H., N. Sougawa, T. Tanaka, T. Hirata, T. Hiroi, K. Tohya, Z. Guo, E. Umemoto, Y. Ebisuno, B.G. Yang, et al. 2006. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J. Immunol. 176:803–810. [DOI] [PubMed] [Google Scholar]
- 41.Ohl, L., M. Mohaupt, N. Czeloth, G. Hintzen, Z. Kiafard, J. Zwirner, T. Blankenstein, G. Henning, and R. Forster. 2004. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 21:279–288. [DOI] [PubMed] [Google Scholar]
- 42.Bursch, L.S., L. Wang, B. Igyarto, A. Kissenpfennig, B. Malissen, D.H. Kaplan, and K.A. Hogquist. 2007. Identification of a novel population of Langerin+ dendritic cells. J. Exp. Med. 204:3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulin, L.F., S. Henri, B. de Bovis, E. Devilard, A. Kissenpfennig, and B. Malissen. 2007. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J. Exp. Med. 204:3119–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ginhoux, F., M.P. Collin, M. Bogunovic, M. Abel, M. Leboeuf, J. Helft, J. Ochando, A. Kissenpfennig, B. Malissen, M. Grisotto, et al. 2007. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J. Exp. Med. 204:3133–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson, N.S., D. El-Sukkari, G.T. Belz, C.M. Smith, R.J. Steptoe, W.R. Heath, K. Shortman, and J.A. Villadangos. 2003. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood. 102:2187–2194. [DOI] [PubMed] [Google Scholar]
- 46.Shortman, K., and S.H. Naik. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7:19–30. [DOI] [PubMed] [Google Scholar]
- 47.Zhang, M., H. Tang, Z. Guo, H. An, X. Zhu, W. Song, J. Guo, X. Huang, T. Chen, J. Wang, and X. Cao. 2004. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat. Immunol. 5:1124–1133. [DOI] [PubMed] [Google Scholar]
- 48.Kabashima, K., T.A. Banks, K.M. Ansel, T.T. Lu, C.F. Ware, and J.G. Cyster. 2005. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 22:439–450. [DOI] [PubMed] [Google Scholar]
- 49.Liu, K., C. Waskow, X. Liu, K. Yao, J. Hoh, and M. Nussenzweig. 2007. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat. Immunol. 8:578–583. [DOI] [PubMed] [Google Scholar]
- 50.Geissmann, F. 2007. The origin of dendritic cells. Nat. Immunol. 8:558–560. [DOI] [PubMed] [Google Scholar]
- 51.Worbs, T., U. Bode, S. Yan, M.W. Hoffmann, G. Hintzen, G. Bernhardt, R. Forster, and O. Pabst. 2006. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 203:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dudda, J.C., J.C. Simon, and S. Martin. 2004. Dendritic cell immunization route determines CD8+ T cell trafficking to inflamed skin: role for tissue microenvironment and dendritic cells in establishment of T cell-homing subsets. J. Immunol. 172:857–863. [DOI] [PubMed] [Google Scholar]
- 53.Dudda, J.C., A. Lembo, E. Bachtanian, J. Huehn, C. Siewert, A. Hamann, E. Kremmer, R. Forster, and S.F. Martin. 2005. Dendritic cells govern induction and reprogramming of polarized tissue-selective homing receptor patterns of T cells: important roles for soluble factors and tissue microenvironments. Eur. J. Immunol. 35:1056–1065. [DOI] [PubMed] [Google Scholar]
- 54.Sheasley-O'Neill, S.L., C.C. Brinkman, A.R. Ferguson, M.C. Dispenza, and V.H. Engelhard. 2007. Dendritic cell immunization route determines integrin expression and lymphoid and nonlymphoid tissue distribution of CD8 T cells. J. Immunol. 178:1512–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Svensson, M., J. Marsal, A. Ericsson, L. Carramolino, T. Broden, G. Marquez, and W.W. Agace. 2002. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J. Clin. Invest. 110:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pabst, O., L. Ohl, M. Wendland, M.A. Wurbel, E. Kremmer, B. Malissen, and R. Forster. 2004. Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. J. Exp. Med. 199:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}