

Abstract
The flavoprotein nitroalkane oxidase (NAO) catalyzes the oxidation of primary and secondary nitroalkanes to the corresponding aldehydes and ketones. The enzyme is a homolog of acyl-CoA dehydrogenase. Asp402 in NAO has been proposed to be the active site base responsible for removing the substrate proton in the first catalytic step; structurally it corresponds to the glutamate which acts as the base in medium chain acyl-CoA dehydrogenase. In the active site of NAO, the carboxylate of Asp402 forms an ionic interaction with the side chain of Arg409. The R409K enzyme has now been characterized kinetically and structurally. The mutation results in a decrease in the rate constant for proton abstraction of 100-fold. Analysis of the three-dimensional structure of the R409K enzyme, determined by X-ray crystallography to a resolution of 2.65 Å, shows that the critical structural change is an increase in the distance between the carboxylate of Asp402 and the positively-charged nitrogen in the side chain of the residue at position 409. The D402E mutation results in a smaller decrease in the rate constant for proton abstraction of 18-fold. The structure of the D402E enzyme, determined at 2.4 Å resolution, shows that there is a smaller increase in the distance between Arg409 and the carboxylate at position 402, and the interaction of this residue with Ser276 is perturbed. These results establish the critical importance of the interaction between Asp402 and Arg409 for proton abstraction by nitroalkane oxidase.
The flavoenzyme nitroalkane oxidase (NAO) catalyzes the oxidation of nitroalkanes to the corresponding aldehydes or ketones with consumption of molecular oxygen and release of nitrite and hydrogen peroxide (Scheme 1) (1). NAO is unusual in that it catalyzes substrate oxidation by removing a substrate proton to form a carbanion intermediate (1, 2), in contrast to the growing evidence that flavoproteins that oxidize carbon-nitrogen and carbon-oxygen bonds typically catalyze loss of the substrate hydrogen as a hydride (3–12). As is generally the case with flavoproteins, the catalytic mechanism of NAO can be divided into oxidative and reductive half reactions (13, 14). In the reductive half-reaction (Scheme 2), an active site base abstracts the α-proton from the neutral nitroalkane substrate to form a nitroalkane anion. The involvement of the substrate anion in the reaction has been demonstrated directly by the ability of the preformed anion to rescue mutant enzymes lacking the active site base (2). The nitroalkane anion then attacks the N5 position of the FAD, forming a flavin adduct that releases nitrite to generate a reactive cationic imine. This flavin imine cation has been trapped with both nitroalkane anion (15, 16) and cyanide (17), confirming its structure and its catalytic competency. During the normal course of catalysis, the electrophilic flavin imine reacts with hydroxide to eventually form reduced FAD and the aldehyde or ketone product. The oxidative half-reaction of NAO is more typical for a flavoprotein oxidase (18); molecular oxygen reacts with the reduced FAD to form hydrogen peroxide and regenerate the oxidized cofactor in a second-order reaction with no detectable intermediates (14).
With the slow substrate nitroethane (19), formation of the substrate anion by NAO is rate-limiting for the reductive half-reaction (20). The nonenzymatic formation of a nitroalkane anion from a nitroalkane has long been studied as a model for proton abstraction from carbon (21, 22). Consequently, analysis of the oxidation of nitroalkanes by NAO has allowed comparison of the enzyme-catalyzed reaction with a well-characterized nonenzymatic reaction (23). NAO catalyzes the ionization of nitroethane 109-fold more effectively than acetate. Analysis of the temperature dependence of the deuterium kinetic isotope effects on the NAO and acetate-catalyzed reactions has established that the contribution of quantum mechanical tunneling to catalysis is comparable and small for both the enzymatic and nonenzymatic reactions.
The cloning and sequencing of NAO led to its unexpected identification as a homolog of the acyl-Co dehydrogenase (ACAD) family of flavoproteins (24). In retrospect, this assignment was consistent with the similarities in the initial catalytic reactions of the two enzymes; in both cases a protein carboxylate abstracts an acidic proton from the substrate (1, 25). The recent determination of the three-dimensional structure of NAO (16) confirmed this assignment and provided additional details regarding the structural basis for the differences in reactivity of NAO and ACAD. Critically, while both the cofactors and the active sites in NAO and ACAD occupy similar locations in the different proteins, the substrates appear to access the active sites from opposite sides. Comparison of the structures shows that NAO Asp402 occupies the same space in the active site as the glutamate residue that is the active site base in the medium and short chain acyl-CoA dehydrogenases (24, 26). The role of Asp402 in NAO has been analyzed previously by site-directed mutagenesis. Mutagenesis to glutamate decreases the rate constant for abstraction of the nitroalkane proton by 15-fold (14), while mutagenesis to alanine or asparagine decreases it by at least 1000-fold (14).
The nearest residues to Asp402 are Arg409, positioned with an ε nitrogen 3.6 Å from a carboxylate oxygen of Asp402, and Ser276, positioned within hydrogen-bonding distance of Asp402 (Figure 1). The distance from Asp402 to Arg409 is appropriate for an electrostatic interaction, suggesting that Arg409 is involved in correctly positioning the active site base for catalysis. We report here the results of kinetic and structural analyses of the effects of site-directed mutagenesis of Arg409 of NAO in order to investigate its contribution to catalysis. In addition, the structure of the D402E enzyme is described, providing further insight into interactions of Asp402 critical for catalysis.
Figure 1.
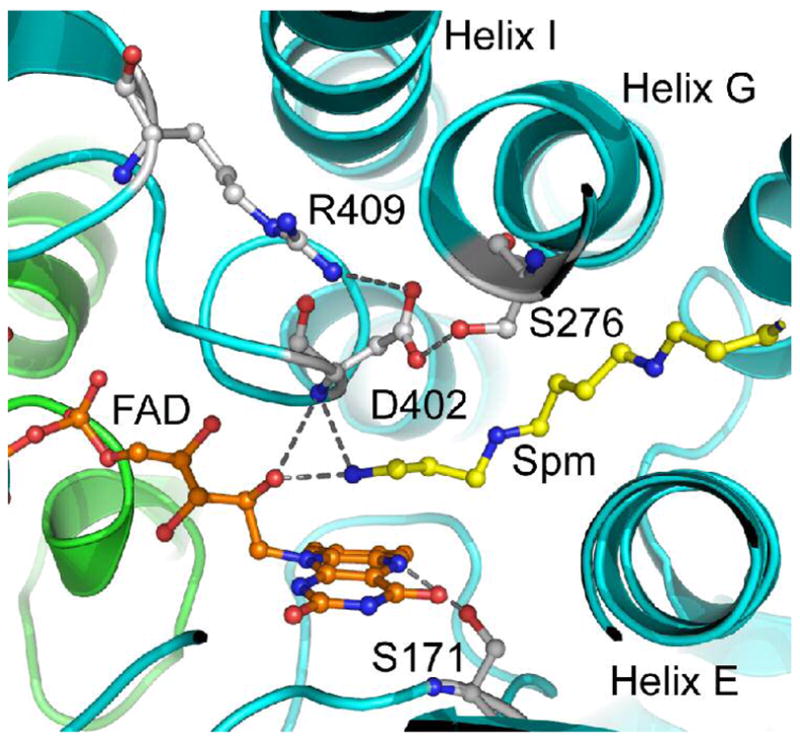
The active site of NAO (PDB file 2C12). The ribbon traces for subunits A and B are colored green and cyan, respectively. The FAD, spermine (Spm) and selected residues are colored with carbon atoms in orange, yellow or gray, respectively. The oxygen and nitrogen are colored in red and blue, respectively. Selected hydrogen bonds are illustrated with gray dashed lines.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were purchased from Sigma-Aldrich Chemical Corp. (Milwaukee, WI). Recombinant nitroalkane oxidase was expressed and purified as previously described (24). The D402E enzyme was purified as previously described (14). The codon for Arg409 was mutated to lysine, alanine, and glutamine using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), and the mutant enzymes were expressed and purified following the protocol for the wild-type enzyme (24). DNA sequencing of the entire coding sequence of each mutant plasmid was performed at the Laboratory for Plant Genome Technologies of Texas A&M University. Enzyme concentrations were determined using an ε446 value of 14.2 mM−1cm−1 (27).
Kinetic Analyses
Enzyme activity was routinely measured in 200 mM HEPES, 0.1 mM FAD, pH 8.0, 30 °C, by monitoring oxygen consumption with a computer-interfaced Hansatech Clark oxygen electrode (Hansatech Instruments, Pentney King’s Lynn, U.K.). When varying the concentration of nitroethane, the steady state kinetic parameters were determined at ambient oxygen concentrations. To vary the concentration of oxygen at a constant concentration of nitroethane, oxygen and argon were combined in different ratios with a MaxBlend low flow air/oxygen blender (Maxtec Inc., Salt Lake City, Utah), and the assay buffer was equilibrated with the gas mixture. To prevent the formation of the anionic form of the substrate, stock solutions of neutral nitroethane were prepared in dimethyl sulfoxide and assays were initiated by the addition of substrate.
Steady-state kinetic data were analyzed using the programs KaleidaGraph (Adelbeck Software, Reading, PA) and Igor Pro (WaveMetrics, Inc., Lake Oswego, OR). Steady state kinetic parameters were determined by fitting the data to the Michaelis-Menten equation or to equation 1, which includes the effect of substrate inhibition by nitroethane. The parameter v is the initial velocity, kcat is the maximal velocity, Km is the Michaelis constant, S is the substrate concentration, and Kai is the substrate inhibition constant. Steady state kinetic isotope effects were calculated from equation 2, where Fi is the fraction of deuterium in the substrate and Dkcat and D(kcat/Km) are the isotope effects on kcat and kcat/Km, respectively.
| (1) |
| (2) |
Structure Determination
Hexagonal rod-shaped crystals of R409K and D402E NAO were obtained using hanging drop vapor-diffusion methods similar to those described previously (16, 28). Briefly, crystals of R409K NAO were grown using a mother liquor solution containing 28–30% (w/v) PEG 4000, 20% (v/v) glycerol, 10% (v/v) 1,6-hexanediol, 100 mM sodium cacodylate, pH 7.5, and 2 mM spermine hydrochloride. Crystals of D402E NAO were grown from 24–30% (w/v) PEG 3350, 25% (v/v) glycerol, 100 mM sodium cacodylate, pH 7.5, and 2 mM spermine hydrochloride. The crystals often appeared within 24 hours at 277 K. They were mounted directly with nylon loops without further cryoprotection and were flash frozen by quick-submersion in liquid nitrogen. X-Ray diffraction data collection was at beam line X26c, X25 or X29 of the National Synchrotron Light Source with crystals held at 100 K. Data were integrated and scaled with either HKL2000 (29) or MOSFLM and SCALA (30). The diffraction data for crystals of both mutant proteins indicated hexagonal unit cells with a = b = 108 Å, c =340 Å. The structures were solved by molecular replacement with either PHASER or MolRep from the CCP4 suit of programs (31). Initially the search models comprised a monomer extracted from the wild-type NAO structure (PDB code 2c0u) (16) from which all solvent and FAD atoms were removed. Each of the potential hexagonal space groups was evaluated by PHASER, searching for at least four monomers. The solution indicated that the crystals were in space group P3221 with one α4 holoenzyme in the asymmetric unit. Model refinement was with REFMAC 5.2 using loose noncrystallographic symmetry restraints for the four monomers in the asymmetric unit. Data collection and model refinement statistics are presented in Table 2. Each structure was confirmed with at least two independent crystals and datasets. Only the highest quality structure for each is presented here.
Table 2.
Data collection and refinement statistics
| NAO Isoform | R409K | D402E |
|---|---|---|
| Wavelength (Å) | 0.9795 | 1.0809 |
| Space group | P3221 | P3221 |
| a (Å), b (Å), c (Å) | 108.3, 108.3, 340.0 | 108.9, 108.9, 337.7 |
| Resolution range (Å) | 50 - 2.65 | 50 - 2.40 |
| High resolution shell (Å) | 2.74 - 2.65 | 2.46 - 2.40 |
| Total observations | 371118 | 144782 |
| Unique observations | 81166 | 81048 |
| Completeness (%) | 97.1 (93.2)a | 88.2 (53.7)a |
| Redundancy | 5.6 (3.4)a | 4.1 (2.2)a |
| Rsym (%) | 12.8 (58.7)a | 8.8 (41.4)a |
| I/σ | 17.1 (1.9)a | 22.3 (1.3)a |
| Model refinement statistics | ||
| Number protein residues | 1720 | 1720 |
| Number ligands/water molecules | 4/0 | 4/56 |
| Resolution range (Å) | 50 - 2.65 | 50 - 2.40 |
| Number of reflections | 73886 | 76997 |
| Rcryst (%) | 22.8 (35.1)a | 20.3 (29.0)a |
| Rfree (%) | 27.7 (39.7)a | 26.3 (39.7)a |
| Ramachandran Statistics | ||
| Most favored | 1377 (91.5%) | 1377 (91.6%) |
| Additional allowed | 118 (7.8%) | 117 (7.7%) |
| Generously allowed | 4 (0.3%) | 6 (0.4%) |
| Disallowed | 6 (0.4%) | 4 (0.3%) B402A |
| Average B values (Å2) | ||
| Protein | 39.84 | 44.07 |
| Water | - | 37.95 |
| FAD | 35.32 | 36.32 |
| R.m.s.d. from ideal geometry | ||
| Bond lengths (Å) | 0.022 | 0.022 |
| Bond angles (°) | 1.986 | 1.985 |
Highest resolution shell.
RESULTS
Kinetic characterization of R409K and D402E NAO
In order to evaluate the contribution of Arg409 of NAO to catalysis, this residue was mutated to alanine, glutamine, and lysine. Only the R409K mutation yielded soluble protein under any of the growth conditions examined, precluding characterization of the effects of the other two mutations. While primary nitroalkanes containing four or more carbons are the best substrates for NAO, with nitroethane as substrate CH bond cleavage is rate-limiting for the reductive half-reaction; binding steps dominate the kinetics of the longer substrates (20). Accordingly, nitroethane was used as substrate to characterize the effects of the mutation. The steady state kinetic behavior for R409K NAO when the concentration of nitroethane is varied resembles that of the wild-type enzyme, in that substrate inhibition is also seen at high concentrations of nitroethane with the mutant protein (32). The steady state kinetic parameters of NAO R409K for both nitroethane and oxygen as substrates are listed in Table 1. The mutation has the largest effect on the kcat/Km value for nitroethane, which is only 1.6 % of the wild-type value. The pH dependence of the kcat/Kne value resembles that of the wild-type protein, in that the activity drops off below a pKa of 6.8 ± 0.1, compared to the value of 6.9 ± 0.1 seen in wild-type NAO (2) (results not shown). The kcat and the kcat/KO2 values are both affected much less by the mutation, decreasing to about one-sixth of the wild-type values. As a further probe of the effects of the mutation on individual rate constants, deuterium kinetic isotope effects were determined with [1,1-2H2]nitroethane. The wild-type enzyme exhibits a large isotope effect on the kcat/Km value for nitroethane and a relatively small effect on the kcat value (14). With the R409K enzyme, the best fit of the data is with identical isotope effects of 8.3 on the kcat and kcat/Kne values (Table 1); this value is not significantly different from the isotope effect on the kcat/Km value for nitroethane for the wild-type enzyme of 9.2 (14). The isotope effects suggest that the mutation has a much greater effect on the reductive half reaction than on subsequent steps, so that CH bond cleavage has become fully rate-limiting for turnover.
Table 1.
Steady state kinetic parameters for R409K and D402E nitroalkane oxidasea
| kinetic parameter | wild-type NAOb | R409K | D402Eb |
|---|---|---|---|
| kcat (s−1) | 15 ± 1 | 2.6 ± 0.3 | 3.07 ± 0.06 |
| kcat/Kne (mM−1s−1) | 6.3 ± 0.4 | 0.098 ± 0.006 | 0.21 ± 0.01 |
| Kne (mM) | 2.3 ± 0.2 | 26.3 ± 4.6 | 14.6 ± 0.8 |
| Kai(mM) | 25 ± 3 | 152 ± 57 | - c |
| kcat/KO2 (mM−1s−1) | 310 ± 30 | 46 ± 12 | 158 ± 7 |
| KO2 (μM) | 82 ± 9 | 42 ± 13 | 19 ± 1 |
| Dkcat | 1.4 ± 0.2 | 8.3 ± 1.8 | 3.6 ± 0.7 |
| D(kcat/Kne) | 9.2 ± 1.1 | 8.3 ± 1.8 | 9.6 ± 2.3 |
Conditions: pH 8.0, 30 °C.
From reference (14), with the exception of the oxygen kinetics for D402E.
Substrate inhibition was not observed with the D402E enzyme.
The effects on individual steps in catalysis of mutating Asp402 of NAO have been described previously (2, 14). The steady state kinetic parameters for the D402E enzyme are summarized in Table 1. As is the case for the R409K mutation, the reductive half-reaction is affected the most, as reflected in the greater decrease in the kcat/Km value for nitroethane compared to the effect on the kcat/Km value for oxygen or the kcat value.
Crystal Structures of R409K and D402E NAO
In order to provide a more complete understanding of the effects of mutating Arg409 and Asp402, we determined the three-dimensional structures of R049K and D402E NAO by X-ray crystallography. The data collection and refinement statistics are reported in Table 2, and the electron density maps for the active site structures of the two mutant proteins are shown in Figure 2. Both mutant enzymes crystallize in space group P3221; however, the unit cells have smaller c axes than the wild-type enzyme (16), so that the asymmetric units contain only one homotetramer, while crystals of the wild-type enzyme contain one tetramer and two subunits of a second. Despite the slightly different packing, the two structures are essentially identical to the structure of the wild-type enzyme (r.m.s.d. 0.29–0.38 Å). The overall active site structure is not perturbed in either mutant protein, and the atoms of the FAD cofactor occupy identical positions in the wild-type and mutant enzymes (Figure 3). Loose noncrystallographic symmetry restraints were applied for the four monomers in the asymmetric unit rather than averaging the structures across all four subunits. As a result, as noted below, there are some differences in the structures of the individual active sites. However, these are limited to the sites of the mutations.
Figure 2.
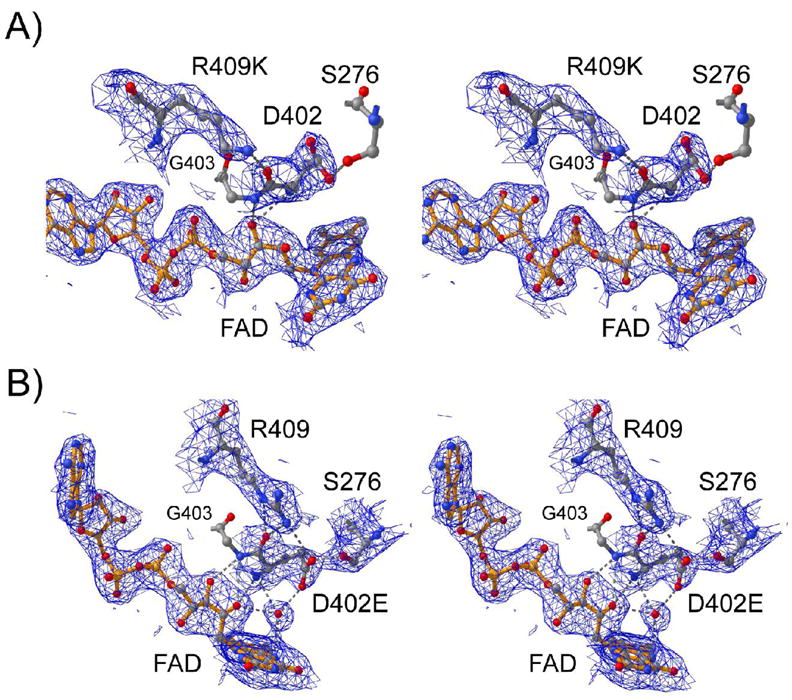
The refined 2Fo−Fc electron density maps (1σ) at 2.65 Å and 2.5 Å resolution for the R409K (A) and D402E (B) enzymes in the active site region of subunit A. The bonds for the FAD are shown in orange, whereas carbon, oxygen and nitrogen atoms are colored in gray, red and blue, respectively. Selected hydrogen bonds are illustrated with gray dashed lines.
Figure 3.
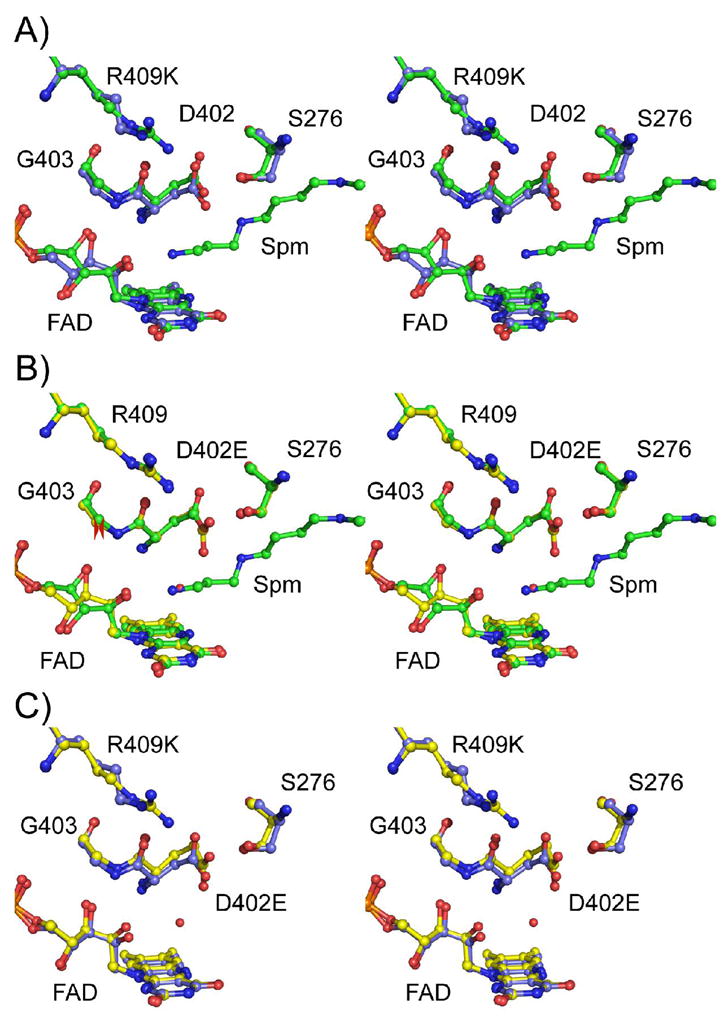
The active site structures of wild-type (PDB code 2c12, subunit D), R409K and D402E NAO in divergent stereo views. A) Overlay of R409K (blue bonds) and wild-type (green bonds) NAO; B) overlay of D402E (yellow bonds) and wild-type (green bonds) NAO; and C) overlay of R409K (blue bonds) and D402E(yellow bonds) NAO.
In the wild-type enzyme, the NH2 of Arg409 is ~3.6 Å from carboxylate OD1 of Asp402, the active site base, an appropriate distance for a strong electrostatic interaction (Figure 4A). In the mutant enzyme the shorter side chain of lysine moves the positive charge more than 1 Å further away from Asp402 (Figure 4B). In the structure of the wild-type enzyme, four of the subunits in each asymmetric unit have a molecule of spermine in the active site, and two have no active site ligand; despite this, the atoms of the active site residues have the same conformation in all six active sites. The distances between the residues given in Figure 4 for the wild-type enzyme are for subunit A, but these vary less than 0.1 Å among all six subunits in the 2.07 Å resolution structure. In contrast, we observe some variability in the relative positions of the lysyl N□ atoms in the four subunits of the R409K enzyme. In all four the β, γ, and δ carbons of lysine occupy the same positions as the corresponding atoms of Arg409 in the wild type enzyme (Figure 3A). In contrast, the lysine Cε occupies slightly different positions in the different subunits, and there is even greater divergence in the positions of the lysyl Nζ atom (Figure S1 in Supplementary Information). In the B and D subunits, the lysyl Nζ atoms project toward Asp402, whereas in the A and C subunits the nitrogen atoms project toward Leu408. However, these differences are most likely due to uncertainties in the positions of these atoms at the present resolution (2.65 Å) and the fewer hydrogen bond interactions with a lysine sidechain compared to an arginine. The averaged electron density maps fit each Lys409 configuration equally well (Figure S1) with an electron density correlation of 0.62, 0.59, 0.62, and 0.58 for the A-D subunits, respectively.2 Consistent with this analysis, the B-factors for the Cε and Nζ atoms average 37.1 ± 1.8 Å2 for the four subunits, whereas the backbone atoms have an average B-factor of 31.6 ± 0.5 Å2 in the four subunits. The distance from the Nζ atom of Lys409 to the carboxylate OD1 of Asp402 varies among the four subunits from 3.9 to 5.2 Å, with an average distance of 4.7 ± 0.6 Å. Thus, while the position of the lysyl Nζ atom is less well-defined than the remaining active site atoms, the shortest possible distance from it to the Asp OD1 is clearly significantly longer than the wild-type distance of ~3.6 Å.
Figure 4.
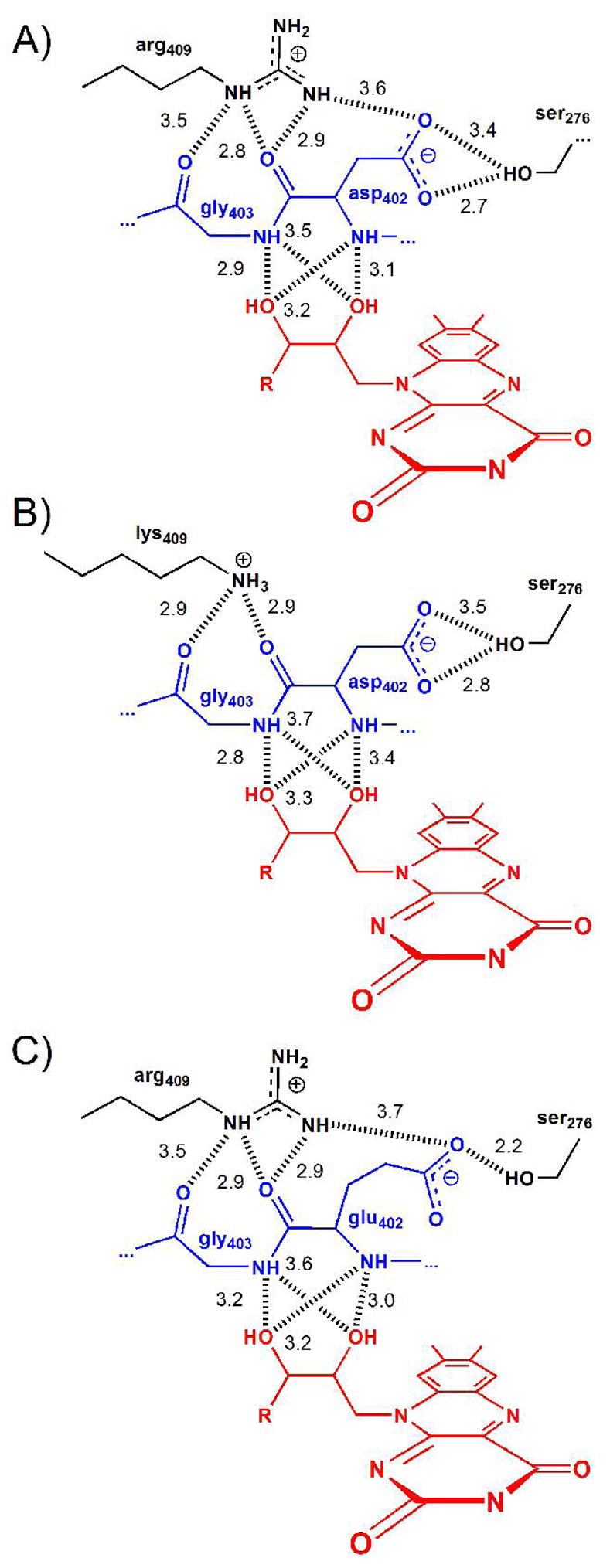
Possible hydrogen bonding interactions and distances deduced from the structures of A) wild-type, B) R409K, and C) D402E NAO.
The hydrogen bonding interactions of the lysyl nitrogen in the R409K enzyme resemble those of the guanidyl Nε in the wild-type enzyme. In the structure of wild-type NAO, the guanidyl group of Arg409 forms hydrogen bonds with the carbonyl oxygen atoms of Gly403 and Asp402 (Figure 4A). In R409K NAO, the average distances from the lysyl nitrogen to these residues in the four subunits are 3.0 ± 0.3 Å to the carbonyl oxygen of Asp402 and 3.8 ± 0.7 Å to the Gly403 oxygen, essentially the same distances seen for Arg409 N□ in the wild-type enzyme, 2.8 and 3.5 Å, respectively. Effectively, the mutation has removed the dimethylamino moiety from arginine. Overall, the R409K mutation conserves the active site structure remarkably well in that the change can be described relatively accurately as a greater distance between the positive charge at position 409 and the carboxylate of Asp402. As a result, the interaction between these residues would be significantly weaker in the mutant protein.
The D402E mutation introduces an additional methylene group instead of truncating a charged side chain. It appears that the former change is easier to accommodate. The position of helix G (Figure 1) restricts the mobility of the side chain of residue 409, so that the carboxylate can only move closer to the flavin to adjust for the additional methylene. As a result, the additional methylene carbon in glutamate increases the distance between the carboxylate oxygen and the guanidyl group of Arg409 somewhat, although less dramatically than the R409K mutation. As is the case with the R409K enzyme, there is some variability in the position of the glutamyl side chain among the different subunits. The most dramatic variation is in subunit B, where the side chain is disordered beyond the Cβ so that no density for the carboxylate can be detected. In the other three subunits, the variability is limited to the angle of rotation of the carboxylate, in that the β, γ and δ carbons of the glutamyl side chain effectively occupy the same positions as the β and γ carbons and carboxylate OD2 of Asp402 (Figures 3 and S1). As a result the distance between the glutamate OE1 and NH2 of Arg409 has increased to an average distance of 4.2 ± 0.6 Å (Figure 4), within error of the wild-type distance of 3.5 Å. Unlike the R409K mutation, replacing Asp402 with glutamate also alters the interaction between the carboxylate of the side chain at that position and Ser276. In the wild-type enzyme there is a good hydrogen bond (2.7 Å) between the hydroxyl of Ser276 and carboxylate OD2 of Asp402. This is the oxygen of the active site base that is most appropriately placed to accept the proton from the substrate. This interaction between Asp402 and Ser276 is unchanged in the R409K enzyme. In contrast, in the D402E enzyme the longer glutamyl side chain results in a good hydrogen bond with OD1, at an average distance of 2.4 ± 0.3 Å, while the distance to OD2 has increased to 3.8 ±0.2.
DISCUSSION
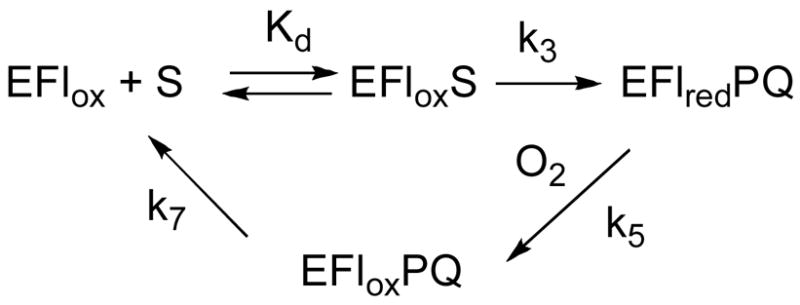
The kinetic mechanism of NAO (Scheme 3) has been established for the wild-type enzyme using a combination of steady state and rapid reaction kinetics (14). This mechanism provides a framework for analysis of the kinetic data for the R409K enzyme. With nitroethane as substrate for the wild-type enzyme, the rate-limiting step in the reductive half reaction is cleavage of the substrate CH bond with rate constant k3 (20). As a result, the deuterium isotope effect on the kcat/Km value for nitroethane, D(kcat/Kne), equals the intrinsic isotope effect for CH bond cleavage, Dk3 (20). Under these conditions, the kcat/Km value for nitroethane equals k3/Kd, with Kd the dissociation constant for nitroethane. For wild-type NAO, overall turnover at saturating concentration of nitroethane and oxygen is limited by product release from the oxidized enzyme, with rate constant k7 (14); equation 3 gives the relationship between the kcat value and k3 and k7. The relationship between the isotope effect on kcat, Dkcat, and the intrinsic isotope effect on the CH bond cleavage step, Dk3, can be derived from equation 3 and is given by equation 4. Because product release is 19-fold slower than reduction with the wild-type enzyme, the isotope effect on the kcat value is small. Oxygen reacts with the reduced enzyme with second-order kinetics, with no indication of saturation at accessible levels of oxygen (14). The value of k5, the rate constant for the reaction of the reduced enzyme with oxygen, is equivalent to the kcat/Km value for oxygen. The KO2 value does not reflect a true binding event; rather, it equals kcat/(kcat/KO2), a collection of rate constants as shown in equation 5.
| (3) |
| (4) |
| (5) |
Equations 3 and 4 allow the data of Table 1 to be used to calculate the values of the individual rate constants in Scheme 3 for the R409K mutant enzyme; these are given in Table 3. For this mutant enzyme, the Dkcat and D(kcat/Kne) values are equivalent and equal to the intrinsic isotope effect on k3 for the wild-type enzyme. Equation 4 shows that the Dkcat value is equal to Dk3 when k7≫k3. Under these conditions, equation 3 simplifies to kcat = k3, setting the value of k3 for the R409K enzyme as 2.6 s-1. The value of k3 for the wild-type enzyme has been measured previously using stopped-flow methods as 247 s−1 (14); thus, the mutation of Arg409 to lysine results in a decrease in the rate constant for CH bond cleavage of almost 100-fold. Because chemistry is rate-limiting with the mutant enzyme, it is only possible to place a lower limit on k7 of 13 based on the precision of the kcat and Dkcat values. This lower limit is close to the value for the wild-type enzyme, suggesting that product release is unaffected by the mutation. Thus, while the mutation only results in a decrease in kcat of about 6-fold, this is due to a much larger decrease in the rate constant for substrate oxidation. The effect on kcat is small because chemistry is not rate-limiting for turnover in the wild-type enzyme. Because kcat equals k3, the Km for the R409K enzyme equals the Kd value; this is close to the Kd value previously determined for the wild-type enzyme (14).
Table 3.
Intrinsic kinetic parameters for R409K and D402E nitroalkane oxidase
| kinetic parameter | wild-type NAO a | R409K | D402Ea |
|---|---|---|---|
| Kd (mM) | 14 ± 1 | 26 ± 5 | 22.8 ± 0.7 |
| k3 (s−1) | 247 ± 5 | 2.6 ± 0.3 | 16.3 ± 0.2 |
| k5 (mM−1s−1) | 310 ± 30 | 46 ± 12 | 158 ± 7 |
| k7 (s−1) | 17 | >13 | 11 ± 3 |
From reference (14).
Mutagenesis of Asp402 to glutamate has previously been shown to decrease the value of k3 by about 16-fold (14), while having little effect on the value of k5 or k7 (Table 3). In contrast, mutation of Asp402 to alanine decreases k3 by at least 1000-fold (2), demonstrating the importance of this residue.
The structures of the mutant proteins establish that these decreased activities are not due to a general disruption of the protein structure. In each case the structural change is localized to the mutated residue. Both the R409K and the D402E mutations alter the interaction between the positively charged residue at position 409 and the negatively charged active site base. The decrease in k3 correlates with the increase in the distance between these two residues. The R409K mutation results in both a larger increase in this distance and a larger decrease in k3 than does the D402E mutation. This is strong evidence for the importance of the interaction between Arg409 and Asp402 to abstraction of the substrate proton by NAO. However, the role of this interaction in catalysis is not clear. It is unlikely to be required for modulating the pKa of Asp402. Moving the positive charge away from Asp402, as in the R409K mutant protein, would be expected to raise the pKa of Asp402, making it more proficient as a base rather than less. In addition, the mutation does not appear to alter the pKa value of Asp402 from the wild-type value of 6.0 The perturbation in the position of the base is greater in the D402E enzyme while the effect is larger, suggesting that the decreased activity of the the R409K enzyme is due to the altered position of the positive charge rather than an undetected change in the position of Asp402.
The D402E mutation has a smaller effect on the distance between Arg409 and the glutamate carboxylate. Indeed, in at least one of the subunits the distance is similar to that seen in the wild-type enzyme, suggesting that the interaction may be intact in a significant fraction of the mutant enzyme. However, extending the side chain of the residue at position 402 has other effects. In contrast to R409K NAO, in the D402E enzyme the interaction with Ser276 is altered, in that the side chain hydroxyl of this residue forms a hydrogen bond with a different carboxylate oxygen in the mutant protein, and the position of the carboxylate oxygen which accepts the substrate proton is altered. Comparison of the structures of the mutant and wild-type proteins suggests that Ser276 is a critical active site residue involved in properly positioning Asp402. A BLAST search of the available protein sequences using the NAO residues from 402–409 as the probe identifies nine other fungal proteins variously annotated as acyl-CoA dehydrogenases or hypothetical proteins. These range from 32–53% identical in sequence to NAO. In all of these, Asp402, Gly403, and Arg409, are conserved, while the residue corresponding to Ser276 is either serine or threonine. Thus, these three residues, Asp402, Arg409, and Ser276, appear to constitute a catalytic triad in NAO. None of these residues is conserved in proteins clearly identifiable as members of the acyl-CoA dehydrogenase family (Figure S2), with the exception that Ser276 is present in the peroxisomal, short chain-specific, acyl-CoA oxidase (ACX4) from Arabidopsis thaliana (34). This suggests that these residues play a significant role in determining the ability of proteins with this structural fold to oxidize nitroalkanes. It appears that the active site structure evolved in NAO so that the side chain of Asp402 is ideally positioned between Arg409 and Ser276 in such a way that proton abstraction from substrate is enhanced. Although the thioester-acyl portion of acyl-CoA resembles the nitro-alkane portion of nitroalkanes, the Michaelis complex in the acyl-CoA dehydrogenase family must differ from the analogous complex in NAO because nitroalkanes lack the CoA moiety. Consequently, the nitroalkane substrates bind to and react with the more compact active site architecture in NAO.
The R409K mutation also affects the rate constant for reoxidation of the reduced enzyme. There is a decrease of 5–6 fold in the kcat/Km value for oxygen, reflecting a comparable decrease in the value of k5, the second order rate constant for the reaction of the reduced enzyme with oxygen. This may reflect the sensitivity of this rate constant to the presence of a nearby positive charge. For example, in glucose oxidase the rate constant for the reaction of the reduced enzyme with oxygen decreases about 2600-fold when an active site histidine residue is deprotonated (35), while there is a 75-fold decrease in this rate constant for choline oxidase when a substrate lacking a positively charged amino moiety is used as substrate (36). The rate-limiting step in the oxidation of reduced flavins by oxygen is the outer-sphere transfer of one electron from the flavin to O2 to form superoxide (18, 35, 37). The presence of a nearby positive charge has been proposed to increase the rate of this reaction by stabilizing the developing negative charge on oxygen (38–40), although recent analyses using Marcus theory suggest that the more important effect of the positive charge is on the reorganization energy of the reaction (41). The decrease in the value of k5 seen here with the R409K enzyme is much smaller than the effect of deleting the charge completely, consistent with this mutation resulting in a different position for the positive charge rather than its loss. The value of k5 only decreases two-fold in the case of the D402E enzyme, consistent with the retention of the positively charged Arg402.
In conclusion, the combination of kinetic and structural data presented here demonstrate the importance of the interaction between Arg409 and Asp402 to the proton abstraction step in the reaction of NAO. Arg409 is important both to position the active site base and increase its reactivity. The effects of mutating Asp402 to glutamate are consistent with this model, but also suggest a critical role for Ser276 in the NAO reaction.
Supplementary Material
Scheme 1.

Scheme 2.

Scheme 3.
Footnotes
This research was supported in part by grants to PFF from the NIH (GM58698) and The Welch Foundation (A-1245) and to AMO from the American Heart Association (Grant in Aid 0555286B), the Offices of Biological and Environmental Research of the US Department of Energy, and the NIH (2 P41 RR012408). Use of the National Synchrotron Light Source at Brookhaven National Laboratory was supported by the U.S. Department of Energy Office of Basic Energy Sciences, under Contract DE-AC02-98CH10886.
The atomic coordinates and structure factors for NAO D402E and R409K have been deposited with the Protein Data Bank with the corresponding file names 2REH, 2rehsf, 2ZAF and rcsb027723, respectively.
Abbreviations: NAO, nitroalkane oxidase; ACAD, acyl-CoA dehydrogenase; Kne, the Km value for nitroethane as the substrate
For comparison, we examined Lys359, which is near Lys409 but is part of helix I. The Lys359 N□ atom forms ionic interactions with the backbone carbonyl oxygen atoms of residues Tyr398, Phe401, Gly403 and the OD1 atom of Asn405. The electron density correlation for Lys359 is 0.70, 0.74, 0.68, and 0.67 for the A–D subunits, respectively.
References
- 1.Fitzpatrick PF, Orville AM, Nagpal A, Valley MP. Nitroalkane oxidase, a carbanion-forming flavoprotein homologous to Acyl-CoA dehydrogenase. Arch Biochem Biophys. 2005;433:157–165. doi: 10.1016/j.abb.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 2.Valley MP, Fitzpatrick PF. Inactivation of nitroalkane oxidase upon mutation of the active site base and rescue with a deprotonated substrate. J Am Chem Soc. 2003;23:8738–8739. doi: 10.1021/ja036045s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menon V, Hsieh CT, Fitzpatrick PF. Substituted alcohols as mechanistic probes of alcohol oxidase. Bioorganic Chem. 1995;23:42–53. [Google Scholar]
- 4.Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Crystal structure of D-amino acid oxidase: A case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J Am Chem Soc. 2000;122:12896–12897. [Google Scholar]
- 6.Fitzpatrick PF. Substrate dehydrogenation by flavoproteins. Acc Chem Res. 2001;34:299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 7.Sobrado P, Fitzpatrick PF. Solvent and primary deuterium isotope effects show that lactate CH and OH bond cleavages are concerted in Y254F flavocytochrome b2, consistent with a hydride transfer mechanism. Biochemistry. 2003;42:15208–15214. doi: 10.1021/bi035546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzpatrick PF. Carbanion versus hydride transfer mechanisms in flavoprotein-catalyzed dehydrogenations. Bioorg Chem. 2004;32:125–139. doi: 10.1016/j.bioorg.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Mechanistic studies of the flavoenzyme tryptophan 2-monooxygenase: Deuterium and 15N kinetic isotope effects on alanine oxidation by an L-amino acid oxidase. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brinkley DW, Roth JP. Determination of a large reorganization energy barrier for hydride abstraction by glucose oxidase. J Am Chem Soc. 2005;127:15720–15721. doi: 10.1021/ja056025l. [DOI] [PubMed] [Google Scholar]
- 11.Ghanem M, Gadda G. On the catalytic role of the conserved active site residue His466 of choline oxidase. Biochemistry. 2005;44:893–904. doi: 10.1021/bi048056j. [DOI] [PubMed] [Google Scholar]
- 12.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Insights into the mechanism of flavoprotein-catalyzed amine oxidation from nitrogen isotope effects on the reaction of N-methyltryptophan oxidase. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heasley CJ, Fitzpatrick PF. Kinetic mechanism and substrate specificity of nitroalkane oxidase. Biochem Biophys Res Commun. 1996;225:6–10. doi: 10.1006/bbrc.1996.1122. [DOI] [PubMed] [Google Scholar]
- 14.Valley MP, Fitzpatrick PF. Reductive half-reaction of nitroalkane oxidase: effect of mutation of the active site aspartate to glutamate. Biochemistry. 2003;42:5850–6. doi: 10.1021/bi034061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gadda G, Edmondson RD, Russel DH, Fitzpatrick PF. Identification of the naturally occurring flavin of nitroalkane oxidase from Fusarium oxysporum as a 5-nitrobutyl-FAD and conversion of the enzyme to the active FAD-containing form. J Biol Chem. 1997;272:5563–5570. doi: 10.1074/jbc.272.9.5563. [DOI] [PubMed] [Google Scholar]
- 16.Nagpal A, Valley MP, Fitzpatrick PF, Orville AM. Crystal structures of nitroalkane oxidase: insights into the reaction mechanism from a covalent complex of the flavoenzyme trapped during turnover. Biochemistry. 2006;45:1138–1150. doi: 10.1021/bi051966w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valley MP, Tichy SE, Fitzpatrick PF. Establishing the kinetic competency of the cationic imine intermediate in nitroalkane oxidase. J Am Chem Soc. 2005;127:2062–2066. doi: 10.1021/ja043542f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J Biol Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 19.Gadda G, Fitzpatrick PF. Substrate specificity of a nitroalkane oxidizing enzyme. Arch Biochem Biophys. 1999;363:309–313. doi: 10.1006/abbi.1998.1081. [DOI] [PubMed] [Google Scholar]
- 20.Gadda G, Choe DY, Fitzpatrick PF. Use of pH and kinetic isotope effects to dissect the effects of substrate size on binding and catalysis by nitroalkane oxidase. Arch Biochem Biophys. 2000;382:138–144. doi: 10.1006/abbi.2000.2009. [DOI] [PubMed] [Google Scholar]
- 21.Bell RP. The Proton in Chemistry. 2. Cornell University Press; Ithaca: 1973. Kinetic isotope effects in proton-transfer reactions; pp. 250–296. [Google Scholar]
- 22.Bernasconi CF. The principle of nonperfect synchronization: More than a qualitative concept? Acc Chem Res. 1992;25:9–16. [Google Scholar]
- 23.Valley MP, Fitzpatrick PF. Comparison of enzymatic and non-enzymatic nitroethane anion formation: thermodynamics and contribution of tunneling. J Am Chem Soc. 2004;126:6244–6245. doi: 10.1021/ja0484606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daubner SC, Gadda G, Valley MP, Fitzpatrick PF. Cloning of nitroalkane oxidase from Fusarium oxysporum identifies a new member of the acyl-CoA dehydrogenase superfamily. Proc Nat Acad Sci USA. 2002;99:2702–2707. doi: 10.1073/pnas.052527799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghisla S, Thorpe C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur J Biochem. 2004;271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 26.Thorpe C, Kim JP. Structure and mechanism of action of the Acyl-CoA dehydrogenases. FASEB J. 1995;9:718–725. doi: 10.1096/fasebj.9.9.7601336. [DOI] [PubMed] [Google Scholar]
- 27.Gadda G, Fitzpatrick PF. Biochemical and physical characterization of the active FAD-containing form of nitroalkane oxidase from Fusarium oxysporum. Biochemistry. 1998;37:6154–6164. doi: 10.1021/bi973085y. [DOI] [PubMed] [Google Scholar]
- 28.Nagpal A, Valley MP, Fitzpatrick PF, Orville AM. Crystallization and preliminary analysis of active nitroalkane oxidase in three crystal forms. Acta Crystallogr D Biol Crystallogr. 2004;60:1456–1460. doi: 10.1107/S0907444904013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Leslie AG. The integration of macromolecular diffraction data. Acta Crystal D. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project. The CCP4 suite: Programs for protein crystallography. Acta Crystal D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Gadda G, Fitzpatrick PF. Iso-mechanism of nitroalkane oxidase: 1. Inhibition studies and activation by imidazole. Biochemistry. 2000;39:1400–1405. doi: 10.1021/bi9922547. [DOI] [PubMed] [Google Scholar]
- 33.Major DT, York DM, Gao J. Solvent polarization and kinetic isotope effects in nitroethane deprotonation and implications to the nitroalkane oxidase reaction. J Am Chem Soc. 2005;127:16374–16375. doi: 10.1021/ja055881u. [DOI] [PubMed] [Google Scholar]
- 34.Mackenzie J, Pedersen L, Arent S, Henriksen A. Controlling electron transfer in Acyl-CoA oxidases and dehydrogenases: a structural view. The Journal of biological chemistry. 2006;281:31012–20. doi: 10.1074/jbc.M603405200. [DOI] [PubMed] [Google Scholar]
- 35.Roth JP, Klinman JP. Catalysis of electron transfer during the activation of O2 by the flavoprotein glucose oxidase. Proc Natl Acad Sci USA. 2003;100:62–67. doi: 10.1073/pnas.252644599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gadda G, Fan F, Hoang JV. On the contribution of the positively charged headgroup of choline to substrate binding and catalysis in the reaction catalyzed by choline oxidase. Arch Biochem Biophys. 2006;451:182–187. doi: 10.1016/j.abb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Eberlein G, Bruice TC. The chemistry of a 1,5-diblocked flavin. 2 Proton and electron transfer steps in the reaction of dihydroflavins with oxygen. J Am Chem Soc. 1983;105:6685–6697. [Google Scholar]
- 38.Massey V, Müller F, Feldberg R, Schuman M, Sullivan PA, Howell LG, Mayhew SG, Matthews RG, Foust GP. The reactivity of flavoproteins with sulfite possible relevance to the problem of oxygen reactivity. J Biol Chem. 1969;244:3999–4006. [PubMed] [Google Scholar]
- 39.Wang R, Thorpe C. Reactivity of medium-chain acyl-CoA dehydrogenase toward molecular oxygen. Biochemistry. 1991;30:7895–7901. doi: 10.1021/bi00246a004. [DOI] [PubMed] [Google Scholar]
- 40.Su Q, Klinman JP. Nature of oxygen activation in glucose oxidase from Aspergillus niger: The importance of electrostatic stabilization in superoxide formation. Biochemistry. 1999;38:8572–8581. doi: 10.1021/bi990044o. [DOI] [PubMed] [Google Scholar]
- 41.Roth JP, Wincek R, Nodet G, Edmondson DE, McIntire WS, Klinman JP. Oxygen isotope effects on electron transfer to O2 probed using chemically modified flavins bound to glucose oxidase. J Am Chem Soc. 2004;126:15120–15131. doi: 10.1021/ja047050e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.