

Abstract
His373 in flavocytochrome b2 has been proposed to act as an active site base during the oxidation of lactate to pyruvate, most likely by removing the lactate hydroxyl proton. The effects of mutating this residue to glutamine have been determined to provide further insight into its role. The kcat and kcat/Klactate values for the mutant protein are 3–4 orders of magnitude smaller than the wild-type values, consistent with a critical role for His373. Similar effects are seen when the mutation is incorporated into the isolated flavin domain of the enzyme, narrowing the effects to lactate oxidation rather than subsequent electron transfers. The decrease of 3500-fold in the rate constant for reduction of the enzyme-bound FMN by lactate confirms this part of the reaction as that most effected by the mutation. The primary deuterium and solvent kinetic isotope effects for the mutant enzyme are significantly smaller than the wild-type values, establishing that bond cleavage steps are less rate-limiting in H373Q flavocytochrome b2 than in the wild-type enzyme. The structure of the mutant enzyme with pyruvate bound, determined at 2.8 Å, provides a rationale for these effects. The orientation of pyruvate in the active site is altered from that seen in the wild-type enzyme. In addition, the active site residues Arg289, Asp 292, and Leu 286 have altered positions in the mutant protein. The combination of an altered active site and the small kinetic isotope effects is consistent with the slowest step in turnover being a conformational change involving a conformation in which lactate is bound unproductively.
Flavocytochrome b2 from Saccharomycescerevisae is a lactate dehydrogenase that catalyzes the transfer of a hydride equivalent from its hydroxy acid substrate to the enzyme-bound flavin and thence to proteins of the mitochondrial electron transport chain via a bound cytochrome (1). This enzyme is the paradigm for the flavoproteins which catalyze the oxidation of α-hydroxy acids, a family of enzymes with conserved three-dimensional and active site structures found in plants, animals, and bacteria (2–8). The two cofactors of flavocytochrome b2 are in different domains of the protein (9). Residues 1-99 make up the heme binding domain and resemble cytochrome b5 in structure. Residues 100-486 contain the FMN in a TIM barrel. This flavin domain is homologous to the other α-hydroxy acid-oxidizing flavoproteins (6) and can catalyze lactate oxidation in the absence of the heme domain (10). Figure 1 shows the arrangements of amino acids in the active site of flavocytochrome b2 in relation to the FMN cofactor and bound pyruvate product (9). Of the seven amino acid residues in Figure 1, six are conserved in all members of the family to date, while Tyr143 is conserved in most (Figure 2). Site-directed mutagenesis has confirmed the importance of several of these residues for substrate oxidation by this family of flavoproteins (11–21).
Figure 1.
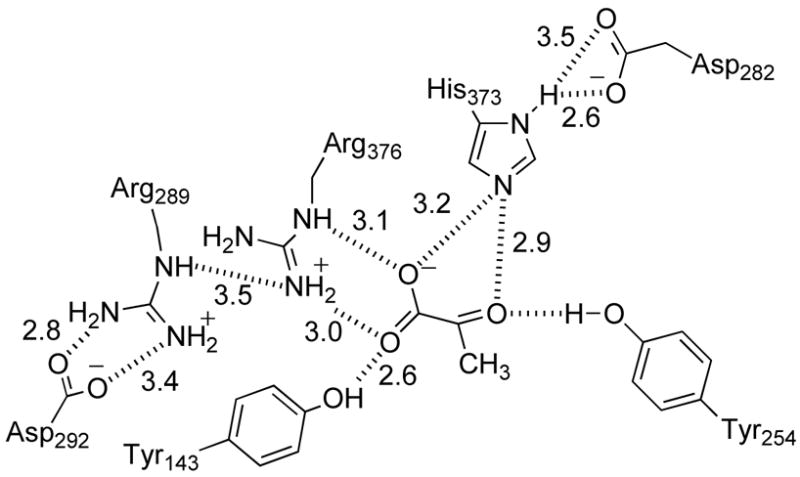
Interactions between active site residues in wild-type flavocytochrome b2 and pyruvate, based on PDB file 1KBI (39).
Figure 2.

Alignment of sequences of flavoprotein hydroxy acid oxidases and dehydrogenases, showing conservation of active site residues. The residue numbering is for flavocytochrome b2.
The details of the mechanism by which these enzymes transfer a hydride equivalent from the α-hydroxy acid substrate to the flavin have been controversial (22–24). Results of mechanistic studies have been interpreted as evidence for removal of the α-hydrogen both as a proton, followed by subsequent transfer of electrons, and as a hydride, so that the proton and two electrons are simultaneously transferred to the flavin. A similar controversy exists for the related flavoprotein amine oxidases, for which there is accumulating support for a hydride transfer mechanism (24–27). Recently, deuterium and solvent kinetic isotope effects on wild-type flavocytochrome b2 and the Y254F mutant enzyme led to a variation on a mechanism involving hydride transfer (Figure 3) (19). In this mechanism, His373 acts as a base to remove the hydroxyl proton of the substrate and form an alkoxide. The α-hydrogen is then transferred to the FMN as a hydride. The previous results could not distinguish whether the alkoxide is a true intermediate or the reaction is highly asynchronous with proton transfer essentially complete in the transition state for CH bond cleavage. The role of His373 in this proposal is different from that in which the α-hydrogen is removed as a proton to form a carbanion, where this residue is the base that abstracts the α-proton (28).
Figure 3.

Proposed mechanism for oxidation of lactate by flavocytochrome b2 (19).
Site-directed mutagenesis of His373 in flavocytochrome b2 or of the corresponding residue in other members of this family of flavoproteins results in a large decrease in activity. In the case of flavocytochrome b2, the kcat and kcat/Km values of the mutant enzyme have been reported to be 8000 and 3500-fold lower than the wild-type values (16). Because the Km value for lactate only changed 2 to 3-fold in the mutant protein, the low residual activity was attributed to contamination with wild-type enzyme, leading to the conclusion that the mutant enzyme is at least 10,000-fold less active than the wild-type enzyme. In the case of lactate monooxygenase from Mycobacterium smegmatis, the H290Q enzyme cannot be reduced by lactate, consistent with a decrease of 7–8 orders of magnitude in activity (14). Similarly, mutation of His274 in mandelate dehydrogenase from Pseudomonas putida to glycine, alanine, or asparagine yields enzyme with no detectable activity, consistent with decreases of at least 100,000-fold (18). Decreases in activity of this magnitude would be unexpected for the mechanism of Figure 3, where the histidine is not involved in a rate-limiting bond cleavage. While large decreases in the activities of the mutant proteins would be more consistent with a mechanism in which the base removes a proton from carbon in a rate-limiting step, the effects are quite large even for such a role. Consequently, we have re-examined H373Q flavocytochrome b2, using a combination of kinetic and structural approaches. The results of these analyses are described here.
Experimental Procedures
Materials
Lithium L-lactate and D,L-lactate were from Sigma (St. Louis, MO). Sodium L-[2-2H]-lactate (98%) and deuterium oxide (99.9%) were purchased from Cambridge Isotope Co., Andover, MA. Hydroxyapatite was from Bio-Rad Laboratories (Hercules, CA).
Enzyme preparation
Site-directed mutagenesis of His373 to glutamine was performed following the procedure the QuikChange protocol (Stratagene) with the primers 5′-GGTGGTTCTATCCAATCAAGGTGGTAGACAATTAGA- 3′ and 5′-TCTAATTGTCTACCACCTTGATTGGATAGAACCACC -3′. The H373Q mutation was introduced into the genes for the wild-type enzyme in pET21d and for the flavin domain in pDSb2 (29). The expression and purification of the wild-type enzyme and mutant enzymes were as described previously (19, 29). Enzyme concentrations were determined using an ε423 value of 183 mM−1 cm−1 and an ε413 value of 129.5 mM−1cm−1 for the reduced and oxidized full length enzymes, respectively (30), and an ε453 value of 11.1 mM−1cm−1 for the flavin domain (31). Purified enzymes were stored in 100 mM potassium phosphate, 1 mM EDTA, 20 mM D,L-lactate, pH 7.5, at −70 °C.
To remove the lactate used for storage, the day of use an aliquot was precipitated with 70% ammonium sulfate in 100 mM potassium phosphate, 1 mM EDTA, pH 7.5, at 4 °C. The pellet obtained by centrifugation at 15,000 × g for 5 min was resuspended in 100 mM potassium phosphate, 1 mM EDTA, pH 7.5. This procedure was repeated twice. The final enzyme concentration was about 3 μM. This treatment also resulted in oxidation of the cofactors. For stopped-flow experiments, enzyme was passed through a Sephadex G-25 column in 100 mM potassium phosphate, 1 mM EDTA, pH 7.5, to remove the lactate.
Enzyme assays
Initial rate assays were performed in 100 mM potassium phosphate, 5 mM EDTA, 1 mM potassium ferricyanide, pH 7.5, 25 °C, with varied concentrations of L-lactate or L-[2-2H]lactate, following the decrease in absorbance at 420 nm, and using an ε420 value for ferricyanide of 1.04 mM−1cm−1. For the flavin domain, 3 mM potassium ferricyanide was used. Concentrations of lactate were determined by end point assay with wild-type flavocytochrome b2.
For pH studies, the buffers were 50 mM Bis-Tris at pH 5.5–7.0, 150 mM HEPES at pH 7.0–8.5, and 50 mM ethanolamine at pH 8.5–10.0. All assays were done in the presence of 1 mM potassium ferricyanide at 25 °C. Solvent isotope effects were determined in 100 mM phosphate, 1 mM EDTA, pH 7.5 or pD 8.0, with 1 mM potassium ferricyanide at 25 °C. Assays were initiated by the addition of 5 μl of enzyme in H2O into a final volume of 1 ml.
Rapid reaction kinetics
Rapid reaction kinetic measurements were performed with a Applied Photophysics SX.18MV stopped-flow spectrophotometer. Enzyme in 100 mM potassium phosphate and 1 mM EDTA at pH 7.5 was mixed with an equal volume of the same buffer containing 10 mM lactate at 25 °C. Enzyme reduction was monitored at 438 nm.
Data Analysis
Kinetic data were analyzed using the programs KaleidaGraph (Adelbeck Software, Reading, PA) and Igor (Wavemetrics, Lake Oswego, OR). Initial rate data were fit to the Michaelis-Menten equation to obtain kcat, kcat/Klactate, and Klactate values. Primary deuterium and solvent isotope effects were calculated using equations 1–3. Equation 1 describes separate isotope effects on the kcat and kcat/Klactate values; equation 2 describes an isotope effect on the kcat value only; and equation 3 describes an isotope effect on the kcat/Klactate value only. Here, Fi is the fraction of heavy atom substitution in the substrate, Ev is the isotope effect on the kcat value minus 1, and Evk is the isotope effect on the kcat/Klactate value minus 1. Stopped-flow traces were fit to equation 4 which describes a single exponential decay: A is the absorbance at time t, A∞ is the final absorbance, and k is the first order rate constant.
| (1) |
| (2) |
| (3) |
| (4) |
Crystallization
Initial crystallization of H373Q flavocytochrome b2 (7 mg ml-1 plus 50 mM pyruvate) was carried out at 18 °C by vapor diffusion with the hanging drop method (32) using a sparse matrix kit (Crystal Screen I and II, Hampton Research, CA). Well-diffracting crystals were obtained in 4 to 8 days at 18 °C from 4 μl hanging drops consisting of a 1:1 mixture of H373Q flavocytochrome b2 and a crystallization buffer containing 100 mM sodium citrate, pH 4.6, 17% polyethylene glycol 3000, and 0.2 mM sucrose monolaurate.
Data Collection
Crystals mounted in cryo loops were flash-cooled in a N2 stream (120 K) after brief soaks in 2 μl of the mother liquor plus 2 μl of 50 % glycerol. The data from the H373Q crystals were recorded on Advanced Photon Source beam line 23ID using a CCD detector. The diffraction data were reduced using DENZO, and intensities were scaled with SCALEPACK (33). The reflections were indexed as primitive tetragonal (a = b = 163.68 Å, c = 112.01 Å) with Laue symmetry 3m1. Examination of the integrated and scaled data indicated the trigonal space group P3221. Solvent content calculations (34) indicated the presence of the homodimer in the asymmetric unit. The crystallographic data collection, statistics and refinement parameters are summarized in Table 1.
Table 1.
Data collection and refinement statistics for H373Q flavocytochrome b2
| Data collection | |
| Resolution, Å | 2.80 to 30.00 |
| Total no. of reflections | 69,254 |
| No. of unique reflections | 36,191 |
| Completeness | 88 % |
| I/[(I)] | 14.5 |
| I/[(I)] in outer shell | 1.9 |
| R merge | 0.26 % |
| Refinement | |
| R-factor | 0.21 |
| R-free | 0.27 |
| Number of protein atoms | 7,077 |
| Number of water molecules | 18 |
| Rmsd from ideal values | |
| Bond lengths (Å) | 0.05 |
| Bond angels (deg) | 2.02 |
| Ramachandran analysis | |
| Most favored (%) | 74% |
| Additionally allowed (%) | 24.3% |
Structure Determination and Refinement
The three dimensional structure of H373Q flavocytochrome b2 complexed with pyruvate was solved by molecular replacement using EPMR (35) with the wild-type structure file 1FCB as the search model for the data extending from 25 to 3.5 Å. After model building and fitting, bias minimized electron density maps were obtained using the Shake and Warp protocol (36). The refinement continued with structure factors measuring 1.0 sigma or better within the resolution range 30 to 2.8 Å to reduce the effects of poorly measured reflections. After repeated cycles of refinement and manual model building, water molecules were added to the structure using Xfit (37). The final model consists of two protein molecules, subunit A and subunit B. Subunit A contains the ordered heme domain as well as the flavin domain. Subunit B shows no interpretable density for the heme domain, so it has been omitted from the model. In both subunits, no electron density was observed for residues 298 to 317, suggesting that they are highly disordered.
Results
Kinetic characterization of H373Q flavocytochrome b2
To determine the effects of mutating His373 flavocytochrome b2 to glutamine, the steady-state kinetic parameters of the mutant protein were determined. The results of these analyses are summarized in Table 2. Ferricyanide was used as the electron acceptor because lactate oxidation is rate-limiting with this substrate, unlike the situation with cytochrome c as substrate (1). The mutant protein still has some activity as a lactate dehydrogenase, although there is a substantial decrease in activity from that of the wild-type enzyme. With the intact protein, the kcat/Km value for lactate is 48,000-fold lower than the wild-type value, while the kcat value is 12,000-fold lower. In addition, the Km values are altered slightly. The Km value for ferricyanide increases to 0.11 ± 0.01 mM (results not shown) from a reported value of less than 0.1 mM for the wild-type enzyme (38). The kcat and kcat/Klactate values for the mutant protein are comparable to those previously reported by Gaume et al.(16) with dichlorophenol indophenol as the electron acceptor. The mutation does not affect the Km value for cytochrome c significantly, in that the mutant protein has a Kcytc value of 210 ± 42 μM compared to a value of 180 μM for the wild-type enzyme (1), but the kcat value of the mutant protein is only one-half as large with cytochrome c as the acceptor compared to the value with ferricyanide (results not shown).
Table 2.
Steady State Kinetic Parameters for Wild-Type and Mutant Flavocytochrome b2
| intact protein | flavin domain | |||||||
|---|---|---|---|---|---|---|---|---|
| kinetic parameter | wild-typea | H373Q | wild-typea | H373Q | ||||
| eq | χ2 | eq | χ2 | |||||
| kcat, s−1 | 372 ± 12 | 0.031 ± 0.001 | 200 ± 7 | 0.057 ± 0.001 | ||||
| kcat/Klactate, mM−1s−1 | 2300 ± 220 | 0.048 ± 0.003 | 543 ± 32 | 0.23 ± 0.02 | ||||
| Klactate, mM | 0.16 ± 0.02 | 0.65 ± 0.06 | 0.36 ± 0.03 | 0.25 ± 0.03 | ||||
| Dkcat | 2.9 ± 0.1 | 1.71 ± 0.07 | 1 | 0.000204 | 3.7 ± 0.3 | 1.42 ± 0.08 | 1 | 0.000152 |
| 1.78 ± 0.05 | 2 | 0.000208 | 1.47 ± 0.06 | 2 | 0.000158 | |||
| D(kcat/Klactate) | 3.0 ± 0.3 | 1.16 ± 0.14 | 1 | 3.5 ± 0.2 | 1.18 ± 0.25 | 1 | ||
| 3.2 ± 0.4 | 3 | 0.000591 | 2.80 ± 0.65 | 3 | 0.000454 | |||
| D2Okcat | 1.38 ± 0.07 | 0.99 ± 0.05 | 1 | 0.000576 | 1.18 ± 0.05 | ndb | ||
| 1.04 ± 0.04 | 2 | 0.000589 | ||||||
| D2vO(kcat/Klactate) | 0.90 ± 0.04 | 1.21 ± 0.16 | 1 | 0.44 ± 0.05 | nd | |||
| 1.18 ± 0.10 | 2 | 0.00578 | ||||||
Data from reference (29).
nd, not determined.
The complete reaction catalyzed by flavocytochrome b2 includes lactate oxidation and electron transfer steps. To determine if the effects of the mutation were due to a decrease in the rate constants for electron transfer from the flavin to the heme, the mutation was also incorporated into the isolated flavin domain of the enzyme. With this protein electrons must be transferred directly from the reduced flavin to the exogenous electron acceptor rather than being passed through the bound cytochrome. The kinetic parameters for the flavin domain of the mutant protein are slightly higher than those of the intact protein (Table 2), but the kcat and kcat/Klactate values are still several thousand-fold less than the wild-type values. Thus, the effects of the mutation are primarily in steps for lactate oxidation.
As a more direct measure of the effect of the mutation on the rate constant for lactate oxidation, the mutant enzyme was mixed with 5 mM lactate in the stopped-flow spectrophotometer in the absence of an electron acceptor. The resulting spectral change was monophasic, with reduction of both the flavin and the heme occurring with the same rate constant of 0.15 ± 0.04 s−1. This is a decrease of 3500-fold from the wild-type value of 520 s−1 (29). Comparable results were obtained for the isolated flavin domain, with a rate constant for reduction by 5 mM lactate of 0.09 ± 0.02 s−1. Thus, the effect of the mutation can be attributed to a substantial decrease in the rate constant for flavin reduction. The simultaneous reduction of both cofactors in the intact mutant protein is consistent with electron transfer from reduced flavin to heme being much more rapid than reduction of the flavin by lactate.
With wild-type flavocytochrome b2, there is no solvent isotope effect on lactate oxidation, but there is a significant primary deuterium isotope effect (29). This is consistent with rapid and reversible removal of the hydroxyl proton followed by much slower cleavage of the carbon-hydrogen bond of lactate. If the role of His373 is to abstract the hydroxyl proton as shown in Figure 3, there should be a significant solvent isotope effect in the mutant protein as this step becomes rate-limiting; the magnitude of the primary isotope effect may also be altered, depending on the relative magnitudes of the rate constants for OH and CH bond cleavage. Consequently primary and solvent isotope effects were determined for H373Q flavocytochrome b2. Primary deuterium isotope effects were determined for both the intact mutant protein and for the flavin domain. In both cases, there was a slightly better fit of the data to equation 1, which applies for significant and nonidentical isotope effects on both the kcat and the kcat/Klactate values, than to either equation 2, which describes an effect on kcat only or equation 3, which describes an effect on kcat/Klactate only. The resulting isotope effects on the kcat values are significantly smaller than the effects seen for the wild-type enzyme, but still substantially greater than unity. The primary isotope effect on the kcat/Klactate value is also significantly smaller than the wild-type value. For both the intact protein and the flavin domain, the D(kcat/Klactate) value is only marginally greater than unity statistically. However, when the data are analyzed using equation 2, which applies when there is only an isotope effect on the kcat value, the resulting χ2 values are slightly larger than is the case with equation 1.
Prior to measurement of the solvent isotope effects, the pH-dependence of the kcat and kcat/Klactate values of the mutant protein were determined. Both kinetic parameters are independent of pH over the pH range 5.5–10 (results not shown). Solvent isotope effects were then determined at pH 7. With wild-type flavocytochrome b2, there is a small solvent isotope effect on the kcat due to effects on the oxidative half-reaction and a slightly inverse effect on the kcat/Klactate value, attributed to a conformational change (29). In contrast, H373Q flavocytochrome b2 exhibits no significant solvent isotope effect on the kcat value and a very small normal effect on the kcat/Klactate value (Table 2). Taken together the isotope effects suggest that both hydrogen abstraction steps have become less rate-limiting in the mutant protein.
Structural characterization of H373Q flavocytochrome b2
To obtain further insight into the effects of the mutation on the properties of the enzyme, the crystal structure of H373Q flavocytochrome b2 was determined at 2.8 Å. The two subunits in the asymmetric unit are not equivalent in the structure of the mutant protein. The electron density map clearly shows the absence of the heme domain in subunit B and the presence of bound pyruvate at the active site of subunit B only (Figure 4A). This asymmetry is also observed in structures of the wild-type enzyme (9, 39). The mutation of His373 to glutamine does not result in any change in the overall protein structure from that seen for the wild-type enzyme. The mutant protein shows the largest divergence from the wild-type structure for residues 292-298 and 317-324 (r.m.s.d. 1.78 Å and 1.70 Å, respectively), residues adjacent to a surface loop which is not seen in any available structure, and residues 100-114 of subunit B, which connect to the missing heme domain in that subunit.
Figure 4.
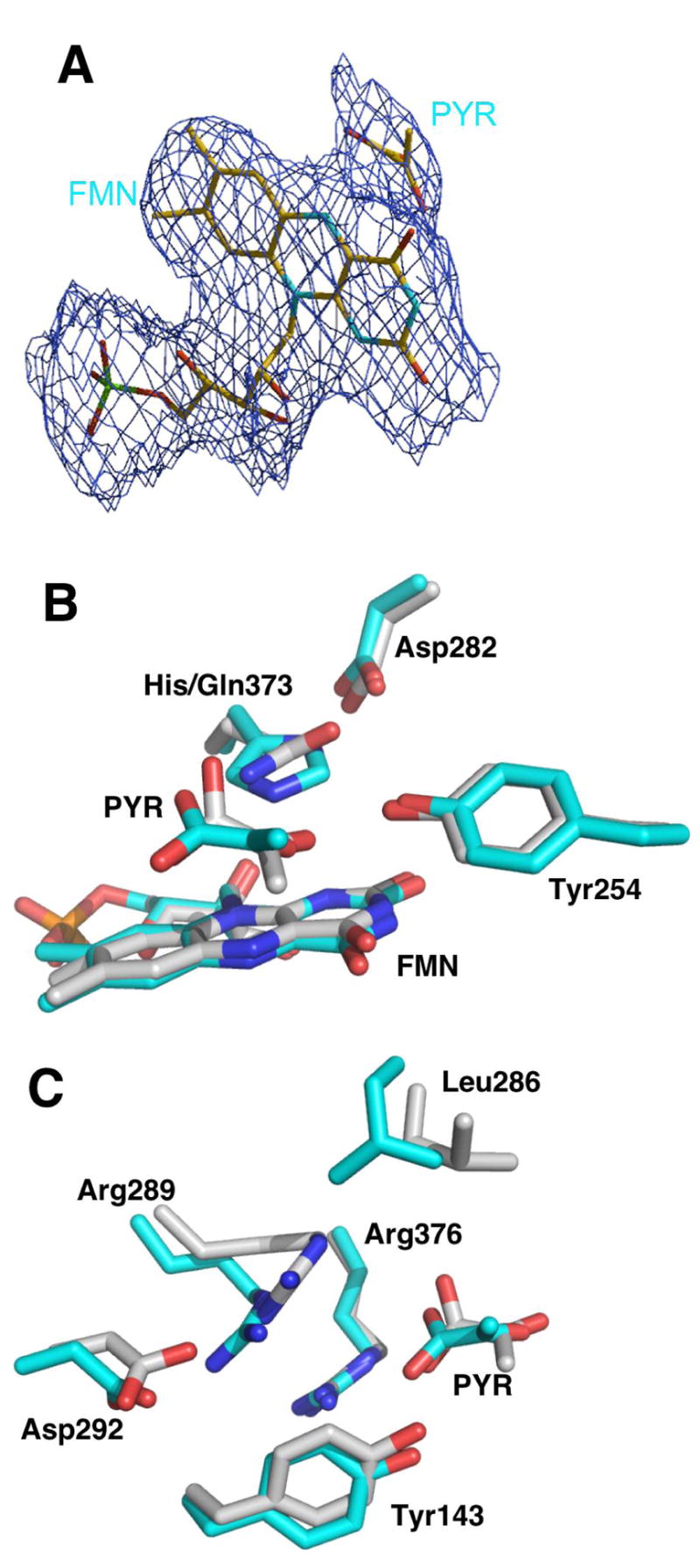
A. Electron density of ligands complexed with H373Qflavocytochrome b2. The electron density of Snw omit maps is contoured at the 1 σ level. B and C: Comparison of the active sites of wild-type and H373Q flavocytochrome b2. The residues and pyruvate (PYR) with carbons in cyan represent the wild-type enzyme structure, while the carbon atoms for the mutant structure are in gray. The wild-type enzyme structure is from PDB file 1KBI (39).
Figure 4 compares the active site chain B of H373Q flavocytochrome b2 with that of the wild-type enzyme, while Figure 5 illustrates the interactions between the active site of the mutant protein and the bound pyruvate. Omit electron density (36) maps of the mutant protein show clear electron density for the side chain of Gln373 in the mutant protein. The glutaminyl side chain of the mutant protein overlaps with the side chain of His373 in the wild-type enzyme (Figure 4B). However, the heteroatoms of the side chains of the glutamine in the mutant protein are twisted by 90° relative to the imidazole nitrogens in His373, and this results in altered interactions with Asp282 and pyruvate. In the mutant enzyme, the distance from the glutaminyl side chain to OD1 in Asp282 is comparable to the distance to the histidinyl side chain in the wild-type protein at 2.6 Å. In contrast, the distance to OD2 in Asp282 has increased from 3.5 Å in the wild-type enzyme to 4.1 Å in the mutant protein due to the different relative positions of the heteroatoms in the histidinyl and glutaminyl side chains (Figure 5). In addition, in the wild-type enzyme, the NE2 of His373 is 2.9 Å from the carbonyl oxygen of pyruvate and 3.2 Å from one of the pyruvate carboxylate oxygens; in the mutant enzyme the corresponding atom of the glutaminyl side chain is closer to the pyruvate carboxylate oxygen (2.8 Å) than the carbonyl (3.1 Å) (Figure 1). The different positions of the heteroatoms involved in this interaction in the wild-type and mutant proteins shift the pyruvate 0.7 Å relative to its position in the wild-type enzyme. The pyruvate is also twisted in the mutant enzyme relative to its position in the wild-type protein, so that the interactions of the carboxylate with Arg376 and Tyr143 are altered, even though the positions of these residues are not (Figure 4C). In the wild-type enzyme, the pyruvate CO1 interacts with the NE of Arg376 while CO2 interacts with Arg376 NH2 and the phenolic oxygen of Tyr143 (Figure 1). The interaction of Arg376 with Tyr143 is only slightly longer in the mutant enzyme, but the distance from the pyruvate carboxylate CO2 to Arg376 has increased from 3.0 to 4.7 Å, so that the primary interaction of this oxygen is now with Tyr143 (Figure 5).
Figure 5.
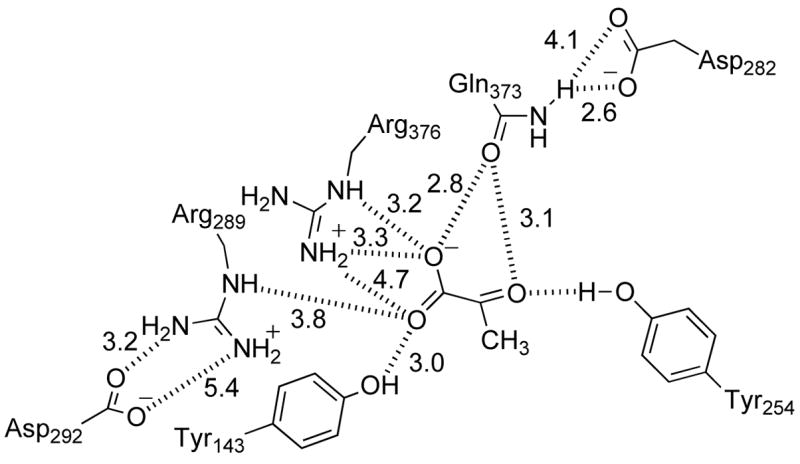
Interactions between active site residues in H373Q flavocytochrome b2 and pyruvate.
More dramatic changes are seen in the positions of other active site residues in the mutant protein, most notably Arg289, Leu286, and Asp292 (Figure 4C). In the wild-type structure, NE of Arg289 is too far from the pyruvate carboxylate for an electrostatic interaction; in contrast, in the mutant protein the position of the side chain of Arg289 has shifted by 2 Å, so that NE of Arg289 is close enough (3.8 Å) to exert some electrostatic influence on the pyruvate carboxylate CO2. This movement of Arg289 also results in a displacement of the side chain of Leu286 by over 2 Å. Finally, in the wild-type structure the guanidinium nitrogens of Arg289 are at appropriate distances (2.8 Å and 3.4 Å) from the carboxylate oxygens of Asp292 for ionic interactions (Figure 1). In H373Q flavocytochrome b2 both NH1 and NH2 of Arg289 are farther from the carboxylate oxygens of Asp292, 3.4 Å and 5.4 Å, respectively, so that only one oxygen of Asp292 is close enough to interact (Figure 5). As a result, the electropotential surface of the active site is altered so that acidic charge of Asp 292 is neutralized by Arg289 in the wild-type enzyme but not in the mutant protein (Figure 6).
Figure 6.
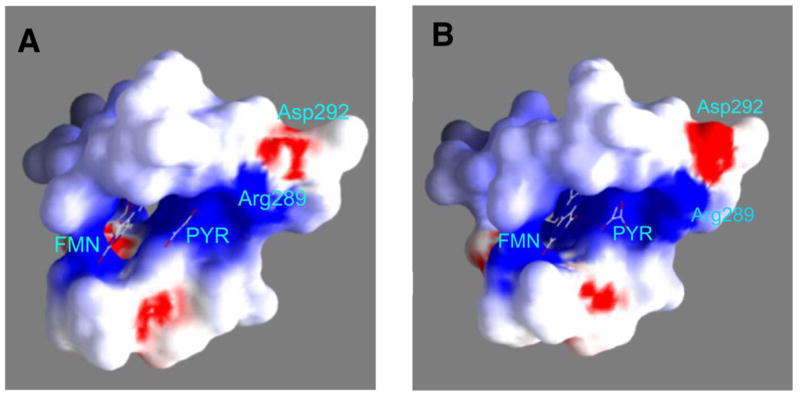
The eletrostatics of the substrate binding pockets of wild-type (A) and H373Q (B) flavocytochrome b2 (red, -9.8 KT/e; white, neutral; and blue, +3.4 KTe).
Similar changes are seen in the active site of chain A, although there is no pyruvate in the active site in this case. The glutamine at position 373 occupies the same position as His 373 of the wild-type enzyme, as does Asp282. As is the case in subunit B, the side chains of Leu286, Arg289, and Asp292 are displaced. As a result, the interactions of Arg289 with both Asp292 and Arg376 are weaker in this subunit also.
Discussion
The effects of mutation of His373 to glutamine are fully consistent with this residue having an important role in the flavocytochrome b2 reaction. Both the kcat and kcat/Klactate values decrease by 3–4 orders of magnitude, in line with the expectations for mutation of an active site base. The steady-state kinetic properties of the intact enzyme with ferricyanide as the electron acceptor agree with a previous study of this mutant by Gaume et al. (16). However, the additional kinetic data presented here do not support the previous conclusion that the residual activity seen with preparations of the mutant protein is due to small amounts of the wild-type enzyme produced by ribosomal incorporation of histidine at position 373. If that were the case the isotope effects on the mutant enzyme would be identical to those on the wild-type enzyme, and the rate constant for flavin reduction would not show a similar decrease. Instead, the kinetic parameters in Table 2 directly reflect the effects of the mutation on the enzyme. The kinetic data establish that mutation of His373 to glutamine primarily alters flavin reduction. The effects of the mutation on the intact protein are mirrored in the effects on the isolated flavin domain, for which the kcat value clearly does not include electron transfer to the heme. Even more explicitly, the decrease of 3,500-fold in the rate constant for flavin reduction measured in single turnover analyses directly identifies the decrease in this kinetic parameter as the critical effect of the mutation.
While there has been substantial controversy regarding the mechanism of lactate oxidation by flavocytochrome b2 and the other flavoprotein hydroxy acid oxidases (22, 24), the mechanism shown in Figure 4 accommodates the vast majority of kinetic and structural studies to date. Here, the role of His373 is to abstract the lactate hydroxyl proton prior to hydride transfer; the negative charge which develops on the oxygen is stabilized by Tyr254. The effects of mutating Tyr254 to phenylalanine (19) provide a benchmark for the expected effects of mutating His373. In the Y254F enzyme, the rate constant for flavin reduction decreases 35-fold. More informatively in terms of insight into the catalytic details, that mutant enzyme exhibits a primary deuterium isotope effect on kcat of 4.5 and a solvent isotope effect of 1.4, while there is no effect of D2O on flavin reduction in the wild-type enzyme (19). These results were previously interpreted as resulting from a change in mechanism in the mutant enzyme to a more synchronous cleavage of the OH and CH bonds, as the loss of the interaction stabilizing the alkoxide results in a delay in OH bond cleavage until hydride transfer is initiated. Mutagenesis of the base responsible for abstracting the lactate hydroxyl proton would be anticipated to have qualitatively the same effect on the reaction, in that loss of the lactate hydroxyl proton would not occur until the decreasing electron density at the alpha carbon due to transfer of the hydride decreased the pKa of the oxygen sufficiently for deprotonation to occur in the absence of the histidine. This synchronous cleavage of the two bonds should be reflected in solvent and primary kinetic isotope effects with values close to those seen with the Y254F enzyme. Indeed, this is the result upon mutagenesis of the histidine in choline oxidase which abstracts the substrate hydroxyl proton prior to hydride transfer (40). However, the isotope effects summarized in Table 2 do not agree with this prediction. The primary isotope effects on both the intact mutant protein and on the flavin domain are significantly smaller than the wild-type values, while the solvent isotope effects are very close to unity.
The isotope effects suggest that the greatest effect of the mutation is not on a bond cleavage step. The structure of the mutant enzyme provides an explanation for the altered activity. While the glutamine occupies the same space in the mutant protein that the histidine does in the wild-type enzyme, the mutation has significant effects on the active site structure. The binding of pyruvate is altered in the mutant protein, suggesting that the binding of the substrate lactate is also altered. The mutation results in a change in the interaction between Asp282 and the glutamine at position 373 which is propagated down the polypeptide backbone, such that several active site residues have altered positions in the mutant protein. Of the residues showing altered positions in the mutant protein, Arg289 is probably the most critical. This residue is conserved throughout the family of flavoproteins which oxidize α-hydroxy acids. The side chain of Arg289 interacts with that of Arg376, the key residue for binding the carboxylate of the substrate (21). The conservative mutation of Arg289 to lysine has been described previously and provides a measure of the effect on activity which any change in this residue will have. The R289K mutation results in a decrease in the kcat value of about 30-fold and a decrease in the kcat/Klactate value of about 90-fold. Based on the structure of the mutant, the decrease in activity in the mutant protein was attributed to altered electrostatic interactions in the active site (21). The H373Q enzyme described here also shows altered electrostatics in the active site (Figure 6).
A straightforward explanation activity of the H373Q enzyme is that there are two contributions to the decrease in activity. The mutation of the activity site base which removes the hydroxyl proton would be expected to decrease the rate constant for lactate oxidation by 1–2 orders of magnitude, based on the properties of the Y254F enzyme. The altered binding of the substrate and the altered electrostatics in the active site result in a further decrease in activity. However, the kinetic data are not compatible with the minimal model in which these changes simply result in a decreased rate constant for lactate oxidation. In such a case, this step would become rate-limiting, resulting in expression of the intrinsic isotope effects for CH and OH bond cleavage. Instead, the primary and solvent isotope effects are much smaller than those seen with the Y254F enzyme. The data can be explained by a model in which the binding mode seen in the structure is not the reactive form. Instead, the substrate binds to this unreactive form and a conformational change is required to yield a reactive conformation. This introduces an internal commitment which suppresses the isotope effect. The rate constant for the reversal of the conformational change must be at least 10-fold faster than CH bond cleavage to decrease the D(kcat/Km) from the intrinsic value of 5 to the observed value. Similarly, the forward rate constant for the conformational change must be about 7-fold slower than CH bond cleavage to reduce the Dkcat value of the flavin domain to 1.5. H373Q flavocytochrome b2 thus provides an example of a mutation in which the structural effects of the mutation on catalysis are comparable to the effects on chemical steps. This observation raises the possibility that the larger effects of mutating this residue in other members of this family of flavoproteins are due primarily to structural perturbations.
Footnotes
This work was supported in part by grants from the NIH to PFF (GM 58698) and from The Welch Foundation to PFF (A-1245) and JCS (A-0015). The atomic coordinates and structure factors (PDB code 2OZ0) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
References
- 1.Lederer F. Flavocytochrome b2. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. II. CRC Press; Boca Raton: 1991. pp. 153–242. [Google Scholar]
- 2.Urban P, Chirat I, Lederer F. Rat kidney L-2-hydroxyacid oxidase. Structural and mechanistic comparison with flavocytochrome b2 from Baker’s yeast. Biochemistry. 1988;27:7365–7371. doi: 10.1021/bi00419a029. [DOI] [PubMed] [Google Scholar]
- 3.Tsou AY, Ransom SC, Gerlt JA, Buechter DD, Babbitt PC, Kenyon GL. Mandelate pathway of Pseudomonas putida: Sequence relationships involving mandelate racemase, (S)-mandelate dehydrogenase, and benzoylformate decarboxylase and expression of benzoylformate decarboxylase in Escherichia coli. Biochemistry. 1990;29:9856–9862. doi: 10.1021/bi00494a015. [DOI] [PubMed] [Google Scholar]
- 4.Ghisla S, Massey V. L-Lactate oxidase. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. II. CRC Press; Boca Raton: 1991. pp. 243–289. [Google Scholar]
- 5.Giegel DA, Williams CH, Jr, Massey V. L-Lactate 2-monooxygenase from Mycobacterium smegmatis. Cloning, nucleotide sequence, and primary structure homology within an enzyme family. J Biol Chem. 1990;265:6626–6632. [PubMed] [Google Scholar]
- 6.Lindqvist Y, Brändén CI, Mathews R, Lederer F. Spinach glycolate oxidase and yeast flavocytochrome b2 are structurally homologous and evolutionarily related enzymes with distinctly different function and flavin mononucleotide binding. J Biol Chem. 1991;266:3198–3207. [PubMed] [Google Scholar]
- 7.Maeda-Yorita K, Aki K, Sagai H, Misaki H, Massey V. L-Lactate oxidase and L-lactate monooxygenase: mechanistic variations on a common structural theme. Biochimie. 1995;77:631–42. doi: 10.1016/0300-9084(96)88178-8. [DOI] [PubMed] [Google Scholar]
- 8.Cunane LM, Barton JD, Chen ZW, Le KH, Amar D, Lederer F, Mathews FS. Crystal structure analysis of recombinant rat kidney long chain hydroxy acid oxidase. Biochemistry. 2005;44:1521–1531. doi: 10.1021/bi048616e. [DOI] [PubMed] [Google Scholar]
- 9.Xia Z-x, Mathews FS. Molecular structure of flavocytochrome b2 at 2.4 Å resolution. J Mol Biol. 1990;212:837–863. doi: 10.1016/0022-2836(90)90240-M. [DOI] [PubMed] [Google Scholar]
- 10.Balme A, Brunt CE, Pallister RL, Chapman SK, Reid GA. Isolation and characterization of the flavin-binding domain of flavocytochrome b2 expressed independently in Escherichia coli. Biochem J. 1995;309:601–605. doi: 10.1042/bj3090601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reid GA, White S, Black MT, Lederer F, Mathews FS, Chapman SK. Probing the active site of flavocytochrome b2 by site-directed mutagenesis. Eur J Biochem. 1988;178:329–33. doi: 10.1111/j.1432-1033.1988.tb14454.x. [DOI] [PubMed] [Google Scholar]
- 12.Macheroux P, Kieweg V, Massey V, Soderlind E, Stenberg K, Lindqvist Y. Role of tyrosine 129 in the active site of spinach glycolate oxidase. Eur J Biochem. 1993;213:1047–1054. doi: 10.1111/j.1432-1033.1993.tb17852.x. [DOI] [PubMed] [Google Scholar]
- 13.Rouviere-Fourmy N, Capeillere-Blandin C, Lederer F. Role of tyrosine 143 in lactate dehydrogenation by flavocytochrome b2. Primary kinetic isotope effect studies with a phenylalanine mutant. Biochemistry. 1994;33:798–806. doi: 10.1021/bi00169a022. [DOI] [PubMed] [Google Scholar]
- 14.Müh U, Williams CH, Jr, Massey V. Lactate monooxygenase II. Site-directed mutagenesis of the postulated active site base histidine 290. J Biol Chem. 1994;269:7989–7993. [PubMed] [Google Scholar]
- 15.Müh U, Williams CH, Jr, Massey V. Lactate monooxygenase III. Additive contributions of active site residues to catalytic efficiency and stabilization of an anionic transition state. J Biol Chem. 1994;269:7994–8000. [PubMed] [Google Scholar]
- 16.Gaume B, Sharp RE, Manson FDC, Chapman SK, Reid GA, Lederer F. Mutation to glutamine of histidine 373, the catalytic base of flavocytochrome b2 (L-lactate dehydrogenase) Biochimie. 1995;77:621–630. doi: 10.1016/0300-9084(96)88177-6. [DOI] [PubMed] [Google Scholar]
- 17.Lehoux IE, Mitra B. Role of arginine 277 in (S)-mandelate dehydrogenase from Pseudomonas putida in substrate binding and transition state stabilization. Biochemistry. 2000;39:10055–10065. doi: 10.1021/bi000161f. [DOI] [PubMed] [Google Scholar]
- 18.Lehoux IE, Mitra B. (S)-Mandelate dehydrogenase from Pseudomonas putida: Mutation of the catalytic base histidine-274 and chemical rescue of activity. Biochemistry. 1999;38:9948–9955. doi: 10.1021/bi9907532. [DOI] [PubMed] [Google Scholar]
- 19.Sobrado P, Fitzpatrick PF. Solvent and primary deuterium isotope effects show that lactate CH and OH bond cleavages are concerted in Y254F flavocytochrome b2, consistent with a hydride transfer mechanism. Biochemistry. 2003;42:15208–15214. doi: 10.1021/bi035546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanders SA, Williams CH, Jr, Massey V. The roles of two amino acid residues in the active site of L-lactate monooxygenase. J Biol Chem. 1999;274:22289–22295. doi: 10.1074/jbc.274.32.22289. [DOI] [PubMed] [Google Scholar]
- 21.Mowat CG, Beaudoin I, Durley RCE, Barton JD, Pike AD, Chen ZW, Reid GA, Chapman SK, Mathews FS, Lederer F. Kinetic and crystallographic studies on the active site Arg289Lys mutant of flavocytochrome b2 (yeast L-lactate dehydrogenase) Biochemistry. 2000;39:3266–3275. doi: 10.1021/bi9925975. [DOI] [PubMed] [Google Scholar]
- 22.Lederer F. The mechanism of flavoprotein-catalyzed alpha-hydroxy acid dehydrogenation, revisited. In: Stevenson KJ, Massey V, Williams CH, editors. Flavins and Flavoproteins 1996. University of Calgary Press; 1996. pp. 545–553. [Google Scholar]
- 23.Fitzpatrick PF. Substrate dehydrogenation by flavoproteins. Acc Chem Res. 2001;34:299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 24.Fitzpatrick PF. Carbanion versus hydride transfer mechanisms in flavoprotein-catalyzed dehydrogenations. Bioorg Chem. 2004;32:125–139. doi: 10.1016/j.bioorg.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J Am Chem Soc. 2000;122:12896–12897. [Google Scholar]
- 26.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Mechanistic studies of the flavoenzyme tryptophan 2-monooxygenase: Deuterium and 15N kinetic isotope effects on alanine oxidation by an L-amino acid oxidase. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ralph EC, Fitzpatrick PF. pH and kinetic isotope effects on sarcosine oxidation by N-methyltryptophan oxidase. Biochemistry. 2005;44:3074–3081. doi: 10.1021/bi047716h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubois J, Chapman SK, Mathews FS, Reid GA, Lederer F. Substitution of Tyr254 with Phe at the active site of flavocytochrome b2: Consequences on catalysis of lactate dehydrogenation. Biochemistry. 1990;29:6393–6400. doi: 10.1021/bi00479a008. [DOI] [PubMed] [Google Scholar]
- 29.Sobrado P, Daubner SC, Fitzpatrick PF. Probing the relative timing of hydrogen abstraction steps in the flavocytochrome b2 reaction with primary and solvent deuterium isotope effects and mutant enzymes. Biochemistry. 2001;40:994–1001. doi: 10.1021/bi002283d. [DOI] [PubMed] [Google Scholar]
- 30.Labeyrie F, Baudras A, Lederer F. Flavocytochrome b2 or L-Lactate cytochrome c reductase from yeast. Methods Enzymol. 1978;53:238–256. doi: 10.1016/s0076-6879(78)53030-9. [DOI] [PubMed] [Google Scholar]
- 31.Iwatsubo I, Mevel-Ninio M, Labeyrie F. Rapid kinetic studies of partial reactions in the heme free derivative of L-lactate cytochrome c oxidoreductase (flavocytochrome b2); the flavodehydrogenase function. Biochemistry. 1977;16:3558–3566. doi: 10.1021/bi00635a009. [DOI] [PubMed] [Google Scholar]
- 32.McPherson A. Preparation and analysis of protein crystals. John Wiley; New York: 1982. [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 35.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystal D. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 36.Kantardjieff KA, Hochtl P, Segelke BW, Tao FM, Rupp B. Concanavalin A in a dimeric crystal form: revisiting structural accuracy and molecular flexibility. Acta Crystal D. 2002;58:735–743. doi: 10.1107/s0907444901019588. [DOI] [PubMed] [Google Scholar]
- 37.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 38.Miles CS, Rouviere-Fourmy N, Lederer F, Mathews FS, Reid GA, Black MT, Chapman SK. Tyr-143 facilitates interdomain electron transfer in flavocytochrome b2. Biochem J. 1992;285:187–192. doi: 10.1042/bj2850187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunane LM, Barton JD, Chen ZW, Welsh FE, Chapman SK, Reid GA, Mathews FS. Crystallographic study of the recombinant flavin-binding domain of baker’s yeast flavocytochrome b2: comparison with the intact wild-type enzyme. Biochemistry. 2002;41:4264–4272. [PubMed] [Google Scholar]
- 40.Ghanem M, Gadda G. On the catalytic role of the conserved active site residue His466 of choline oxidase. Biochemistry. 2005;44:893–904. doi: 10.1021/bi048056j. [DOI] [PubMed] [Google Scholar]