

Abstract
The T box transcription antitermination system is a riboswitch found primarily in Gram-positive bacteria which monitors the aminoacylation of the cognate tRNA and regulates a variety of amino acid-related genes. Novel 4,5-disubstituted oxazolidinones were identified as high affinity RNA molecular effectors that modulate the transcription antitermination function of the T box riboswitch.
Identifying RNA ligands that modulate transcription regulation is an important area for drug discovery that has been only minimally explored to date. One potential therapeutic target is the T box transcription antitermination mechanism. This mechanism regulates many amino acid-related genes, including aminoacyl-tRNA synthetase genes, and is found predominantly in Gram-positive bacteria.1 The T box RNAs are members of the “riboswitch” family in which nascent RNAs directly sense effector molecules to control gene expression.2–4 The T box genes contain a complex set of structural elements within the 5′ untranslated region of their mRNAs (the “leader region”). These elements include a transcription termination signal that abrogates synthesis of the full-length mRNA and a competing antiterminator element. Read-through of the terminator, and expression of the downstream gene, is dependent on binding of a specific uncharged tRNA to the nascent RNA transcript; each gene in the T box family responds independently to the cognate uncharged tRNA.5 The T box antitermination mechanism can function in the absence of additional cellular factors,6 and the antiterminator RNA element is a critical component of the mechanism.5 The leader RNA-tRNA interaction stabilizes the antiterminator element, thereby preventing formation of the competing terminator element (Figure 1). The antiterminator element is highly conserved and has been extensively characterized by genetic, biochemical and structural biology approaches.7–9
Figure 1.
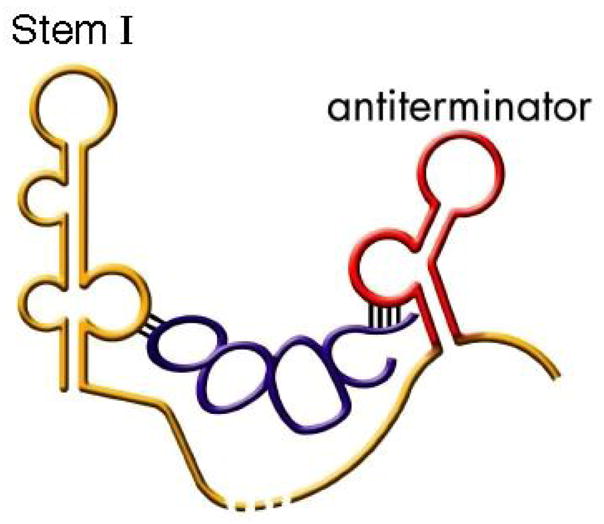
Schematic of tRNA (blue) binding to T box leader RNA. The tRNA anticodon loop base pairs with the Specifier Sequence in Stem I while the tRNA acceptor end base pairs with nucleotides in the antiterminator (red).1
A significant challenge in rational ligand design for RNA structure-specific binding is to achieve both high affinity and excellent tertiary structure specificity. Aminoglycosides, the most widely studied RNA ligands, bind primarily in divalent cation binding sites.10–12 The electrostatic attraction between the multiple protonated amino groups and the negatively charged RNA phosphate backbone leads to very high affinities. However, due to the ubiquitous presence of divalent cation binding sites in RNA, primarily for tertiary fold stabilization,13 the aminoglycosides readily bind many RNAs14 thus reducing their utility for RNA structure-specific ligand design. A variety of other RNA ligands have been investigated,15–21 but few have met the challenge of achieving high specificity for unique RNA structures and high affinity without relying on significant electrostatic interactions. A preliminary investigation of 4,5-disubstituted oxazolidinones22 demonstrated that these ligands may have the potential for specific structural recognition of T box antiterminator RNA.
Functionally relevant models of the T box antiterminator element have been developed.8 AM1A is a model of the wild-type antiterminator and is fully functional in vivo in the context of the full-length leader sequence.7 C11U is a reduced function variant; the corresponding C to U change in the full T box leader sequence results in a significant decrease in antitermination efficiency in vivo23 and in vitro (N.G. and T.H., unpublished). The C11U model RNA exhibits reduced affinity for tRNA in vitro, indicating a strong correlation between structural recognition in the tRNA complex in vitro and antitermination efficiency.8,24 The reduction in activity and affinity along with evidence for structural differences compared to AM1A9 make C11U an excellent specificity control.
We recently reported a novel class of 4,5-disubstituted oxazolidinones (1)25 that bind RNA without extensive reliance on electrostatic interactions.22 These compounds exhibited good binding specificity and affinity for AM1A compared to C11U, but they have relatively poor water solubility. We sought a method to improve water solubility and retain the affinity and specificity previously observed. Our solution was to link a series of alkyl/aryl groups to the oxazolidinone ring via a basic amine (e.g. 2). We predicted that this could be accomplished via the reaction of aziridine 3 with an appropriate amine. Reaction of aziridine 325 with a secondary amine provided oxazolidinones 4a – 4d in good yields. A small group of amines was initially chosen. As our initial lead compound had a simple phenyl ring in place of the NR2 group, we wanted to include aromatic rings on the amine. This group of amines includes N-methyl aniline, N-methyl phenethyl amine, N-phenyl piperazine and a single non-aryl substituted amine, morpholine. Reaction of 4 with an excess of an acid chloride provided compounds 2a, 2b, 2c, 2e and 2f directly.26 For the majority of these compounds we chose R′ = PhCH2 as this was the optimal ester in our previous lead compound. We chose two other R′ groups, and one R′ = nC7H15 was linked to the N-methyl aniline derivative 2c to provide another direct analogue of 1. We also included a carbamate in place of the ester to improve water solubility and potentially provide additional sites for noncovalent contacts with the RNA target. This compound (2d) was readily prepared by treatment of 2a with TFA, followed by neutralization and reaction with 4-acetylphenylisocyanate. All compounds were converted to their hydrochloride salts prior to biological evaluation.
These compounds differ significantly in substitution pattern from other known oxazolidinone RNA ligands (e.g., 3,5-disubstituted oxazolidinones such as the antibiotic linezolid).27–29 While there have been structure activity relationship studies of the antibacterial activity of linezolid analogs,30–34 little has been reported regarding RNA recognition.
RNAs 3′-Fl-18-Rh-AM1A and 3′-Fl-18-Rh-C11U were prepared and a fluorescence resonance energy transfer (FRET) binding assay22,35 performed (Table 1). Compounds 2a and 2b are the closest structurally to 1 with only an amine inserted between the oxazolidinone and the phenyl ring. These two compounds differ at the ester substitution, with 2a retaining the phenyl acetate ester of 1 and 2b having an octanoate ester. Compound 2a shows similar affinity to C11U as 1 while showing somewhat worse affinity to AM1A. Compound 2b shows excellent and roughly equal affinity to both AM1A and C11U. The phenylpiperazine substituted oxazolidinones (2c, 2d) both show excellent affinity to AM1A. Oxazolidinones 2b, 2c and 2d bound T box antiterminator model RNA with low micromolar to nanomolar Kd values. These Kd values rival those of the aminoglycosides for binding to AM1A (neomycin Kd = 8 μM).35 This high affinity without extensive reliance on electrostatic attraction (only two protonatable amines vs. six for neomycin) points to the importance of non-electrostatic interactions in the binding of oxazolidinones to RNA. Compound 2c bound to AM1A with eight-fold greater affinity than C11U, while 2d bound to both model RNAs with roughly similar affinity. While structural differences between 2c and 2d are minimal, the carbamate moiety of 2d may provide both additional hydrogen bonding capabilities and restricted rotation around the C-N bond relative to 2c. Compound 2e in which the ring of the N-substitution is an N-methyl phenethyl amine can be viewed as a conformationally less constrained N-phenyl piperazine. This compound showed substantially less affinity to AM1A relative to either 2c or 2d. Compound 2f in which the aromatic ring on the amine is completely gone shows reasonable affinity to AM1A. Given the good affinity and selectivity of 2c and the good affinity and poor selectivity of 2d, we chose to further examine both of these compounds in further studies. These two molecules differ only in the acyl substitution suggesting that significant non-covalent contacts are mediated through this substituent.
Table 1.
Oxazolidinone affinity for antiterminator model RNAa
With the FRET-labeled antiterminator RNA used in the binding studies, the maximal change in relative fluorescence (Frel) correlates with structure-specific conformational changes in the RNA induced upon ligand binding.35 There was a significant difference in the maximal Frel for 2c binding to AM1A (max. Frel = 0.2) compared to 2d (max. Frel = 0.3), strongly indicating that the two oxazolidinones bind AM1A in different manners.
Further evidence for different binding modes was observed in enzymatic cleavage assay patterns (Figure 3). No significant change in the RNase A cleavage of AM1A was observed in the presence of oxazolidinone 2c. However, in the presence of 2d the relative band intensities decreased for positions 6 (37%), 7 (69%) and 8 (47%). With RNase T1 cleavage of AM1A in the presence of 2c, there was enhanced cleavage at position 15 (70%) and reduced cleavage in several locations, most notably positions 7 (44%), 8 (41%) and 9 (57%). With 2d, however, there was an overall decrease in RNase T1 enzymatic cleavage. The differential enzymatic cleavage patterns of 2c compared to 2d may be due to different RNA binding modes.
Figure 3.
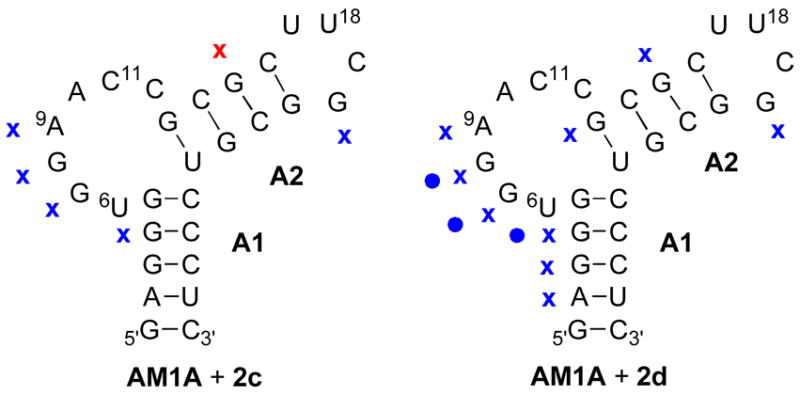
Summary of enzymatic probing assays of AM1A in the presence of 2c and 2d with RNase T1 (x) and RNase A (circle) indicating enhanced (red) or reduced (blue) cleavage in the presence of the ligand.
The effect of 2c and 2d on antiterminator RNA function was tested using an in vitro transcription antitermination system. Transcription of the Bacillus subtilis glyQS T box gene with B. subtilis RNA polymerase in the absence of tRNA resulted in efficient termination (Figure 4); addition of uncharged tRNAGly resulted in a 20-fold increase in read-through of the termination site, as previously reported.6 Addition of 2c (1.3 mM) caused a 40% reduction in the efficiency of tRNAGly-dependent antitermination, but had no effect on transcription of a non-T box DNA template (data not shown). The reduction in antitermination is likely to be due to competitive inhibition since the affinity of 2c for binding to AM1A decreased five-fold in the presence of cognate tRNA (1.25 μM) to a Kd of 64 ±16 μM (R2 = 0.9). In contrast, 2d (0.8 mM) resulted in increased antitermination in the presence or absence of tRNA, suggesting that this compound stabilizes the antiterminator element, obviating the requirement for tRNA binding. The different effects of 2c and 2d on antitermination are consistent with different modes of interaction with the antiterminator model RNAs. The relatively high concentration of 2c required to compete with the acceptor end of tRNA for binding the antiterminator and to inhibit antitermination is reasonable given the strong tRNA-model antiterminator RNA affinity (Kd = 0.02 μM)24 and given that the tRNA can pre-bind the leader at the Specifier Sequence before the antiterminator is transcribed,36 thus likely further enhancing affinity in the context of the in vitro transcription antitermination assay.
Figure 4.
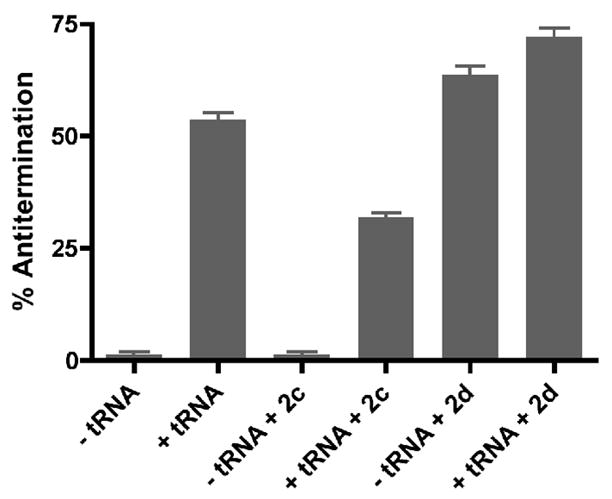
Effect of 2c and 2d on B. subtilis glyQS antitermination activity in vitro (n = 3).
In this report, we have identified 4,5-disubstituted oxazolidinones that both bind to the T box antiterminator RNA element and directly affect antitermination. These compounds are amine substituted analogues of previously reported oxazolidinones. The inclusion of the basic amine in the compound significantly enhances the affinity. The affinity and structural specificity was sufficiently high that 2c acted as an inhibitory molecular effector and disrupted in vitro antitermination. Compound 2d acted as an enhancing molecular effector leading to tRNA-independent antitermination in vitro. The differing binding modes of 2c and 2d with antiterminator RNA provides strong evidence that rational drug design strategies can be used to selectively develop high affinity ligands (rivaling the nM affinity observed with 2d) that retain the antitermination inhibitory activity of 2c.
Supplementary Material
Figure 2.
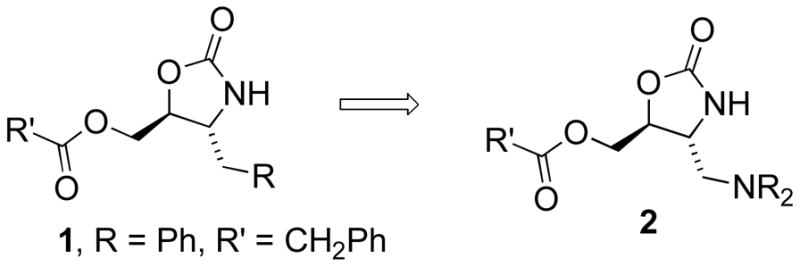
Previously prepared oxazolidinone RNA binding agents (1) and proposed new analogues 2.
Scheme 1.
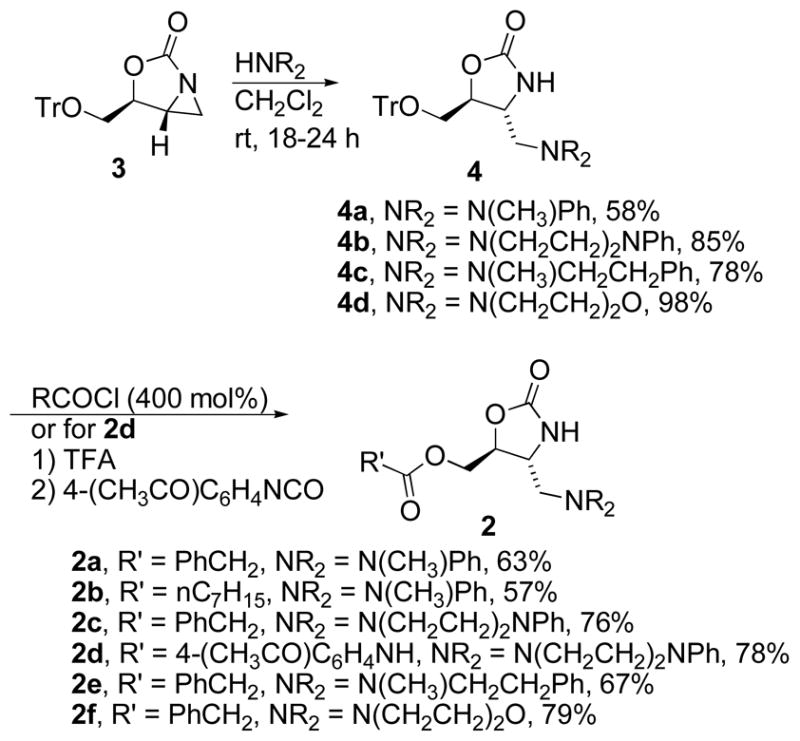
Synthesis of amine substituted oxazolidinones 3a – 3f.
Acknowledgments
We thank the NIH (JVH, Grants GM61048, GM073188; TMH, Grant GM47823), the Southeastern Ohio Science and Technology Commercialization Initiative (SOSCI) and Ohio University, through the NanoBioTechnology Initiative (NBTI), for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference and Notes
- 1.Grundy FJ, Henkin TM. Front Biosci. 2003;8:d20. doi: 10.2741/908. [DOI] [PubMed] [Google Scholar]
- 2.Grundy FJ, Henkin TM. Curr Opin Microbiol. 2004;7:126. doi: 10.1016/j.mib.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Henkin TM, Grundy FJ. Cold Spring Harbor Symp Quant Biol. 2006;71:1. doi: 10.1101/sqb.2006.71.020. [DOI] [PubMed] [Google Scholar]
- 4.Tucker BJ, Breaker RR. Curr Opin Struct Biol. 2005;15:342. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Grundy FJ, Henkin TM. Cell. 1993;74:475. doi: 10.1016/0092-8674(93)80049-k. [DOI] [PubMed] [Google Scholar]
- 6.Grundy FJ, Winkler WC, Henkin TM. Proc Natl Acad Sci USA. 2002;99:11121. doi: 10.1073/pnas.162366799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grundy FJ, Moir TR, Haldeman MT, Henkin TM. Nucleic Acids Res. 2002;30:1646. doi: 10.1093/nar/30.7.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerdeman MS, Henkin TM, Hines JV. Nucleic Acids Res. 2002;30:1065. doi: 10.1093/nar/30.4.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerdeman MS, Henkin TM, Hines JV. J Mol Biol. 2003;326:189. doi: 10.1016/s0022-2836(02)01339-6. [DOI] [PubMed] [Google Scholar]
- 10.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Science. 1996;274:1367. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 11.Hermann T, Westhof E. J Mol Biol. 1998;276:903. doi: 10.1006/jmbi.1997.1590. [DOI] [PubMed] [Google Scholar]
- 12.Vicens Q, Westhof E. Chembiochem. 2003;4:1018. doi: 10.1002/cbic.200300684. [DOI] [PubMed] [Google Scholar]
- 13.Draper DE. Ann Rev Biophys Biomol Struct. 2005;34:221. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 14.Hermann T. Biochimie. 2002;84:869. doi: 10.1016/s0300-9084(02)01460-8. [DOI] [PubMed] [Google Scholar]
- 15.Gelus N, Bailly C, Hamy F, Klimkait T, Wilson WD, Boykin DW. Bioorg Med Chem. 1999;7:1089. doi: 10.1016/s0968-0896(99)00041-3. [DOI] [PubMed] [Google Scholar]
- 16.Hansen LH, Mauvais P, Douthwaite S. Mol Microbiol. 1999;31:623. doi: 10.1046/j.1365-2958.1999.01202.x. [DOI] [PubMed] [Google Scholar]
- 17.Mayer M, James TL. J Am Chem Soc. 2004;126:4453. doi: 10.1021/ja0398870. [DOI] [PubMed] [Google Scholar]
- 18.Tor Y. Chembiochem. 2003;4:998. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]
- 19.Sinha R, Hossain M, Kumar GS. Biochemica et biophysica acta. 2007;1770:1636. doi: 10.1016/j.bbagen.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 20.Soonsil H, Lee KH, Yu J. Bioorg Med Chem Lett. 2006;16:4757. doi: 10.1016/j.bmcl.2006.06.094. [DOI] [PubMed] [Google Scholar]
- 21.Kazuhiko N, Horie S, Goto Y, Kobori A, Hagihara S. Bioorg Med Chem. 2006;14:5384. doi: 10.1016/j.bmc.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 22.Means JA, Katz SJ, Nayek A, Anupam R, Hines JV, Bergmeier SC. Bioorg Med Chem Lett. 2006;16:3600. doi: 10.1016/j.bmcl.2006.03.068. [DOI] [PubMed] [Google Scholar]
- 23.Rollins SM, Grundy FJ, Henkin TM. Mol Microbiol. 1997;25:411. doi: 10.1046/j.1365-2958.1997.4851839.x. [DOI] [PubMed] [Google Scholar]
- 24.Means JA, Wolf S, Agyeman A, Burton JS, Simson CM, Hines JV. Chem Biol Drug Design. 2007;69:139. doi: 10.1111/j.1747-0285.2007.00476.x. [DOI] [PubMed] [Google Scholar]
- 25.Katz SJ, Bergmeier SC. J Comb Chem. 2002;4:162. doi: 10.1021/cc010050f. [DOI] [PubMed] [Google Scholar]
- 26.Bergmeier SC, Arason KM. Tetrahedron Lett. 2000;41:5799. [Google Scholar]
- 27.Matassova N, Rodnina M, Endermann R, Kroll H, Pleiss U, Wild H, Wintermeyer W. RNA. 1999;5:939. doi: 10.1017/s1355838299990210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong L, Kloss P, Douthwaite S, Andersen N, Swaney S, Shinabarger D, Mankin A. J Bacteriol. 2000;182:5325. doi: 10.1128/jb.182.19.5325-5331.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barbachyn MR, Ford CW. Angew Chem Int Ed. 2003;42:2010. doi: 10.1002/anie.200200528. [DOI] [PubMed] [Google Scholar]
- 30.Das B, Rudra S, Yadav A, Ray A, Rao AVS, Raja S, Soni A, Saini S, Shukla S, Pandya M, Bhateja P, Malhotra S, Mathur T, Arora SK, Rattan A, Mehta A. Bioorg Med Chem Lett. 2005;15:4261. doi: 10.1016/j.bmcl.2005.06.063. [DOI] [PubMed] [Google Scholar]
- 31.Das J, Rao CVL, Sastry T, Roshaiah M, Sankar PG, Khadeer A, Kumar MS, Mallik A, Selvakumar N, Iqbal J, Trehan S. Bioorg Med Chem Lett. 2005;15:337. doi: 10.1016/j.bmcl.2004.10.073. [DOI] [PubMed] [Google Scholar]
- 32.Dixit PP, Nair PS, Patil VJ, Jain S, Arora SK, Sinha N. Bioorg Med Chem Lett. 2005;15:3002. doi: 10.1016/j.bmcl.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 33.Gravestock MB. Curr Opin Drug Discovery Dev. 2005;8:469. [PubMed] [Google Scholar]
- 34.Reck F, Zhou F, Girardot M, Kern G, Eyermann CJ, Hales NJ, Ramsay RR, Gravestock MB. J Med Chem. 2005;48:499. doi: 10.1021/jm0400810. [DOI] [PubMed] [Google Scholar]
- 35.Means JA, Hines JV. Bioorg Med Chem Lett. 2005;15:2169. doi: 10.1016/j.bmcl.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Grundy FJ, Yousef MR, Henkin TM. J Mol Biol. 2005;346:73. doi: 10.1016/j.jmb.2004.11.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.