

Abstract
Desmosdumotin C (1) and its analogs previously showed potent, selective in vitro anticancer activity. To explore structure-activity-relationships of 1 and further increase potency and selectivity, fifteen novel analogs (7–15 and 21–26) were synthesized, and evaluated for cytotoxity against several human tumor cell lines, as well as inhibition of human endothelial (HUVEC) replication. 4-Bromo-3′,3′,5′-tripropyl analog 26 showed significant cytotoxity against A549, A431, 1A9, and HCT-8 with ED50 values of 1.0, 1.2, 0.9, and 1.3 μg/mL respectively. Compound 26 also strongly inhibited the growth of matched tumor cells, KB-VIN and its parent cell KB. Furthermore, analogs 13 and 21 were over fivefold more potent against KB-VIN than KB. Bromination of ring-B and tripropyl functionalization of ring-A enhanced activity, while alkylation of ring-B promoted KB-VIN/KB selectivity. 2-Furyl analog 16 showed selective activity against HUVEC, suggesting that it may have potential as a new prototype for angiogenesis inhibition.
Introduction
Natural products are a significant source of drug candidates. An impressive number of modern drugs have been developed from natural sources, especially from plants used as traditional folk medicines.1 Thus, drug discovery from medicinal plants plays an important role in the treatment and prevention of various human diseases, and the continuous importance of natural products in modern medicine has been highlighted in several recent reviews and reports.2–6 Our research interest is the discovery and development of novel anticancer drugs from natural plants. Currently, about three-quarters of anticancer drugs come from either natural products or their derivatives.7 Cancer is a leading cause of death worldwide accounting for thirteen percent of all deaths in 2005,8 even though many effective and diverse cancer treatments have been approved and are used. Major problems associated with cancer chemotherapy include undesirable toxic side effects and multidrug resistance. Therefore, a pressing need to develop more effective antitumor drugs still remains.
Desmos dumosus (Roxb.) is a climber plant found in alluvial forests in southern Asia, and has been used in Chinese folk medicine as an antimalarial, insecticidal, antirheumatic, antispasmodic, and analgesic agent.9 Recently, some novel bioactive flavonoids, named desmosdumotins B and C, were isolated from the plant root.10, 11 Desmosdumotin C (1) has a distinctive chalcone skeleton with an unusual non-aromatic A ring possessing a gem-dimethyl group on C-3′ and methyl group on C-5′. It showed significant and selective in vitro cytotoxicity against 1A9 (ovarian cancer) and A549 (human lung carcinoma) cell lines with IC50 values of 4.0 and 3.5 (μg/mL, respectively.10 In addition, it was more active against drug-resistant KB-VIN cells than against the parent KB (epidermoid nasopharyngeal carcinoma) cell line. Thus, 1 is a promising new lead for further new antitumor analog development. We previously achieved a simple first total synthesis of 111 as well as some modifications of the A- and B-rings and reported the cytotoxic activity data against four tumor cell lines, 1A9, A549, KB and KB-VIN.12 Among the tested compounds, 4-bromodesmosdumotin C (2) showed two- to three-fold enhanced activity compared with 1. This promising result encouraged us to continue the modification of this series to develop novel anticancer drug candidates. In this paper, we describe further modifications of the A- and B-rings as well as evaluation of newly and previously synthesized analogs against seven human tumor cell lines, A549, 1A9, KB, KB-VIN, A431 (epidermoid skin carcinoma), HCT-8 (colon adenocarcinoma), and PC-3 (prostate cancer), as well as HUVEC.
Chemistry
The simplicity of our accomplished synthesis11 of 1 allows easy modification of the A-ring, by using another alkyl iodide rather than methyl iodide in the first step, and the B-ring, by using a different aromatic aldehyde from benzaldehyde in the final step (Scheme 1). First, nine B-ring modified analogs, 7–15, were synthesized from intermediate 29 using 4-fluorophenylaldehyde, o- and p-tolualdehyde, 4-ethylbenzaldehyde, 2,6- and 3,5-dimethylbenzaldehyde, 2,4,6-trimethylbenzaldehyde, 2-vinylbenzaldehyde, and 1-naphthaldehyde under basic aldol conditions. Second, modification of the A-ring to introduce an ethyl or propyl group at the C-3 and C′-5′ positions was accomplished by the following method. Treatment of trihydroxyacetophenone 28 with 3 mol eq. of ethyl or propyl iodide in the presence of sodium methoxide provided the corresponding trialkylacetophenones (32 and 33, respectively). Similarly, treatment of 3012 with 1 mol eq. of ethyl iodide afforded 31. Selective methylation of the resulting trialkylacetophenones, 31–33, with an excess of TMSCHN2 at low temperature, followed by aldol reaction with benzaldehyde gave 21, 22, and 25. The aldol reaction of 35 and 36 with 4-methyl, 4-ethyl, or 4-bromobenzaldehyde gave A- and B-ring analogs, 23, 24, and 26, respectively. All synthesized compounds, except for 7, 12, and 15, exist as a mixture of two tautomeric isomers as discussed in our prior papers.12–13 Compounds 1–6,16–19, 20 and 27 were synthesized previously.12
Scheme 1.

Syntheses of 1-Analogs: B-ring Modifications
a See reference 11.b Based on recovered starting material.c See reference 12.
Results and Discussion
Together with 1 and previously synthesized analogs, all newly synthesized compounds were evaluated for in vitro anticancer activity against several human tumor cell lines, A549, A431, 1A9, HCT-8, PC-3, KB and KB-VIN, and against HUVEC, a normal cell useful for assessing anti-angiogenic potential. The average ED50 values (μg/mL) are listed in Table 1.
Table 1.
Cytotoxic Activity Data for 1-Analogs
| ED50 (μg/mL)a | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | A549 | A431 | 1A9 | HCT-8 | PC-3 | KB | KB-VIN | HUVEC |
| 1 | 3.5 | 5.1 | 3.5 | 3.7 | 11.1 | 4.0 | 3.0 | 2.1 |
| 2 | 1.4 | 9.0 | 1.1 | 7.4 | 17.7 | 1.7 | 1.9 | 3.6 |
| 3 | 3.0 | 9.2 | 2.8 | 5.6 | 18.2 | 3.6 | 2.9 | 2.5 |
| 4 | 2.8 | 4.5 | 2.9 | 2.9 | 14.3 | 3.3 | 2.4 | 1.6 |
| 5 | 2.4 | 6.6 | 2.5 | 4.1 | 15.6 | 3.3 | 2.8 | 1.7 |
| 6 | 3.1 | 14.9 | 2.9 | 14.8 | NAb | 3.8 | 3.8 | 4.0 |
| 7 | 3.3 | 4.1 | NTC | 3.6 | 7.2 | 3.7 | 2.7 | 2.0 |
| 8 | 4.3 | 5.1 | NT | 4.1 | 8.2 | 3.9 | 1.9 | 2.4 |
| 9 | 4.6 | 5.2 | NT | 4.0 | 9.0 | 4.1 | 1.6 | 2.3 |
| 10 | 6.1 | 12.0 | 5.3 | 10.5 | 15.1 | 6.9 | 1.9 | NT |
| 11 | 8.3 | 10.1 | NT | 7.8 | 17.1 | 7.1 | 2.1 | 3.0 |
| 12 | 4.3 | 4.0 | NT | 2.9 | 8.8 | 3.1 | 1.6 | 2.5 |
| 13 | 8.0 | 20.0 | 6.2 | 8.4 | NA | NA | 1.84 | NT |
| 14 | 2.2 | 2.6 | NT | 2.1 | 4.6 | 1.9 | 1.1 | 1.1 |
| 15 | 3.6 | 4.1 | NT | 2.3 | 9.0 | 2.8 | 1.3 | 1.8 |
| 16 | 10.0 | 14.7 | 7.9 | 17.5 | 20.6 | 9.9 | 8.8 | 2.4 |
| 17 | 4.5 | 10.8 | 4.0 | 8.6 | 18.2 | 5.5 | 3.4 | 2.5 |
| 18 | 3.5 | 6.0 | 3.5 | 5.1 | 10.8 | 4.3 | 3.0 | 1.6 |
| 19 | >5 | 10.5 | 4.6 | 5.0 | 11.1 | 7.3 | 3.8 | 2.0 |
| 20 | >5(48) | 7.0 | 3.6 | 3.3 | 14.2 | 4.1 | 2.9 | NA |
| 21 | 5.5 | 13.8 | 3.8 | 9.4 | 18.9 | 4.3 | 0.3 | NT |
| 22 | 3.4 | 5.4 | 2.1 | 2.8 | 7.7 | 2.8 | 1.2 | NT |
| 23 | 4.6 | 10.3 | 4.3 | 9.4 | 15.0 | 3.0 | 1.0 | NT |
| 24 | 3.8 | 9.8 | 3.8 | 8.5 | 15.0 | 3.6 | 2.6 | NT |
| 25 | 1.4 | 1.9 | 1.5 | 1.4 | 3.3 | 0.6 | 0.8 | NT |
| 26 | 1.0 | 1.2 | 0.9 | 1.3 | 2.3 | 0.9 | 0.9 | NT |
| 27 | 9.0 | 14.0 | 8.0 | 15.8 | 17.8 | 10.5 | 6.5 | 2.1 |
See Experimental “Cytotoxic Activity Assay” for cell line descriptions.
Not active: >20 μg/mL
Not tested.
Compound 1 showed activity against A549, 1A9, HCT-8 and HUVEC with ED50 values of 3.5, 3.5, 3.7, and 2.1 μg/mL, respectively, but was less active against A431 and PC-3 cells. Compound 1 was also 1.3-fold more active against KB-VIN cells (ED50 3.0 μg/mL) than the parent KB cell line. Except for 14, 25 and 26, most 1-analogs showed similar activity patterns, as described later.
Significantly enhanced cytotoxic activities were observed with 2, 2-vinyl desmosdumotin C (14), 3′,3′,5′-tripropyl desmosdumotin C (25), and 4-bromo-3′,3′,5′-tripropyl desmosdumotin C (26). Especially, 26 displayed enhanced in vitro antitumor activity against all cell lines with ED50 values of 0.9–2.3 μg/mL. In addition, 26 was not affected by multidrug resistant expressing P-glycoprotein, since it showed similar and potent cell growth inhibition of KB and KB-VIN cell replication (ED50 0.9 μg/mL). The strong activity of 26 could be predicted since it combines the structural features of 4-bromo 2 and 3′,3′,5′-tripropyl 25, which also were quite potent against A549, 1A9, KB, and KB-VIN cell lines. Notably, 25 strongly inhibited the growth of KB and KB-VIN cells with ED50 values of 0.6 and 0.8 μg/mL, respectively.
Interestingly, while most analogs, including 2, were generally less active against PC-3 replication, three compounds, 14, 25 and 26, displayed enhanced activity against PC-3 with ED50 values of 4.6, 3.3 and 2.3 μg/mL, respectively. These three analogs also inhibited the growth of A431 cells with ED50 values of 2.6, 1.9 and 1.2 μg/mL, respectively. Furthermore, in the PC-3, A431, and HCT-8 cell lines, bromination at C-4 (2) decreased cytotoxic activity relative to 1, while fluorination (7) increased activity.
From comparison of the data for 1 with that of 3–5 and 8–9, the insertion of a methoxy or methyl group on the phenyl B-ring had either no effect on or slightly decreased activity, regardless of the substitution position. The 4-ethyl (10), 2,6-dimethyl (11), and trimethyl (13) derivatives also showed decreased activity against all cell lines, although 1 and the 3,5-dimethyl derivative (12) had comparable potencies. However, it is noteworthy that the introduction of an alkyl group on the B-ring enhanced the growth inhibition of the KB-VIN cell line resulting in a two- to five-fold ratio of KB/KB-VIN selectivity (see 8–14). However, the insertion of hetero aromatic ring rather than the phenyl ring did not affect the cytotoxic activity (see 16–18).
For the A-ring analogs, tetramethyl analog 19, dimethyl analog 20 (ceroptene),14 3′,3′-dimethyl-5′-ethyl analog 21, and 4′-demethyl analog 27 were less potent against all cell lines than the parent compound 1. However, 21 showed a remarkably high KB/KB-VIN selectivity ratio of 14.3. Among the triethyl compounds, 23 and 24 were less active than 1, while 22, without a substituent on the B-ring, showed slightly enhanced activity against most cell lines, except for A549 and A431.
Angiogenesis is necessary for tumor growth and metastasis; therefore, it presents another target for cancer treatment. Selected compounds were also evaluated in a standard anti-angiogenesis assay using HUVEC. All tested compounds, except 20, showed potent activity with ED50 values less than 4.0 μg/mL. Compound 14 showed the highest potency with an ED50 value of 1.1 μg/mL. However, this compound also showed strong activity against other tumor cell lines suggesting that the activity against HUVEC may be non-specific. However, analogs 16–18, in which the phenyl B-ring was replaced with 5-membered heteroaromatic rings, demonstrated significant activity against HUVEC with less activity against tumor cell replication. In particular, 2-furyl analog 16 possessed the highest differential with an ED50 value of 2.4 μg/mL against HUVEC compared to ED50 values of 7.9–20.6 μg/mL against the other tumor cell lines. This selective activity suggests that 16 may have potential as a new prototype as an angiogenesis inhibitor.
In summary, the preliminary SAR studies led to the following observations: 1) a bromide function on the B-ring enhanced activity (1 vs 2 and 25 vs 26); 2) the order of potency for C-3′ and -5′ functionalities was 3′,3′,5′-tripropyl > 3′,3′,5′-trimethyl > 3′,3′,5′-triethyl > 3′,3′-dimethyl-5′-ethyl > 3′,3′-dimethyl > 3′,3′,5′,5′-tetramethyl; 3) an alkyl group on the B-ring promoted KB-VIN/KB selectivity; 4) a C-4′methoxy group is required for activity; 5) 5-membered hetero B-rings increased the differential angiogenesis activity. From all results, 26 showed significant promise as a new antitumor drug candidate because of its potent cytotoxity against tumor cell lines and lack of multidrug cross resistance. Focused studies on the KB-VIN/KB selectivity of desmosdumotin series are currently ongoing and the results will be reported elsewhere in the near future.
Experimental Section
Materials and Methods
Melting points were measured with a Fisher Johns melting apparatus without correction. Proton nuclear magnetic resonance (1H NMR) spectra were measured on a 300MHz Varian Gemini 2000 spectrometer using TMS as internal standard. All chemical shifts are reported in ppm. Mass spectra were measured on a PE-SCIEX API 3000 instrument with turbo ion spray source or Agilent-1100, LC/MSD-Trap. Thin-layer chromatography (TLC) and preparative TLC were performed on precoated silica gel GF plates purchased from Merck, Inc. Biotage Flash™ or Isco Companion systems were used for medium pressure column chromatography. Silica gel (200–400 mesh) from Aldrich, Inc. was used for column chromatography. All other chemicals were obtained from Aldrich, Inc.
General Procedures for the Aldol Reactions
A solution of acetyl compound (29 or 34–36) in EtOH-50% aq. KOH (1:1, v/v) and an appropriate aldehyde (excess) was stirred at room temperature. After the reaction was complete by TLC analysis, the mixture was poured into ice-cold 1N HCl, then extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was chromatographed on silica gel with CH2Cl2–hexane as eluent to afford the target compound, which was crystallized from CH2Cl2-hexane.
4-Fluoro desmosdumotin C (7)
Yellow prisms, mp 109–110 °C (CH2Cl2-hexane). 1H-NMR (CDCl3): δ 19.18 (s, 1H, chelated-OH), 8.27 (d, 1H, J = 15.6 Hz, trans-olefinic proton), 7.89 (d, 1H, J = 15.6 Hz, trans-olefinic proton), 7.71-7.60 (m, 2H, Ar-2,6-H), ), 7.12-7.00 (m, 2H, Ar-3,5-H), 3.96 (s, 3H, OCH3), 2.00 (s, 3H, 5′-CH3), 1.38 (s, 6H, 3′-CH3×2). MS m/z 331 (M++1). Anal. (C19H19FO4) C, H, O.
4-Methyl desmosdumotin C (8)
Yellow prisms, mp 110–111 °C (CH2Cl2-hexane). 1HNMR (300 MHz, CDCl3): δ = 19.32 and 18.92 (3:1, each s, 1H, chelated-OH), 8.52 and 8.31 (1:3, each d, 1H, J = 15.7 Hz, olefin), 7.95 and 7.93 (1:3, each d, 1H, J = 15.7 Hz, olefin), 7.62-7.54 (m, 2H, Ar-2′, 6′-H), 7.24-7.17 (m, 2H, Ar-3, 5-H), 3.96 and 3.89 (3:1, each s, 3H, OCH3), 2.00 and 1.96 (3:1, each s, 3H, 5′-CH3), 1.47 and 1.38 (1:3, each s, 6H, 3′-CH3×2). MS m/z 327 (M++1). Anal. (C20H22O4) C, H, O.
2-Methyl desmosdumotin C (9)
Yellow prisms, mp 93–94 °C (CH2Cl2-hexane). 1H NMR (300 MHz, CDCl3): δ = 19.32 and 18.62 (4:1, each s, 1H, chelated-OH), 8.34 (s, 2H), 8.05-7.94 (m, 1H), 7.51-7.25 (m, 3H, Ar-H), 4.10 and 4.03 (4:1, each s, 3H, OCH3), 2.63 (s, 3H, Ar-CH3), 2.15 and 2.01 (4:1, each s, 3H, 5′-CH3), 1.52 (s, 6H, 3′-CH3×2). MS m/z 327 (M++1). Anal. (C20H22O4) C, H, O.
4-Ethyl desmosdumotin C (10)
Yellow prisms, mp 90–91 °C (CH2Cl2-hexane). 1H NMR (300 MHz, CDCl3): δ 19.19 and 18.80 (2:1, each s, 1H, chelated-OH), 8.52 and 8.32 (1:2, each d, 1H, J = 15.9 Hz, olefin), 7.96 and 7.94 (1:2, each d, 1H, J = 15.9 Hz, olefin), 7.62 and 7.60 (1:2, each d, 2H, J = 7.9 Hz, Ar-2, 6-H), 7.23 and 7.22 (1:2, each d, 2H, J = 7.9 Hz, Ar-3, 5-H), 3.95 and 3.89 (2:1, each s, 3H, OCH3), 2.68 (q, 2H, J = 7.7 Hz, Ar-CH2CH3), 2.00 and 1.96 (2:1. each s, 3H, 5′-CH3), 1.47 and 1.38 (1:2, each s, 6H, 3′-CH3×2), 1.25 (t, 3H, J = 7.7 Hz, Ar-CH2CH3). MS m/z 339 (M+−1). Anal. (C21H24O) C, H, O.
2,6-Dimethyl desmosdumotin C (11)
Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.14 and 18.80 (2:1, each s, 1H, chelated-OH), 8.17 (d, 1H, J = 15.7 Hz, olefin), 7.87 (d, 1H, J = 15.7 Hz, olefin), 7.20-7.01 (m, 3H, Ar-3,4,5-H), 3.95 and 3.88 (2:1, each s, 3H, OCH3), 2.44 (s, 6H, Ar-2,6-CH3), 2.00 and 1.92 (2:1, each s, 3H, 5′-CH3), 1.48 and 1.35 (1:2, each s, 6H, 3′-CH3×2). MS m/z 341 (M++1). Anal. (C21H24O4) C, H, O.
3,5-Dimethyl desmosdumotin C (12)
Yellow prisms, mp 105–106 °C (CH2Cl2-hexane). 1H NMR (300 MHz, CDC13): δ 19.20 (s, 1H, chelated-OH), 8.31 (d, 1H, J = 15.7 Hz, olefin), 7.91 (d, 1H, J = 15.7 Hz, olefin), 7.30 (s, 2H, Ar-2′, 6′-H), 7.03 (s, 1H, Ar-4-H), 3.96 (s, 3H, OCH3), 2.34 (s, 6H, Ar-3,5-CH3), 2.00 (s, 3H, 5′-CH3), 1.39 (s, 6H, 3′-CH3×2). MS m/z 341 (M++1). Anal. (C21H24O4) C, H, O.
2,4,6-Trimethyl desmosdumotin C (13)
Yellow prisms, mp 87–88 °C (AcOEt-hexane). 1H NMR (300 MHz, CDC13): δ 19.17 and 18.48 (3:1, each s, 1H, chelated-OH), 8.21-8.09 (m, 1H, olefin), 7.96 and 7.90 (1:3, each d, 1H, J = 15.5 Hz, olefin), 6.92 and 6.90 (1:3, each s, 2H, Ar-3, 5-H), 3.95 and 3.88 (3:1, each s, 3H, OCH3), 2.43 and 2.41 (3:1, each s, 6H, Ar-2,6-CH3), 2.30 and 2.29 (1:3, each s, 3H, Ar-4-CH3), 2.00 and 1.92 (3:1. each s, 3H, 5′-CH3), 1.47 and 1.35 (1:3, each s, 6H, 3′-CH3×2). MS m/z 355 (M++1). Anal. (C22H26O4 · 1/8H2O) C, H.
3-Vinyl desmosdumotin C (14)
Yellow prisms, mp 76–77 °C (CH2Cl2-hexane). 1H NMR (300 MHz, CDC13): δ 19.19 and 18.79 (3:1, each s, 1H, chelated-OH), 8.54 and 8.34 (1:3, each d, 1H, J = 15.9 Hz, olefin), 7.96 and 7.93 (1:3, each d, 1H, J = 15.9 Hz, olefin), 7.67 and 7.65 (1:3, each br s, 1H, Ar-2-H), 7.60 and 7.58 (1:3, each d, 1H, J = 7.4 Hz, Ar-4 or 6-H), 7.47 and 7.45 (1:3, each d, 1H, J = 7.4 Hz, Ar-4 or 6-H), 7.37 and 7.36 (1:3, each t, 1H, Ar-5-H), 6.74 (dd, 1H, J = 10.8 and 17.4 Hz, Ar-CH=CH2), 5.81 (d, 1H, J = 17.4 Hz, Ar-CH=CH2), 5.31 (d, 1H, J = 10.8 Hz, Ar-CH=CH2), 3.96 and 3.89 (3:1, each s, 3H, OCH3), 2.00 and 1.96 (3:1, each s, 3H, 5′-CH3), 1.48 and 1.39 (1:3, each s, 6H, 3′-CH3×2). MS m/z 359 (M++1). Anal. (C21H22O4) C, H, O.
2-[1-Hydroxy-3-(naphthalen-1-yl)allyidene]-5-methoxy-4,6,6-trimethylcyclohex-4-ene-1,3-dione (15)
Yellow prisms, mp 128–129 °C (CH2Cl2-hexane). 1H NMR (300 MHz, CDCl3): δ 19.2 (s, 1H, chelated-OH), 8.83 (d, 1H, J = 15.4 Hz, olefin), 8.42 (d, 1H, J = 15.4 Hz, olefin), 8.28 (d, 1H, J = 7.5 Hz, naphthyl-2 or 8-H), 8.04 (d, 1H, J = 7.5 Hz, naphthyl-2 or 8-H), 7.96-7.84 (m, 2H, naphthyl-H), 7.64-7.46 (m, 3H, naphthyl-H), 7.37 and 7.36 (1:3, each t, 1H, Ar-5-H), 6.74 (dd, 1H, J = 10.8 and 17.4 Hz, Ar-CH=CH2), 5.81 (d, 1H, J = 17.4 Hz, Ar-CH=CH2), 5.31 (d, 1H, J = 10.8 Hz, Ar-CH=CH2), 3.96 and 3.89 (3:1, each s, 3H, OCH3), 2.00 and 1.96 (3:1, each s, 3H, 5′-CH3), 1.48 and 1.39 (1:3, each s, 6H, 3′-CH3×2). MS m/z 363 (M++1). Anal. (C23H22O4) C, H, O.
3′,3′-Dimethyl-5′-ethyl desmosdumotin C (21)
75% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.37 and 18.90 (2:1, each s, 1H, chelated-OH), 8.67 and 8.47 (1:2, each d, 1H, J = 15.9 Hz, olefin), 8.10 and 8.07 (1:3, each d, 1H, J = 15.9 Hz, olefin), 7.89-7.76 (m, 2H, Ar-2,6-H), 7.59-7.48 (m, 3H, Ar-3,4,5-H), 4.12 and 4.06 (2:1, each s, 3H, OCH3), 2.65 and 2.62 (2:1, each q, J = 7.4 Hz, 2H, 5′-CH2CH3), 1.62 and 1.53 (1:2, each s, 6H, 3′-CH3×2), 1.32 and 1.29 (2:1, each t, J = 7.4 Hz, 3H, 5′-CH2CH3). MS m/z 325 (M+−1). Anal. (C20H22O4) C, H, O.
3′,3′,5′-Triethyl desmosdumotin C (22)
57% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.30 and 18.89 (3:2, each s, 1H, chelated-OH), 8.52 and 8.44 (2:3, each d, 1H, J = 15.3 Hz, olefin), 7.96 and 7.93 (2:3, each d, 1H, J = 15.3 Hz, olefin), 7.73-7.66 (m, 2H, Ar-2,6-H), 7.43-7.36 (m, 3H, Ar-3,4,5-H), 4.03 and 3.96 (3:2, each s, 3H, OCH3), 2.60 and 2.56 (3:2, each q, 2H, J = 7.4 Hz, 5′-CH2CH3), 2.04-1.76 (m, 4H, 3′-CH2CH3×2), 1.20 and 1.16 (3:2, each t, 3H, J = 7.4 Hz, 5′-CH2CH3), 0.71 and 0.70 (2:3, each t, 3H, J = 7.4 Hz, 3′-CH2CH3×2). MS m/z 355 (M++1). Anal. (C22H26O4) C, H, O.
4-Methyl-3′,3′,5′-triethyl desmosdumotin C (23)
63% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.30 and 18.91 (3:2, each s, 1H, chelated-OH), 8.50 and 8.41 (2:3, each d, 1H, J= 15.3 Hz, olefin), 7.96 and 7.93 (2:3, each d, 1H, J = 15.3 Hz, olefin), 7.64-7.55 (m, 2H, Ar-2,6-H), 7.25-7.14 (m, 2H, Ar-3,5-H), 4.03 and 3.95 (3:2, each s, 3H, OCH3), 2.60 and 2.56 (3:2, each q, 2H, J = 7.4 Hz, 5′-CH2CH3), 2.38 and 2.35 (3:2, each s, 3H, Ar-4-CH3), 2.07-1.90 and 1.90-1.72 (3:2, each m, 4H, 3′-CH2CH3×2), 1.30-1.21 (m, 3H, 5′-CH2CH3), 0.70 (t, 6H, J = 7.4 Hz, 3′-CH2CH3×2). MS m/z 367 (M+−1). Anal. (C22H24O4) C, H.
4-Methyl-3′,3′,5′-triethyl desmosdumotin C (24)
61% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.30 and 18.93 (2:1, each s, 1H, chelated-OH), 8.50 and 8.42 (1:2, each d, 1H, J = 15.3 Hz, olefin), 7.97 and 7.93 (1:2, each d, 1H, J = 15.3 Hz, olefin), 7.68-7.56 (m, 2H, Ar-2,6-H), 7.30-7.18 (m, 2H, Ar-3,5-H), 4.03 and 3.95 (2:1, each s, 3H, OCH3), 2.55-2.50 (m, 4H, CH2CH3×2), 2.08-1.72 (m, 4H, CH2CH3×2), 1.33-1.10 (m, 6H, CH2CH3×2), 0.80-0.64 (m, 6H, CH2CH3×2). MS m/z 381 (M+−1). Anal. (C24H30O4) C, H, O.
3′,3′,5′-Tripropyl desmosdumotin C (25)
76% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.29 and 18.90 (2:1, each s, 1H, chelated-OH), 8.49 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.95 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.74-7.65 (m, 2H, Ar-2,6-H), 7.44-7.34 (m, 3H, Ar-3,4,5-H), 4.00 and 3.92 (2:1, each s, 3H, OCH3), 2.56-2.42 (m, 2H, 5′-CH2Et), 2.00-1.65 (m, 4H, 3′-CH2Et×2), 1.65-1.48 (m, 2H, 3′-CH2CH2CH3), 1.16-0.96 (m, 7H, 5′-CH2CH2CH3×2 and 3′-CH2CH2CH3), 0.90-0.77 (m, 6H, 3′-CH2CH2CH3×2). MS m/z 395 (M+−1). Anal. (C25H32O4) C, H, O.
4-Bromo-3′,3′,5′-tripropyl desmosdumotin C (26)
38% Yield. Yellow prisms, mp 114–115 °C. 1H NMR (300 MHz, CDCl3): δ 19.28 and 18.81 (2:1, each s, 1H, chelated-OH), 8.48 and 8.40 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.86 and 7.84 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.60-7.47 (m, 5H, Ar-H), 4.00 and 3.92 (2:1, each s, 3H, OCH3), 2.58-2.41 (m, 2H, 5′-CH2Et), 2.00-1.80 (m, 4H, 3′-CH2Et×2), 1.80-1.46 (m, 2H, 3′-CH2CH2CH3), 1.18-0.93 (m, 7H, 5′-CH2CH2CH3×2 and 3′-CH2CH2CH3), 0.93-0.77 (m, 6H, 3′-CH2CH2CH3×2). MS m/z 475 and 477 (M+−1). Anal. (C25H31O4Br) C, H, O.
Synthesis of Intermediates 31–36
2-Acetyl-6-ethyl-3,5-dihydroxy-4,4-dimethylcyclohexa-2,5-dienone (31)
A solution of 2-acetyl-3,5-dihydroxy-4,4-dimethylcyclohexa-2,5-dienone (30, 713 mg, 3.6 mmol) and sodium methoxide (2.0 mL, 9.3 mmol, 25% MeOH solution) in anhydrous MeOH (3 mL) containing ethyl iodide (0.3 mL, 3.8 mmol) was refluxed for 4 h. After removal of volatile solvent, the residue was partitioned between EtOAc and in aqueous HCl. The water phase was extracted with EtOAc (×3). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel with EtOAc–hexane (1:9 to 1:4, v/v) as an eluent to provide 31 (196 mg, 24%) along with recovered starting material. Colorless prisms, mp 153–154 °C (EtOAc–hexane). 1H NMR (300 MHz, DMSO-d6): δ 19.00 (br s, 1H, chelated-OH), 2.48 [s, 3H, C(O)CH3], 2.35 (q, 2H, J = 7.2 Hz, CH2CH3), 1.29 (s, 6H, 4-CH3×2), 0.95 (t, 3H, 6-CH2CH3). MS m/z 223 (M+−1). Anal. (C12H16O4·1/8H2O) C, H, O.
2-Acetyl-4,4,6-triethyl-3,5-dihydroxycyclohexa-2,5-dienone (32)
A solution of 2,4,6-trihydroxyacetophenone (1.117 g, 6.7 mmol) and sodium methoxide (4.8 mL, 22.2 mmol, 25% MeOH solution) in anhydrous MeOH (5 mL) containing ethyl iodide (1.65 mL, 20.6 mmol) was refluxed for 7 h. The reaction mixture was cooled to 0 °C and acidified with in aqueous HCl, then extracted with EtOAc (×3). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel with EtOAc–hexane (1:9 to 1:4, v/v) as an eluent to provide 32 (723 mg, 43%). Colorless prisms, mp: 149–150 °C (EtOAc-hexane). 1H NMR (300 MHz, DMSO-d6): δ 18.98 (br s, 1H, chelated-OH), 2.51 [s, 3H, C(O)CH3], 2.42 (q, 2H, J = 7.2 Hz, 6-CH2CH3), 1.81 (q, 4H, J = 7.4 Hz, 4-CH2CH3×2), 0.97 (t, 3H, J = 7.2 Hz, 6-CH3CH3), 0.57 (t, 3H, J = 7.2 Hz, 4-CH2CH3×2). MS m/z 251 (M++1). Anal. (C21H22O4·1/16H2O) C, H, O.
2-Acetyl-3,5-dihydroxy-4,4,6-tripropylcyclohexa-2,5-dienone (33)
The procedure was identical to that described above except with propyl iodide instead of ethyl iodide to provide 33 (41%). Colorless prisms, mp: 95–96 °C (EtOAc-hexane). 1H NMR (300 MHz, CD3OD): δ 2.53 [s, 3H, C(O)CH3], 2.42 (t, 2H, J = 7.5 Hz, 6-CH2Et), 1.94-1.74 (m, 4H, 4-CH2Et×2), 1.54-1.38 (m, 2H, , 6-CH3CH2CH3), 1.16-0.90 (m, 4H, 4- CH3CH2CH3×2), 0.94 (t, 3H, J = 7.4 Hz, 6-CH3CH2CH3), 0.81 (t, 6H, J = 7.1 Hz, 6-CH3CH2CH3×2). MS m/z 293 (M+−1). Anal. (C17H26O4) C, H, O.
2-Acetyl-6-ethyl-3-hydroxy-5-methoxy-4,4-dimethylcyclohexa-2,5-dienone (34)
To a solution of 31 (124 mg, 0.55 mmol) in anhydrous EtOAc–MeOH (5:1, 1.8 mL), a solution of TMSCHN2 (2 mL, 4.0 mmol, 2 M solution in diethyl ether) was added slowly at −78 °C under argon atmosphere and the mixture was stirred for 3 h. Acetic acid was then added to destroy the excess TMSCHN2. The mixture was concentrated in vacuo, and the residue was purified by silica gel column chromatography with EtOAc–hexane (1:9 to 1:4 v/v) as an eluent to obtain 34 (117 mg, 89%). yellow oil. 1H NMR (300 MHz, CDCl3): δ 18.97 and 18.14 (2:1, each s, 1H, chelated-OH), 3.97 and 3.90 (2:1, each s, 3H, OCH3), 2.71 and 2.61 [1:2, each s, 3H, C(O)CH3], 2.48 and 2.43 (2:1, each q, 2H, J = 7.4 Hz, 6-CH2CH3), 1.45 and 1.34 (1:2, each s, 6H, 4-CH3), 1.16 and 1.11 (2:1, each t, 3H, 6-CH2CH3×2). MS m/z 237 (M+−1).
2-Acetyl-4,4,6-triethyl-3-hydroxy-5-methoxycyclohexa-2,5-dienone (35)
The same procedure was employed to obtain 35 (80%) from 32. yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.00 and 18.12 (3:2, each s, 1H, chelated-OH), 4.02 and 3.93 (3:2, each s, 3H, OCH3), 2.71 and 2.64 [2:3, each s, 3H, C(O)CH3], 2.57 and 2.51 (3:2, each q, 2H, J = 7.4 Hz, 6-CH2CH3), 2.03-1.72 (m, 4H, 4- CH2CH3×2), 1.18 and 1.12 (3:2, each t, 3H, 6-CH2CH3), 0.67 and 0.65 (2:3, each t, 6H, 4-CH2CH3×2). MS m/z 267 (M++1).
2-Acetyl-3-hydroxy-5-methoxy-4,4,6-tripropylcyclohexa-2,5-dienone (36)
The same procedure was employed to obtain 36 (53%) from 33 along with recovered starting material (32%). yellow oil. 1H NMR (300 MHz, CDCl3): δ 18.99 and 18.15 (3:2, each s, 1H, chelated-OH), 3.98 and 3.90 (3:2, each s, 3H, OCH3), 2.67 and 2.63 [2:3, each s, 3H, C(O)CH3], 2.53-2.40 (m, 2H, 6-CH2Et), 1.95-1.42 (m, 6H, 6-CH2CH2CH3 and 4-CH2Et×2), 1.10-0.95 (m, 7H, 6-CH2CH2CH3 and 4-CH2CH2CH3×2), 0.83 and 0.80 (3:2, each t, 6H, 4-CH2CH2CH3×2). MS m/z 307 (M+−1).
Cytotoxic Activity Assay15
All stock cultures were grown in T-25 flasks. Freshly trypsinized cell suspensions were seeded in 96-well microtitre plates at densities of 1500–7500 cells per well with compounds added from DMSO-diluted stock. After 3 days in culture, attached cells were fixed with cold 50% trichloroacetic acid and then stained with 0.4% sulforhodamine B. The absorbency at 562 nm was measured using a microplate reader after solubilizing the bound dye. The mean ED50 is the concentration of agent that reduces cell growth by 50% under the experimental conditions and is the average from at least three independent determinations that were reproducible and statistically significant. The following human tumor cell lines were used in the assay: A549 (human lung carcinoma), A431 (epidermoid skin carcinoma), 1A9 (human ovarian carcinoma), HCT-8 (colon adenocarcinoma), PC-3 (prostate cancer), KB (nasopharyngeal carcinoma), KB-VTN (vincristine-resistant KB subline), HUVEC (human endothelial). All cell lines were obtained from the Lineberger Cancer Center (UNC-CH) or from ATCC (Rockville, MD) and were cultured in RPMI-1640 medium supplemented with 25 mM HEPES, 0.25% sodium bicarbonate, 10% fetal bovine serum, and 100 μg/mL kanamycin.
Angiogenesis Assay
Method is according to the NCI's Angiogenesis Resource Center protocol. HUVEC were purchased from Cambrex Bio-science. HUVEC (1.5×103) were plated in a 96-well plate in 100 μl of EGM-2 (Clonetec H CC3162). After 24 h (day zero), the test compound (100 μl) was added to each well at 2× the desired concentration in EGM-2 medium. One plate was stained with 0.5% crystal violet in 20% MeOH for 10 minutes, rinsed with water, and air-dried. The remaining plates were incubated for 72 h at 37 °C. After 72 h, plates were stained with 0.5% crystal violet in 20% MeOH, rinsed with water and air-dried. The stain was eluted with 1:1 solution of EtOH:0.1 M sodium citrate, and absorbance was measured at 540 nm with an ELISA reader.
Supplementary Material
Elemental analysis data for compounds 7-15, 21-26, and 31-33. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.
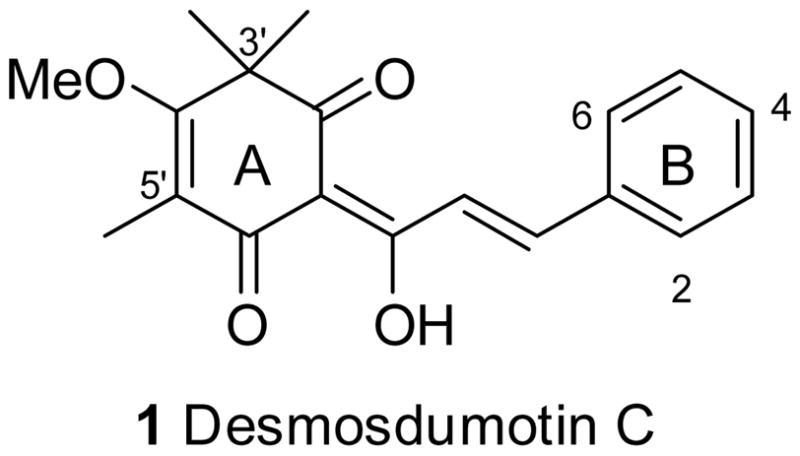
Structure of 1
Scheme 2.

Syntheses of 1-Analogs: A/B-ring Modifications
Acknowledgments
This work was supported by a NIH grant CA17625 from the National Cancer Institute, awarded to K. H. Lee.
Abbreviations
- HUVEC
human umbilical vein endothelial cell
- KB
vincristine-resistant
- KB-VIN
expressing P-glycoprotein, cell subline
References
- 1.Balunas MJ, Kinghorn AD. Drug discovery from medicinal plants. Life Science. 2005;78:431–441. doi: 10.1016/j.lfs.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Rollinger JM, Langer T, Stuppner H. Strategies for efficient lead structure discovery from natural products. Curr Med Chem. 2006;13:1491–1507. doi: 10.2174/092986706777442075. [DOI] [PubMed] [Google Scholar]
- 3.Jones WP, Chin YW, Kinghorn AD. The role of pharmacognosy in modern medicine and pharmacy. Curr Drug Targets. 2006;7:247–264. doi: 10.2174/138945006776054915. [DOI] [PubMed] [Google Scholar]
- 4.Butler MS. Natural products to drugs: natural product derived compounds in clinical trials. Nat Prod Rep. 2005;22:162–195. doi: 10.1039/b402985m. [DOI] [PubMed] [Google Scholar]
- 5.Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 6.Paterson I, Anderson EA. The renaissance of natural products as drug candidates. Science. 2005;310:451–53. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- 7.Tan G, Gyllenhaal C, Soejarto DD. Biodiversity as a source of anticancer drugs. Curr Drug Targets. 2006;7:265–277. doi: 10.2174/138945006776054942. [DOI] [PubMed] [Google Scholar]
- 8.http://www.who.int/mediacentre/factsheets/fs297/en/
- 9.Jiang Su New Medical College, Ed. Chung Yao Da Tzu Dien (Dictionary of Chinese Materia Medicia) Vol. 2. Shanghai Science & Technology Press; Hong Kong: 1977. p. 1919. [Google Scholar]
- 10.Wu JH, McPhail AT, Bastow KF, Shiraki H, Ito J, Lee KH. Tetrahedron Lett. 2002;43:1391–1393. [Google Scholar]
- 11.Nakagawa-Goto K, Wu JH, Lee KH. First total synthesis of desmosdumotin C. Syn Commun. 2005;35:1735–1739. [Google Scholar]
- 12.Nakagawa-Goto K, Wu JH, Bastow KF, Wu CC, Lee KH. Antitumor agents 243. Syntheses and cytotoxicity of desmosdumotin C derivatives. Bioorg Med Chem. 2005;13:2325–2330. doi: 10.1016/j.bmc.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 13.Bick IRC, Horn DHD. Nuclear magnetic resonance studies. V. The tautomerism of tasmanone and related β-triketones. Aust J Chem. 1965;18:1405–1410. [Google Scholar]
- 14.a) Wollenweber E, Dietz VH, Schilling G, Favre-Bonvin J, Smith DM. Flavonoids from chemotypes of the goldback fern, Pityrogramma tiangularis. Phytochemistry. 1985;24:965–972. [Google Scholar]; b) Sbit M, Dupont L, Dideberg O, Vilain C. Structure of ceroptene. Acta Crystallogr Sect C. 1987;C43:2204–2206. [Google Scholar]
- 15.Nakanishi Y, Chang FR, Liaw CC, Wu YC, Bastow KF, Lee KH. Acetogenins as selective inhibitors of the human ovarian 1A9 tumor cell line. J Med Chem. 2003;46:3185. doi: 10.1021/jm020548b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data for compounds 7-15, 21-26, and 31-33. This material is available free of charge via the Internet at http://pubs.acs.org.