

Abstract
In connection with an ongoing investigation of indenoisoquinoline topoisomerase I (Top1) inhibitors as potential therapeutic agents, the intercalation pharmacophore possessing di(methoxy) and methylenedioxy substituents was held constant and new derivatives were synthesized with nitrogen heterocycles appended to the lactam side chain. Compounds were evaluated for Top1 inhibition and for cytotoxicity in the National Cancer Institute’s human cancer cell screen. Some of the more potent derivatives were also screened for in vivo activity in a hollow fiber assay. The results of these studies indicate that lactam substituents possessing nitrogen heterocycles can provide highly cytotoxic compounds with potent Top1 inhibition. Molecular modeling of these compounds in complex with DNA and Top1 suggests that some of the lactam substituents are capable of interacting with the DNA base pairs above and below the site of intercalation and/or with Top1 amino acid residues, resulting in increased biological activity.
Introduction
In the 1990’s, the National Cancer Institute (NCI) developed a COMPARE algorithm to facilitate the prediction of biological targets from data generated by the 60 cell human cancer screen.1 Utilizing the COMPARE algorithm, it was predicted that indenoisoquinoline 1 (NSC 314622, Figure 1) would be a topoisomerase I (Top1) inhibitor.2 Subsequent in vitro testing confirmed this prediction and more potent derivatives (such as 2) have been developed.2–9
Figure 1.

Representative indenoisoquinoline and camptothecin Top1 inhibitors.
Irinotecan (3) and topotecan (4) are the only current Top1 inhibitors approved by the Food and Drug Administration (FDA) for the treatment of cancer and they validate Top1 as a therapeutic target for anticancer drug development. However, these camptothecin derivatives are not ideal drug molecules. Camptothecins are compromised by the reversibility of the Top1-DNA cleavage complex, which necessitates long infusion times for maximum activity,10 and they are inherently unstable and suffer from lactone ring opening to form a hydroxy acid that has a high affinity for human serum albumin.11 As a result of the pharmacokinetic problems of the camptothecins, there is great interest in the development of non-camptothecin Top1 inhibitors as anticancer agents. Top1 inhibitors such as the indenoisoquinolines are not limited by the same pharmacokinetic problems that limit the camptothecins. Some of the more potent indenoisoquinolines stabilize the Top1-DNA cleavage complex to a greater extent than camptothecin, they are more stable chemically, and the in vitro biological activities of certain derivatives are comparable.1,12 Furthermore, it is known that clinically useful topoisomerase II (Top2) inhibitors have preferential activity for different cancers, and it can be expected that different Top1 inhibitors will display different spectra of anticancer activity as well.13
The present investigation was undertaken to explore novel functionalities appended to the lactam side chain of the indenoisoquinolines, specifically focusing on incorporation of heterocyclic ring systems. Previous work with an indenoisoquinoline system lacking substituents on the aromatic rings demonstrated that potent biological activity (cytotoxicity and Top1 inhibition) could be achieved.5 However, the research demonstrated a small and consistent benefit to having the di(methoxy) and methylenedioxy substituents appended to the isoquinoline and indenone rings, and this has led to their incorporation in the present investigation.5 Previous work has also led to the realization that amino, morpholine, and imidazole motifs appended to the lactam side chain further enhanced biological activity with respect to compounds lacking these nitrogenous substituents.5,7,9 To explain this enhancement, hypotheses such as increased aqueous solubility, DNA targeting, transporter hijacking, and hydrogen bonding of the side chain amines with amino acid residues in the ternary cleavage complex have been proposed and may indeed be involved for some molecules.5,7
Several recent developments have occurred that allow further speculation and necessitate a reinvestigation of indenoisoquinolines bearing heterocyclic lactam side chains. The crystal structures of complexes have been reported with small molecule inhibitors such as topotecan,14 camptothecin,15 an indolocarbazole,15 an indenoisoquinoline,15 and a norindenoisoquinoline3,16 intercalated within duplex DNA bound to human Top1. Using these structures, models of various indenoisoquinolines in ternary complex with Top1 and cleaved duplex DNA have been generated and suggest incorporating ammonium cations and heterocycles on the lactam side chain. If the models are credible, then appropriate functionalities may be capable of increasing Top1 inhibition, and ultimately cellular cytotoxicity, by interacting with amino acid residues and/or the DNA base pairs at the site of intercalation. Thus, the present investigation represents a new effort toward the development of indenoisoquinoline Top1 inhibitors where the di(methoxy) and methylenedioxy substituents on the aromatic nucleus are combined with previously utilized and novel heterocyclic lactam side chains in an effort to improve the anticancer activity of the indenoisoquinolines.
Chemistry
Indenoisoquinolines 6–18 were prepared according to the methods summarized in Scheme 1. Utilizing compound 517 as a key advanced intermediate, nucleophilic displacement of the alkyl bromide functionality with a variety of primary and secondary amines readily provided the corresponding analogues possessing heterocyclic lactam substituents. In general, the conditions utilizing potassium carbonate in 1,4-dioxane to effect displacement were readily amenable to most of the analogues synthesized (compounds 7 and 8 being the only exceptions). However, treatment of the heterocyclic precursors to 7 and 8 with sodium hydride in dimethylformamide readily effected the desired transformation. Ultimately, compounds 6, 8–10, and 13–16 were treated with a solution of hydrochloric acid in diethyl ether to provide the corresponding hydrochloride salts. This was found to both aid purification and provide an improved solubility profile for the biological assays.
Scheme 1. Reagents and Conditions.
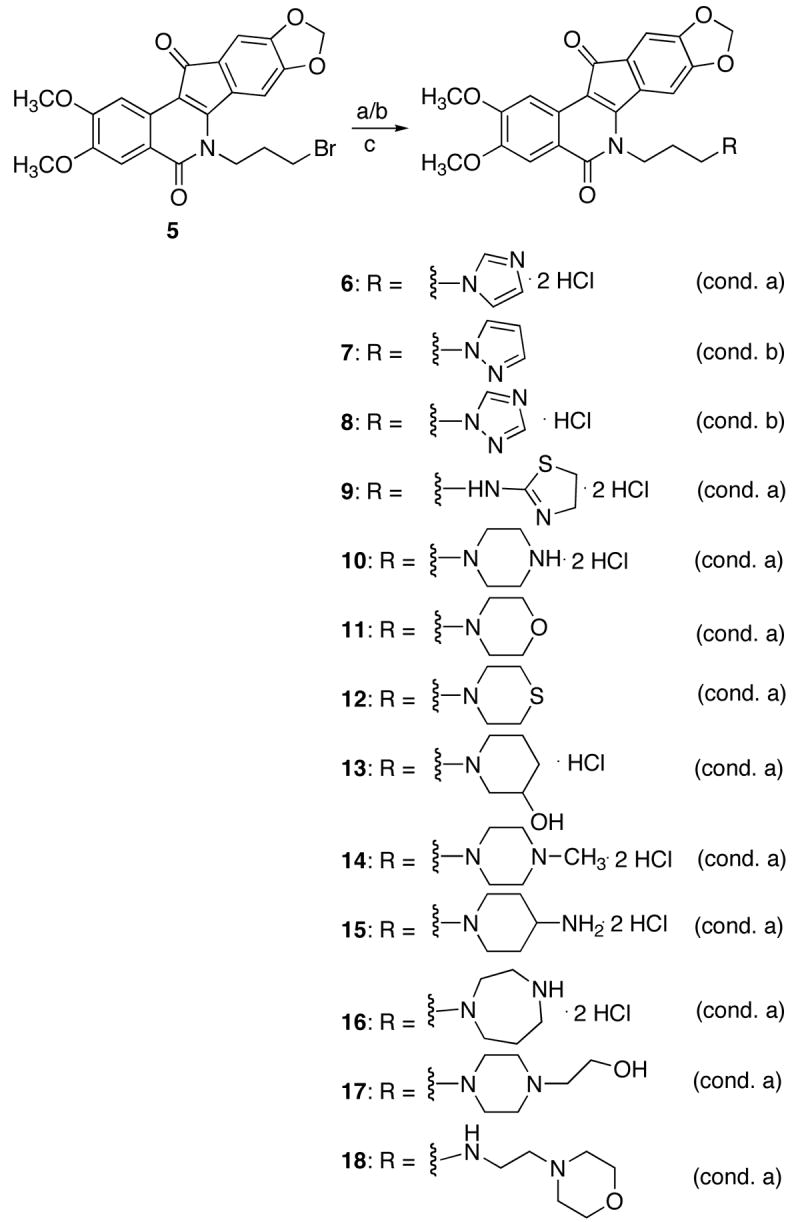
Reagents and Conditions: a) amine, K2CO3, 1,4-dioxane, 100 °C; b) amine,NaH, DMF, 60 °C; c) 2 M HCl in ether, CHCl3.
Biological Results and Discussion
The indenoisoquinolines were examined for antiproliferative activity against the human cancer cell lines in the National Cancer Institute screen, in which the activity of each compound was evaluated in approximately 55 different cancer cell lines of diverse tumor origins. The GI50 values obtained in selected cell lines, along with the mean graph midpoint (MGM) values, are summarized in Table 1. The MGM is based on a calculation of the average GI50 for all of the cell lines tested (approximately 55) in which GI50 values below and above the test range (10−8 to 10−4 molar) are taken as the minimum (10−8 molar) and maximum (10−4 molar) drug concentrations used in the screening test. For comparison purposes, the activities of the previously reported lead compound 12, its more potent derivative 217, and previously reported imidazole-appended analogues 19,5 20,9 and 219 are also included in the table. The relative potencies of the compounds towards the production of topoisomerase I-mediated DNA cleavage are also listed in the table. The potencies are expressed semiquantitatively as follows: +: weak activity; ++: similar activity as the parent compound 1; +++ and ++++: greater activity than the parent compound 1; ++++: similar activity as 1 μM camptothecin.
Table 1.
Cytotoxicities and Topoisomerase I Inhibitory Activities of Indenoisoquinoline Analogues.
| cytotoxicity (GI50 in μM)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | lung
HOP-62 |
colon
HCT-116 |
CNS
SF-539 |
melanoma
UACC-62 |
ovarian
OVCAR-3 |
renal
SN12C |
prostate
DU-145 |
breast
MDA-MB-435 |
MGMb | Top 1
Cleavagec |
| 1 | 1.30 | 35.0 | 41.0 | 4.20 | 73.0 | 68.0 | 37.0 | 96.0 | 20.0 | ++ |
| 2 | 0.06 | 0.13 | 0.26 | 0.25 | 0.31 | 0.31 | 0.04 | 1.21 | 0.16 ± 0.02 | +++ |
| 6 | <0.010 | <0.010 | 0.037 | <0.010 | 0.085 | <0.010 | <0.010 | 0.020 | 0.079 ± 0.023 | ++++ |
| 7 | 0.447 | 1.99 | 0.398 | 0.269 | 56.2 | 0.316 | 0.363 | 7.08 | 2.16 ± 0.24 | ++ |
| 8 | 0.079 | 1.91 | 0.288 | <0.010 | 61.7 | 0.085 | 0.085 | >100 | 3.55 | +++ |
| 9 | <0.010 | <0.010 | <0.010 | 0.014 | 0.041 | <0.010 | <0.010 | <0.010 | 0.112 ± 0.066 | ++++ |
| 10 | <0.005 | 0.575 | <0.005 | 1.20 | 2.04 | 0.091 | 0.015 | 4.57 | 0.382 ± 0.119 | ++ |
| 11 | 1.78 | 1.15 | 0.040 | 0.030 | 74.1 | 0.813 | 0.155 | 67.6 | 4.64 ± 1.25 | ++++ |
| 12 | 26.3 | 72.4 | 18.2 | 37.2 | 34.7 | NT | >100 | >100 | 50.1 | ++ |
| 13 | <0.005 | <0.005 | <0.005 | 5.01 | 5.75 | 0.126 | <0.005 | 0.977 | 0.243 ± 0.088 | +++ |
| 14 | 18.2 | 1.48 | 17.8 | 15.1 | 15.1 | 11.5 | 10.7 | >100 | 12.0 | + |
| 15 | 0.427 | 0.120 | 0.100 | 1.29 | 0.832 | 0.257 | 0.182 | 1.74 | 0.766 ± 0.254 | + |
| 16 | <0.005 | 0.214 | 0.145 | 0.457 | 5.01 | 0.145 | 0.081 | 2.63 | 0.715 ± 0.335 | +++ |
| 17 | 9.77 | 2.34 | 1.44 | 1.23 | 15.1 | >100 | 0.275 | >100 | 7.86 ± 0.27 | ++ |
| 18 | <0.005 | <0.005 | 0.550 | 0.162 | 0.525 | 1.48 | 0.603 | 1.95 | 1.27 ± 0.84 | +++ |
| 19 | 2.69 | 1.41 | 2.34 | 0.79 | 1.66 | 1.66 | 1.41 | 2.75 | 1.86 | ++++ |
| 20 | < 0.010 | < 0.010 | < 0.010 | < 0.010 | < 0.010 | < 0.010 | < 0.010 | 0.014 | 0.059 ± 0.026 | +++ |
| 21 | 0.19 | 0.274 | 0.016 | 0.012 | 0.864 | 0.015 | 0.017 | 2.17 | 0.370 ± 0.28 | ++++ |
The cytotoxicity GI50 values are the concentrations corresponding to 50% growth inhibition.
Mean graph midpoint for growth inhibition of all human cancer cell lines successfully tested. Compounds with standard deviations listed for the MGM values were tested twice, while those without standard deviations for the MGM values were tested once.
The compounds were tested at concentrations ranging up to 10 μM. The activity of the compounds to produce Top1-mediated DNA cleavage was expressed semi-quantitatively as follows: +: weak activity; ++: similar activity as the parent compound 1; +++ & ++++: greater activity than the parent compound 1; ++++: similar activity as 1 μM camptothecin.
An increase in the biological activity of indenoisoquinolines resulting from an aminoalkyl chain attached to the lactam nitrogen has been identified previously.7,17 As the next logical step, it seemed prudent to investigate the effects of replacing the amino group with various nitrogen heterocycles (Scheme 1, 6–18) by evaluating the cytotoxicity and Top1 inhibition of the newly synthesized analogues (Table 1). With multiple heteroatoms present in these newly synthesized analogues, along with a flexible three-carbon linker, the development of an adequate hypothetical structural model that rationalized all of the biological activity results proved difficult. Depending on the nature of the heterocyclic side chain, molecular modeling indicated it could be capable of interacting with the DNA base pairs and Top1 amino acid residues in several different orientations within the ternary complex. This variability made the rationalization of biological activities resulting from minor changes in the structure of the heterocycle increasingly difficult. However, the results listed in Table 1 indicate that heterocycles possessing a heteroatom capable of serving as a hydrogen bond acceptor at physiological pH generally provide the best results regarding Top1 inhibition and cytotoxicity (compounds 6, 8, 9, 11, and 13). Attempts to rationalize this trend using molecular modeling were generally unsuccessful due to the abundance of both hydrogen bond donors and acceptors surrounding the drug intercalation site and lactam side chain in the ternary complex.
With regard to compounds 6–9, each of which possess a five-membered heterocycle appended to the lactam side chain, the biological results suggest that the types of ring systems involved still confer significant activity. Compound 6, with its imidazole motif, was the most potent compound synthesized in the present investigation with an MGM of 0.079 μM and Top1 inhibition of ++++, making it comparable to 1 μM camptothecin in the assay. This outcome could be expected with the activities of compounds 19, 20, and 21 serving as a precedent (Table 1). These previously reported compounds are potent Top1 inhibitors that display unusually potent cytotoxicity, with the exception of unsubstituted compound 19.5,9 As mentioned above, there is a small, yet consistent benefit to possessing dimethoxy- and methylenedioxy-substituted isoquinoline and indenone rings.5 Gratifyingly, incorporating this substitution pattern in the present study produced an analogue 6 that retained the potent Top1 inhibition seen with compound 19, but improved the cytotoxicity making it similar to compounds 20 and 21 (Figure 2).
Figure 2.
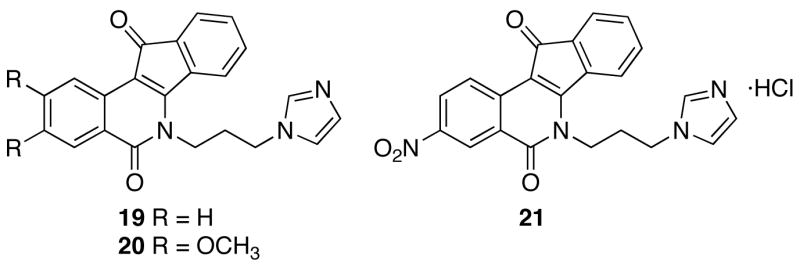
Previously synthesized heterocyclic indenoisoquinoline derivatives.
Altering the 1,3 nitrogen relationship of imidazole in 6 to the 1,2 relationship present in compound 7 resulted in a significant loss of biological activity. Inclusion of an additional nitrogen heteroatom (as for 8) resulted in a decrease in cytotoxicity but still maintained significant Top1 inhibition. A hypothetical model of 6 in ternary complex highlights the importance of the 1,3 nitrogen atom relationship regarding Top1 inhibition that is illustrated by compounds 6, 7, and 8 (Figure 3). The hypothetical model illustrated in Figure 3 was developed utilizing the previously reported crystal structure of an indenoisoquinoline in complex with DNA and Top1.3,15 It postulates that compound 6 has the potential to make three hydrogen bonding contacts with Top1. The first two contacts, in the minor groove with Arg364 and on the scissile side of the DNA with Asn722, have been previously reported for indenoisoquinoline analogues.3,15 However, the model indicates a potential hydrogen bond between the 3-position nitrogen of the lactam heterocycle in compound 6 and Lys436. This hydrogen bond provides a rationale for the inactive Top1 inhibition of 7, which does not have an appropriately placed heteroatom in the heterocycle for hydrogen bonding. The model also explains the Top1 activity of 8, which has the 4-position nitrogen of the 1,2,4-tetrazole motif oriented in a similar position to accept a hydrogen bond from Lys436. The slight decrease in the Top1 inhibitory activity of 8 (Top1 +++) compared to 6 (Top1 ++++) could be due to a decrease in the basicity of the 4-position nitrogen of the 1,2,4-triazole motif and a subsequent decrease in participation as a hydrogen bond acceptor with Lys436. An additional 1,3-nitrogen-substituted analogue 9 (MGM 0.112 μM, Top1 ++++) with unshared electron density between the 1,3-related nitrogen atoms (exocyclic and endocyclic) also provided a potent analogue. These results led to the realization that a 1,3 relationship between hydrogen bond donors/acceptors appears to be optimal for biological activity in 5-membered heterocyclic ring systems.
Figure 3.

Stereoview of compound 6 (red) in ternary complex with DNA and Top1. The lactam side chain is pointing into the major groove and potential hydrogen bonds are indicated in yellow. The diagram is programmed for wall-eyed (relaxed) viewing.
Six-membered heterocyclic rings were also investigated (Table 1, compounds 10–15, 17, and 18), with potent biological activity still observed for some of the compounds. For these ring systems, however, a 1,4-relationship between heteroatoms capable of donating and accepting hydrogen bonds was observed to be necessary for optimal biological activity. Compound 10 (MGM 0.382 μM, Top1 ++), with a pyrazine ring serving as its appended heterocycle, showed potent cytotoxicity, but poor Top1 inhibition. However, replacing the 4-position nitrogen atom in the heterocycle with an oxygen atom (thereby providing morpholine derivative 11) reversed the biological activity profile to provide a potent Top1 inhibitor (++++), but a poorly cytotoxic molecule (MGM 4.64 μM). Incorporation of a sulfur atom, which forms weaker hydrogen bonds than oxygen or nitrogen, in place of the aforementioned heteroatoms in the 4-position, provided compound 12. This compound was a poor Top1 inhibitor (++) and a weakly cytotoxic molecule (MGM 50.1 μM). Thus, compound 12 emphasizes that a 4-position heteroatom capable of strong hydrogen bonding is important for biological activity, but there is an imperfect correlation between cytotoxicity and Top1 inhibition in this series of compounds. Compounds 13 and 14 further demonstrate the importance of the 4-position and further support a specific hydrogen bonding role for atoms in the 4-position. Compound 13 (MGM 0.243 μM, Top1 +++), with an exocyclic alcohol, demonstrated potent cytotoxicity and good Top1 inhibition. Thus, by comparing its activity with 10 and 11, it can be inferred that the 4-position heteroatom needs to be capable of serving as a hydrogen bond donor in order to attain potent cytotoxicity (the ether motif present in 11 can only serve as a hydrogen bond acceptor). However, Top1 inhibition was enhanced by having a 4-position heteroatom capable of serving as a hydrogen bond acceptor (10 is doubly protonated at physiological pH and can only serve as a hydrogen bond donor). The exocyclic alcohol in 13 can act as both a hydrogen bond acceptor and a hydrogen bond donor, and compound 13 performed well in both assays, while compound 12 was inactive in both assays. Extending the length of the lactam side chain had no significant affect on the role of the 4-position of the heterocycle. Compound 18 (MGM 1.27 μM, Top1 +++), with its pendant morpholine ring, would be expected to act better as a Top1 inhibitor than as a cytotoxic agent according to the rational mentioned above, and it indeed supported the general trend.
Alkylation of the 4-position of the heterocycle was found to be detrimental to biological activity, with compounds 14 and 17 being relatively inactive in the assays when compared to several of the other molecules. The cytotoxicities of these molecules were rather interesting since they would have been expected to be more active in light of the 4-position being a hydrogen bond donor at physiological pH.
The relationship between the 4-position of the heterocycle and its effect on biological activity was further investigated with analogues 15 and 16. Expanding the size of the heterocycle by a single carbon atom to afford compound 16 (MGM 0.715 μM, Top1 +++) provided a moderately active Top1 inhibitor and cytotoxic molecule. Comparing the biological activities of analogues 16 and 10, whose lactam side chains are a homopyrazine ring and a pyrazine ring, respectively, a two-fold loss in cytotoxicity was observed for the ring-expanded analogue. However, the loss in cytotoxicity was accompanied by a slight increase in Top1 inhibition. Compound 15 (MGM 0.766 μM, Top1 +) was a moderately cytotoxic analogue and a poor Top1 inhibitor. According to the structure-activity relationships for the studied analogues, this result was consistent since the exocyclic primary amine present in 15 would be expected to be protonated at physiological pH and thereby serve only as a hydrogen bond donor.
All of the newly synthesized compounds were examined for induction of DNA cleavage in the 3′-end-labeled PvuII/HindIII fragment of pBluescript SK(−) phagemid DNA in the presence of Top1.2 The resulting cleavage patterns of some of the more potent indenoisoquinolines are displayed in Figure 4, with the results being compared to 1 μM camptothecin. Differences in cleavage sites and band intensities between camptothecin and the indenoisoquinolines in Figure 4 are noteworthy. For example, the camptothecin band at site 37 was not prominent in the indenoisoquinoline assays (the exception being 8). Conversely, the cleavage site at 44 was intense for most of the indenoisoquinolines. As a result of different cleavage site specificities, it may be possible to selectively target different cancer cell genes using a camptothecin or an indenoisoquinoline. Figure 4 also indicates that several cleavage sites are conserved between camptothecin and the indenoisoquinolines. Cleavage sites 70, 92, 97, and 119 were prominent for camptothecin and most of the indenoisoquinolines, especially compounds 11, 19, and 21. It is also worth highlighting the fact that several cleavage bands were more prominent at intermediate drug concentrations as opposed to high drug concentrations, especially for compounds 8 and 9. This could be due to inhibition of Top1 cleavage activity at higher drug concentrations due to a direct effect on the enzyme, or alternatively, to DNA intercalation, which could result in DNA unwinding to render it a poorer substrate for Top1.
Figure 4.
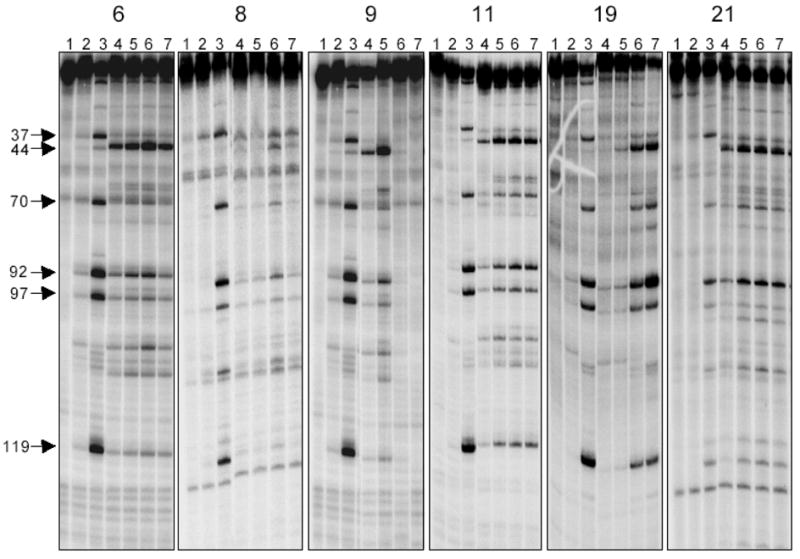
Comparison of the Top1-mediated DNA cleavages at different drug concentrations. The DNA used corresponds to the 3′-end-labeled PvuII/HindIII fragment of pBluescript SK (−) phagemid DNA. Top1 was present in all reaction mixtures except Lane 1. Lane 1: DNA; Lane 2: DNA + Top1; Lane 3: 1 μM camptothecin; Lanes 4–7: inhibitor concentrations of 0.1, 1.0, 10, and 100 μM, respectively. The figure is comprised of 5 gels that are placed side by side to facilitate comparison.
Several of the active analogues (10, 11, 13, 16, and 17) were evaluated as anticancer agents in an in vivo animal model in which polyvinylidene fluoride hollow fibers containing various cancer cell cultures were implanted intraperitoneally (IP) and subcutaneously (SC) into mice, and the compounds were administered by the IP route. The effects of the compounds on the reduction of viable cancer cell mass compared to those of controls were determined. Each compound was tested in the hollow fiber assay against a panel of twelve human tumor cell lines as described previously.18 The compounds were solubilized in 10% DMSO in saline/Tween-80R and administered intraperitoneally once daily for a total of four doses at each of two dose levels. The two doses were selected based on single dose toxicity studies for each derivative. A score of 2 was assigned each time the compound produced a 50% or greater reduction in viable cell mass compared to vehicle-treated controls. The score for each compound was summed for the intraperitoneal fibers and the subcutaneous fibers to provide the total score for each derivative (Table 2). For comparative purposes, the performance of compounds 1 and 2 are included in Table 2. Three of the five newly tested analogues (indenoisoquinolines 2, 10, and 16) were more potent than lead compound 1. All of the newly tested compounds were more active at the IP implant site than at the SC implant site. This could imply problems with absorption, distribution, or metabolism of these compounds. None of the newly synthesized compounds were more potent than 2, but compound 6 is currently undergoing hollow fiber testing and xenograft studies at the National Cancer Institute.
Table 2.
Hollow Fiber Activities of Indenoisoquinoline Analogues.
| compound | IP scorea | SC scorea | total score | cell killb |
|---|---|---|---|---|
| 1 | 2 | 6 | 8 | Y |
| 2 | 16 | 4 | 20 | N |
| 10 | 16 | 2 | 18 | N |
| 11 | 4 | 2 | 6 | N |
| 13 | 6 | 0 | 6 | N |
| 16 | 12 | 2 | 14 | N |
| 17 | 8 | 0 | 8 | N |
The IP and SC scores listed are the sums of all the IP and SC scores for each compound.
A net cell kill at one or more implant sites is indicated with a Y.
In conclusion, several potent indenoisoquinolines have been synthesized with lactam side chains containing nitrogen heterocycles. Several structural motifs have been found to increase the potency of Top1 inhibition with imidazole and morpholine substituents the most active in this aspect of biological evaluation. Furthermore, a hypothetical model has been constructed that rationalizes the Top1 inhibitory activity of the imidazole, pyrazole, and 1,2,4-tetrazole substituted analogues. It was observed that heteroatom placement within the lactam side chain has a significant impact on both Top1 inhibition and cytotoxicity, with 3- and 4-position heteroatoms providing optimal activity for 5- and 6-membered heterocyclic ring systems, respectively. Furthermore, the ability to participate as a hydrogen bonding donor or acceptor at the 4-position was found to differentiate molecules that were either potent Top1 inhibitors or highly cytotoxic agents.
Experimental Section
Melting points were determined in capillary tubes and are uncorrected. Infrared spectra were obtained using CHCl3 as the solvent (unless otherwise specified) and were recorded using a Perkin Elmer 1600 series FTIR. Except where noted, 1H NMR spectra were obtained using CDCl3 as solvent and the residual solvent peak as an internal standard. 1H NMR spectra were recorded on an ARX300 300 MHz Bruker NMR spectrometer. Electrospray mass spectra were determined using a FinniganMATT LCQ (Thermoquest Corp., San Jose, CA) instrument at the Purdue Campus-Wide Mass Spectrometry Center. Microanalyses were performed at the Purdue University Microanalysis Laboratory. Analytical thin-layer chromatography was carried out on Analtech silica gel GF 1000-micron glass plates. Compounds were visualized with short wavelength UV light. Silica gel flash chromatography was performed using 230–400 mesh silica gel.
General Procedure for the Synthesis of Indenoisoquinolines 6 and 9–18
A mixture of indenoisoquinoline 517 (0.500 g, 1.06 mmol), amine (2.11 mmol), and anhydrous K2CO3 (0.584 g, 4.23 mmol) in anhydrous 1,4-dioxane (30 mL) was heated at 100 °C for 4 h. The reaction mixture was cooled and then concentrated. The residue was diluted with water (50 mL), extracted with CHCl3 (2 x 50 mL), washed with 1% aq HCl (50 mL), water (50 mL), sat NaCl (50 mL), and dried over Na2SO4. The crude product was purified by flash column chromatography (SiO2), eluting with a 0–5% gradient of methanol in chloroform, to provide the pure indenoisoquinoline.
3-(Imidazolyl-1-propyl)-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (6)
The desired analogue was obtained as a dark purple solid (245 mg, 63%): mp 316–318 °C. 1H NMR (CDCl3) δ 8.01 (s, 1 H), 7.63 (s, 1 H), 7.60 (s, 1 H), 7.14 (s, 1 H), 7.04 (s, 2 H), 6.40 (s, 1 H), 6.07 (s, 2 H), 4.45 (t, J = 5.8 Hz, 2 H), 4.20 (t, J = 6.6 Hz, 2 H), 4.03 (s, 3 H), 3.98 (s, 3 H), 2.33 (t, J = 6.9 Hz, 2 H); ESIMS m/z (rel intensity) 460 (MH+, 100). Anal. (C25H21N3O6·0.2 H2O) C, H, N. The hydrochloride salt was formed by dissolving the product in chloroform (50 mL) and an anhydrous solution of 2 M HCl in diethyl ether (15 mL, 30.0 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 6 h and the precipitated product was filtered and washed with chloroform (50 mL), methanol (20 mL), and dried over P2O5 for 24 h to afford the product as a dark purple solid (170 mg, 79%): mp 270–272 °C. 1H NMR (DMSO-d6-CD3OD, 2:1) δ 9.07 (s, 1 H), 7.78 (s, 2 H), 7.60 (s, 1 H), 7.42 (s, 1 H), 7.14 (s, 1 H), 6.96 (s, 1 H), 6.13 (s, 2 H), 4.41 (t, J = 6.6 Hz, 2 H), 4.36 (t, J = 7.3 Hz, 2 H), 3.86 (s, 3 H), 3.82 (s, 3 H), 2.35 (t, J = 6.1 Hz, 2 H); ESIMS m/z (rel intensity) 494 (MH+, 100). Anal. (C25H22N3O6Cl) C, H, N.
6-[3-Pyrazolyl-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (7)
Indenoisoquinoline 517 (0.2113 g, 0.448 mmol) was added to sodium hydride (86.8 mg of a 60% suspension in mineral oil, 2.17 mmol) and pyrazole (0.1749 g, 2.57 mmol) in DMF (50 mL) and the reaction mixture was heated at 60 °C for 4 h. The reaction mixture was diluted with water (200 mL) and extracted with chloroform (200 mL). The organic layer was washed with water (7 x 200 mL) and concentrated. Benzene was added (2 x 30 mL) and the mixture was concentrated again. The residue was dissolved in chloroform (4 mL) and diethyl ether (50 mL) was added. The precipitate was washed with diethyl ether (100 mL) and a dark red solid (118.5 mg, 57.6%) was obtained: mp 262–264 °C (dec). IR (film) 3462, 3104, 2918, 1693, 1640, 1557, 1495, 1488, 1430, 1394, 1308, 1284, 1251, 1205, 868, 785, 769 cm−1; 1H NMR (DMSO-d6) δ 7.97 (s, 1 H), 7.65 (d, J = 1.5 Hz, 1 H), 7.61 (s, 1 H), 6.57 (s, 1 H), 6.97 (s, 1 H), 6.68 (s, 1 H), 6.33 (s, 1 H), 6.05 (s, 2 H), 4.40 (m, 4 H), 4.01 (s, 3 H), 3.96 (s, 3 H), 2.45 (m, 2 H); ESIMS m/z (rel intensity) 460 (MH+, 100). Anal. (C25H21N3O6·0.75 H2O) C, H, N.
6-{3-[2-(1,2,4)]-Triazolyl-1-propyl}-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (8)
Indenoisoquinoline 517 (0.2538 g, 0.538 mmol) was added to sodium hydride (124.8 mg of a 60% suspension in mineral oil, 3.12 mmol) and 1,2,4-triazole (0.2673 g, 0.566 mol) in DMF (50 mL) and the reaction mixture was heated at 60 °C for 3 h. The reaction mixture was diluted with water (200 mL) and the precipitate was separated by filtration and washed with water (50 mL). The precipitate was partially dissolved in methanol-chloroform 1:1 (200 mL). Diethyl ether (100 mL) was added and the precipitate was separated by filtration and washed with additional diethyl ether (100 mL) to provide the product as the free base. The residue was dissolved in trifluoroacetic acid (2 mL) and hydrochloric acid (4 mL of a 2 M solution in diethyl ether) was added, followed by more diethyl ether (30 mL). The product was collected as a dark red solid (159.5 mg, 57%): mp > 240 °C. IR (KBr) 3429, 1694, 1647, 1553, 1500, 1487, 1431, 1394, 1311, 1254, 1207, 1032, 928, 873, 800, 786, 722, 617 cm−1; 1H NMR (DMSO-d6) δ 8.56 (s, 1 H), 7.99 (s, 1 H), 7.90 (s, 1 H), 7.52 (s, 1 H), 7.15 (s, 1 H), 7.10 (s, 1 H), 6.19 (s, 2 H), 4.44–4.38 (m, 4 H), 3.90 (s, 3 H), 3.86 (s, 3 H), 2.25 (m, 2 H); ESIMS m/z (rel intensity) 461 (MH+, 53), 392 (MH+ – C2N3H3, 100). High resolution ESIMS m/z (rel intensity) 461.1464 (100, MH+) (calculated mass 461.1461).
6-(3-Thiazolylamino-1-propyl)-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Dihydrochloride (9)
The product (213 mg, 41%) was dissolved in chloroform (50 mL) and treated with an anhydrous solution of 2 M HCl in diethyl ether (15 mL, 30.0 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 6 h and the precipitated product was filtered and washed with chloroform (50 mL), methanol (10 mL), and dried over P2O5 to provide the desired analogue as a pale purple solid (140 mg, 61%): mp 298–300 °C (dec). 1H NMR (DMSO-d6) δ 7.82 (s, 1 H), 7.44 (s, 1 H), 7.38 (s, 1 H), 7.04 (s, 1 H), 6.18 (s, 2 H), 4.42 (bs, 2 H), 4.07 (bs, 2 H), 3.88 (s, 3 H), 3.83 (s, 3 H), 3.76 (bs, 4 H), 2.07 (bs, 2 H); ESIMS m/z (rel intensity) 494 (MH+, 100). Anal. (C25H25N3O6SCl2·0.6 CHCl3) C, H, N.
6-(3-Piperazinyl-1-propyl)-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Dihydrochloride (10)
The product (350 mg, 72%) was dissolved in chloroform and treated with 2 M HCl in diethyl ether (9.0 mL, 18.2 mmol) at room temperature to afford the desired analogue as a pale purple solid (280 mg, 84%): mp 276–278 °C (dec). 1H NMR (D2O) δ 6.63 (bs, 1 H), 6.53 (s, 1 H), 6.47 (bs, 1 H), 6.18 (s, 1 H), 5.91 (s, 2 H), 3.90 (bs, 2 H), 3.51 (s, 3 H), 3.46 (bs, 11 H), 3.20 (bs, 2 H), 2.02 (bs, 2 H); ESIMS m/z (rel intensity) 478 (MH+, 100). Anal. (C26H29Cl2N3O6·2.3 H2O) C, H, N.
3-[(Morpholinyl)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (11)
The product was isolated as a dark purple solid (0.220 g, 72%): mp 290–292 °C. 1H NMR (CDCl3) δ 7.98 (s, 1 H), 7.59 (s, 1 H), 7.36 (s, 1 H), 7.02 (s, 1 H), 6.07 (s, 2 H), 4.48 (t, J = 7.39 Hz, 2 H), 4.02 (s, 3 H), 3.95 (s, 3 H), 3.76 (bs, 4 H), 2.54 (bs, 6 H), 2.01 (bs, 2 H); ESIMS m/z (rel intensity) 479 (MH+, 100). Anal. (C26H26N2O7·0.2 H2O) C, H, N.
3-[(Thiomorpholinyl)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (12)
The product was isolated as a dark purple solid (275 mg, 53%): mp 306–308 °C. 1H NMR (CDCl3) δ 7.80 (s, 1 H), 7.60 (s, 1 H), 7.33 (s, 1 H), 7.04 (s, 1 H), 6.08 (s, 2 H), 4.48 (t, J = 6.4 Hz, 2 H), 4.02 (s, 3 H), 3.96 (s, 3 H), 2.84–2.78 (bs, 8 H), 2.67 (bs, 2 H), 2.09 (bs, 2 H); ESIMS m/z (rel intensity) 495 (MH+, 100). Anal. (C26H26N2O6S·0.3 H2O) C, H, N.
6-[3-(3-Hydroxypiperidinyl)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (13)
The product (220 mg, 0.45 mmol, 70%) was treated with 2 M HCl in diethyl ether (4.0 mL, 6.69 mmol) in chloroform at room temperature to afford the desired analogue as a purple solid (210 mg, 89%): mp 288–290 °C. 1H NMR (D2O) δ 6.54 (bs, 1 H), 6.41 (s, 1 H), 6.29 (bs, 1 H), 6.06 (s, 1 H), 5.88 (s, 2 H), 3.82 (bs, 2 H), 3.45 (s, 3 H), 3.37 (bs, 7 H), 3.15 (bs, 3 H), 1.99 (bs, 4 H), 1.68 (bs, 2 H); ESIMS m/z (rel intensity) 493 (MH+, 100). Anal. (C27H29ClN2O7·1.4 H2O) C, H, N.
3-[(1-Methylpiperazinyl)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (14)
The desired analogue was isolated as a dark purple solid (160 mg, 51%): mp 254–256 °C. 1H NMR (CDCl3) δ 7.99 (s, 1 H), 7.60 (s, 1 H), 7.30 (s, 1 H), 7.03 (s, 1 H), 6.08 (s, 2 H), 4.47 (t, J = 6.0 Hz, 2 H), 4.02 (s, 3 H), 3.96 (s, 3 H), 2.55 (bs, 10 H), 2.30 (s, 3 H), 1.99 (bs, 2 H); ESIMS m/z (rel intensity) 492 (MH+, 100). Anal. (C27H29N3O6·0.5 CHCl3) C, H, N.
6-[3-(4-Aminopiperidinyl)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Dihydrochloride (15)
The product (205 mg, 66%) was dissolved in chloroform (30 mL) and treated with 2 M HCl in diethyl ether (5.2 mL, 10.40 mmol) at room temperature for 8 h. The precipitate was filtered and washed with chloroform (30 mL) to provide the desired analogue as a dark purple solid (165 mg, 85%): mp 262–264 °C (dec). 1H NMR (D2O) δ 6.62 (s, 1 H), 6.50 (s, 1 H), 6.44 (s, 1 H), 6.17 (s, 1 H), 5.92 (s, 2 H), 3.92 (bs, 2 H), 3.64 (bs, 2 H), 3.50 (s, 4 H), 3.45 (s, 3 H), 3.23 (bs, 2 H), 3.08 (bs, 2 H), 2.25 (m, 2 H), 2.06 (bs, 2 H), 1.90 (m, 2 H); ESIMS m/z (rel intensity) 492 (MH+, 70).
6-(3-Homopiperazinyl-1-propyl)-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Dihydrochloride (16)
The obtained product (390 mg, 0.66 mmol, 69%) was dissolved in chloroform and treated with 2 M HCl in diethyl ether (10.0 mL, 19.8 mmol) to afford the desired analogue as a purple solid (305 mg, 82%): mp 264–266 °C (dec). 1H NMR (D2O) δ 6.71 (bs, 1 H), 6.56 (bs, 2 H), 6.21 (bs, 1 H), 5.92 (s, 2 H), 3.98 (bs, 2 H), 3.63–3.57 (bs, 6 H), 3.55 (s, 3 H), 3.50 (s, 3 H), 3.36–3.25 (bs, 4 H), 2.19 (bs, 2 H), 2.09 (bs, 2 H); ESIMS m/z (rel intensity) 492 (MH+, 100). Anal. (C27H31Cl2N3O6·0.7 H2O) C, H, N.
3-[(1-Hydroxyethyl-piperazine)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17)
The desired analogue was isolated as a dark brown solid (258 mg, 47%): mp 262–264 °C. 1H NMR (CDCl3) δ 8.00 (s, 1 H), 7.60 (s, 1 H), 7.32 (s, 1 H), 7.04 (s, 1 H), 6.06 (s, 2 H), 4.52 (bs, 2 H), 4.03 (s, 3 H), 3.96 (s, 3 H), 3.22 (bs, 4 H), 3.13 (bs, 6 H), 2.84 (bs, 2 H), 2.68 (bs, 2 H), 1.73 (bs, 4 H), 1.63 (bs, 4 H), 1.43 (s, 18 H), 1.41 (s, 9 H); ESIMS m/z (rel intensity) 522 (MH+, 100). Anal. (C28H31N3O7·0.8 H2O) C, H, N.
6-[(3-Morpholylethylamino)-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (18)
The desired analogue was isolated as a pale purple solid (245 mg, 59%): mp 215–217 °C. 1H NMR (CDCl3) δ 8.01 (s, 1 H), 7.62 (s, 1 H), 7.42 (s, 1 H), 7.05 (s, 1 H), 6.06 (s, 2 H), 4.52 (bs, 2 H), 4.03 (s, 3 H), 3.97 (s, 3 H), 3.70 (bs, 4 H), 2.81 (bs, 2 H), 2.73 (bs, 2 H), 2.53 (bs, 2 H), 2.46 (bs, 4 H), 2.02 (bs, 2 H); ESIMS m/z (rel intensity) 522 (MH+, 100). Anal. (C28H31N3O7·1.0 H2O) C, H, N.
Top1-Mediated DNA Cleavage Reactions
Human recombinant Top1 was purified from Baculovirus as described previously.19 The 161 bp fragment from pBluescript SK(−) phagemid DNA (Stratagene, La Jolla, CA) was cleaved with the restriction endonuclease Pvu II and Hind III (New England Biolabs, Beverly, MA) in supplied NE buffer 2 (50 μL reactions) for 1 h at 37 °C, and separated by electrophoresis in a 1% agarose gel made in 1X TBE buffer. The 161 bp fragment was eluted from the gel slice using the QIAEX II kit (QIAGEN Inc., Valencia, CA). Approximately 200 ng of the fragment was 3′-end labeled at the Hind III site by fill-in reaction with [alpha-32P]-dGTP and 0.5 mM dATP, dCTP, and dTTP, in React 2 buffer (50 mM Tris-HCl, pH 8.0, 100 mM MgCl2, 50 mM NaCl) with 0.5 units of DNA polymerase I (Klenow fragment). Unincorporated 32P-dGTP was removed using mini Quick Spin DNA columns (Roche, Indianapolis, IN), and the eluate containing the 3′-end-labeled 161 bp fragment was collected. Aliquots (approximately 50,000 dpm/reaction) were incubated with Top1 at 22 °C for 30 min in the presence of the tested drug. Reactions were terminated by adding SDS (0.5% final concentration). The samples (10 μL) were mixed with 30 μL of loading buffer (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue, pH 8.0). Aliquots were separated in denaturing gels (16% polyacrylamide, 7 M urea). Gels were dried and visualized by using a Phosphoimager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Molecular Modeling
The structure of the ternary complex, containing topoisomerase I, DNA, and an indenoisoquinoline was utilized as follows.15 All of the atoms were fixed according to Sybyl® atom types and hydrogens were added and minimized using the MMFF94s force field and MMFF94 charges. The structure of the indenoisoquinoline 6, constructed in Sybyl® and energy minimized with the Tripos force field and MMFF94 charges, was overlapped with the structure of the crystallized indenoisoquinoline according to structural similarities of the aromatic rings in the ternary complex, and the structure of the crystallized indenoisoquinoline was then deleted. The new whole complex was subsequently subjected to energy minimization using the MMFF94s force field with MMFF94 charges. During energy minimization, the structure of the indenoisoquinoline was allowed to move while the structures of the protein and nucleic acids were frozen. The energy minimization was performed using the Powell method with a 0.05 kcal/mol·Å energy gradient convergence criterion and a distance-dependent dielectric constant.
Supplementary Material
Elemental analyses for compounds 6, 7, 9–14, and 16–18. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support of this work with Research Grant UO1 CA89566, Training Grant ST32 CA09634-12, and an American Chemical Society Division of Medicinal Chemistry Predoctoral Fellowship (A.M.). The in vitro and in vivo testing was conducted through the Developmental Therapeutics Program, DCTD, NCI under Contract NO1-CO-56000.
References
- 1.Paull KD, Hamel E, Malspeis L. Cancer Chemotherapeutic Agents. American Chemical Society; Washington, DC: 1995. Prediction of Biochemical Mechanism of Action from the In Vitro Antitumor Screen of the National Cancer Institute; pp. 9–45. [Google Scholar]
- 2.Kohlhagen G, Paull K, Cushman M, Nagafuji P, Pommier Y. Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 3.Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M. Synthesis and Mechanism of Action Studies of a Series of Norindenoisoquinoline Topoisomerase I Poisons Reveal an Inhibitor with a Flipped Orientation in the Ternary DNA-Enzyme-Inhibitor Complex As Determined by X-ray Crystallographic Analysis. J Med Chem. 2005;48:4803–4814. doi: 10.1021/jm050076b. [DOI] [PubMed] [Google Scholar]
- 4.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 5.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 6.Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of Cytotoxic 11-Aminoalkenylindenoisoquinoline and 11-Diaminoalkenylindenoisoquinoline Topoisomerase I Inhibitors. Bioorg Med Chem. 2004;12:5147–5160. doi: 10.1016/j.bmc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 7.Nagarajan M, Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Featuring Polyamine Side Chains on the Lactam Nitrogen. J Med Chem. 2003;46:5712–5724. doi: 10.1021/jm030313f. [DOI] [PubMed] [Google Scholar]
- 8.Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker B, Stewart L, Cushman M. Design, Synthesis, and Biological Evaluation of Cytotoxic 11-Alkenylindenoisoquinoline Topoisomerase I Inhibitors and Indenoisoquinoline-Camptothecin Hybrids. J Med Chem. 2003;46:3275–3282. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]
- 9.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Benz[d]indeno[1,2-b]pyran-5,11-diones: Versatile Intermediates for the Design and Synthesis of Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2006;16:1846–1849. doi: 10.1016/j.bmcl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Pommier Y. Eukaryotic DNA Topoisomerase I: Genome Gate Keeper and Its Intruders, Camptothecins. Semin Oncol. 1996;23:1–10. [PubMed] [Google Scholar]
- 11.(a) Jaxel C, Kohn KW, Wani MC, Pommier Y. Structiure-Activity Study of the Actions of Camptothecin Derivatives on Mammalian Topoisomerase I: Evidence for a Specific Receptor Site and a Relation to Antitumor Activity. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]; (b) Minami H, Beijnen JH, Verweij J, Ratain MJ. Limited Sampling Model for the Area under the Concentration Time Curve of Total Topotecan. Clin Cancer Res. 1996;2:43–46. [PubMed] [Google Scholar]; (c) Danks MK, Pawlik CA, Whipple DO, Wolverton JS. Intermittant Exposure of Medulloblastoma Cells to Topotecan Produces Growth Inhibition equivalent to Continuous Exposure. Curr Topics Med Chem. 1997;3:1731–1738. [PubMed] [Google Scholar]; (d) Haas NB, LaCreta FP, Walczak J, Hudes GR, Brennan JM, Ozols RF, O’Dwyer PJ. PhaseI/Pharmaco-kinetic Study of Topotecan by 24-Hour Continuous Infusion Weekly. Cancer Res. 1994;54:1220–1226. [PubMed] [Google Scholar]
- 12.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani F, Rustum Y, Cushman M, Pommier Y. Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 13.Meng LH, Liao ZY, Pommier Y. Non-Camptothecin DNA Topoisomerase I Inhibitors in Cancer Therapy. Curr Topics Med Chem. 2003;3:305–320. doi: 10.2174/1568026033452546. [DOI] [PubMed] [Google Scholar]
- 14.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analog. Proc Natl Acad Sci USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 16.Marchand C, Antony S, Kohn KW, Cushman M, Ioanoviciu A, Staker BL, Burgin AB, Stewart L, Pommier Y. A Novel Norindenoisoquinoline Structure Reveals a Common Interfacial Inhibitor Paradigm for Ternary Trapping of the Topoisomerase I-DNA Covalent Complex. Mol Cancer Ther. 2006;5:287–295. doi: 10.1158/1535-7163.MCT-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J Med Chem. 2000;43:3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 18.Hollingshead M, Plowman J, Alley M, Mayo J, Sausville E. The Hollow Fiber Assay. Contrib Oncol. 1999;54:109–120. [Google Scholar]
- 19.Pourquier P, Ueng LM, Fertala J, Wang D, Park HJ, Essigman J, Bjornsti MA, Pommier Y. Induction of Reversible Complexes between Eukaryotic DNA Topoisomerase I and DNA-containing Oxidative Base Damages. 7,8-Dihydro-8-Oxoguanine and 5-Hydroxycytosine. J Biol Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analyses for compounds 6, 7, 9–14, and 16–18. This material is available free of charge via the Internet at http://pubs.acs.org.