

Abstract
A novel microfluidic device that can selectively and specifically isolate exceedingly small numbers of circulating tumor cells (CTCs) through a monoclonal antibody (mAB) mediated process by sampling large input volumes (≥1 mL) of whole blood directly in short time periods (<37 min) was demonstrated. The CTCs were concentrated into small volumes (190 nL), and the number of cells captured was read without labeling using an integrated conductivity sensor following release from the capture surface. The microfluidic device contained a series (51) of high-aspect ratio microchannels (35 μm width × 150 μm depth) that were replicated in poly(methyl methacrylate), PMMA, from a metal mold master. The microchannel walls were covalently decorated with mABs directed against breast cancer cells overexpressing the epithelial cell adhesion molecule (EpCAM). This microfluidic device could accept inputs of whole blood, and its CTC capture efficiency was made highly quantitative (>97%) by designing capture channels with the appropriate widths and heights. The isolated CTCs were readily released from the mAB capturing surface using trypsin. The released CTCs were then enumerated on-device using a novel, label-free solution conductivity route capable of detecting single tumor cells traveling through the detection electrodes. The conductivity readout provided near 100% detection efficiency and exquisite specificity for CTCs due to scaling factors and the nonoptimal electrical properties of potential interferences (erythrocytes or leukocytes). The simplicity in manufacturing the device and its ease of operation make it attractive for clinical applications requiring one-time use operation.
Introduction
It is generally accepted that most cancer-related mortalities result from metastatic disease.1 While the process of metastasis is not well understood, a number of chemical, physical, and molecular events occur that ultimately result in the dissemination and deposition of tumor cells into targeted organs using the circulatory system and/or bone marrow as the carrier(s). This “seed and soil” process was proposed as early as 1889 by Paget and was later modified with the caveat that shed tumor cells consist of a heterogeneous population with subpopulations possessing different metastatic potentials. The fundamental entities primarily responsible for spawning metastatic disease are circulating tumor cells (CTCs), which can be produced during early stages of tumorigenesis.4 Elucidating the quantity of CTCs in peripheral blood or bone marrow can serve as an indicator for the clinical management of several cancer-related diseases by providing information on the success/failure of therapeutic intervention and disease stage forecasting.5, The isolation and enumeration of exfoliated CTCs in peripheral blood or bone marrow for a variety of cancer-related diseases has already been reported for a variety of cancer-related diseases, such as breast,79 colorectal,10 prostate,11 renal,12 bladder,13 and nonsmall cell lung14 cancers. As an example of the clinical utility of CTC information, Cristofanilli et al. recently reported a study of 177 breast cancer patients using the amount of CTCs in peripheral blood as an indicator of survival.7 Patients with ≥5 CTCs per 7.5 mL of whole blood possessed a median progression-free survival of 2.7 months versus 7.0 months for those patients containing <5 CTCs in 7.5 mL of their peripheral blood.
The major issue with securing viable clinical information via quantification of CTC levels is the extremely low abundance or rare-event nature of these cells among a high number of spectator cells in peripheral blood.6,15–18 For example, it is clinically useful to quantitatively enumerate 0–10 CTCs in whole blood composed of >109 erythrocytes and >106 leukocytes per mL.7
For sampling rare events in a large population, three important metrics must be assessed: (1) throughput, the number of cell identification or sorting steps per unit time; (2) recovery, an indicator of the fraction of target cells collected from the input sample; and (3) purity, which depends on the number of “interfering” cells excluded from the analysis.19 In addition to these three metrics, highly efficient quantification of the number of enriched cells must be provided as well.
The approaches used to date to enrich CTCs from clinical samples have provided lower-than-desired recoveries with high purity, relatively poor purity but with high recoveries, or, in other cases, highly specialized sample processing and handling whose success is laboratory dependent.20–25 For example, immunomagnetic approaches for CTC enrichment using ferromagnetic micrometer-sized particles coated with molecular recognition elements specific for antigenic-bearing target cells can interrogate diluted blood samples typically yield modest recoveries (~70%) but extremely favorable purity.22 In the case of size-based separations employing nuclear tracked membranes, polycarbonate membranes with varying pore sizes (8–14 μm) can filter large volumes (9.0–18 mL) of blood and recover nearly 85% of the CTCs, but significant numbers of leukocytes are also retained (i.e., low purity) potentially complicating the enumeration process.26 Investigations utilizing reverse-transcription PCR, in which mRNAs are used as surrogates to report CTC levels, have the ability to detect one CTC in an excess of 106 mononucleated cells.27 However, these assays are prone to high interlaboratory variability and require extensive sample handling and manipulation.
Most of the CTC isolation/sorting tools currently in use possess some common procedural characteristics that make them prohibitively difficult to implement, such as the ability to sort only the mononucleated fraction of whole blood requiring density gradient centrifugation prior to enrichment and the use of either flow cytometry or fluorescence microscopy following cell staining to enumerate the enriched CTCs. These additional steps require sample handling and transfer, which can induce cell loss or contamination that can dramatically affect the assay result, especially when dealing with low numbers of targets.
Microfluidics provides a venue for producing integrated systems that can process clinical samples in closed architectures to minimize sample contamination and loss. However, high throughput sampling of relatively large volumes (>1 mL) has not been a mainstay for microfluidics due to the macro-to-micro dilemma resulting from the small dimensional features associated with these devices. For example, exhaustively sampling a 1.0 mL volume input using a microchannel of 30 μm × 30 μm at a linear velocity of 1.0 mm s−1 would require 309 h (12.9 days). This sampling bottleneck was recently addressed by studies with a glass-based microfluidic device fabricated using deep reactive ion etching.28 The high-surface area immunological capture bed consisted of microposts (100 μm diameter × 100 μm tall) arranged in an equilateral triangular format; the device was capable of capturing ~60% of CTCs from untreated whole blood with enumeration achieved by cell staining and microscopic visualization.
Herein we report our efforts aimed toward the development of a self-contained system capable of meticulously separating intact CTCs from peripheral blood and directly quantifying the CTC level upon isolation and enrichment. As a result of careful fluidic design rules and system integration methods, we have successfully employed a microfluidic system capable of exhaustively and rapidly interrogating >1.0 mL of unprocessed whole blood possibly harboring low-abundant CTCs. At the heart of the system were carefully engineered, exceedingly efficient high-aspect ratio capture beds decorated with mABs specific for antigenic integral membrane proteins expressed in CTCs of epithelial origin and a label-free, highly specific single-cell conductivity sensor. The device operational characteristics were achieved by tailoring the dominant CTC capture dynamics, specific device architecture, and suspension linear velocity in a high throughput microsampling unit (HTMSU) containing high-aspect ratio microstructures replicated in a polymeric substrate. Direct single-cell counting of the captured cells was made possible by their release as a result of enzymatic digestion of cell-antigen/antibody-surface complexes. Quantitative assessment of CTC numbers was accomplished using an integrated conductivity sensor capable of specifically detecting CTCs via their electrical signatures without requiring cell staining or microscopic visualization.
Material and Methods
HTMSU Fabrication
Figure 1 shows a schematic of the HTMSU and several micrographs of this polymer-based device. The device consisted of a series of 51 high-aspect ratio channels that were either linear or sinusoidally configured and shared a set of common input/output ports. Devices were replicated from a mold master using hot embossing. See the Supporting Information for details on the fabrication of the mold master and the microreplication. The substrate selected for the HTMSU was PMMA due to its high fidelity of forming structures with high aspect ratio via microreplication, minimal nonspecific adsorption of whole blood components to its surface, and its ability to generate functional surface scaffolds through UV irradiation for the attachment of a variety of biological moieties.29
Figure 1.

Schematics of the HTMSU showing the following: (A) A scaled AutoCAD diagram of the sinusoidally shaped capture channels with brightfield optical micrographs of (B) the integrated conductivity sensor consisting of cylindrical Pt electrodes that were 75 μm in diameter with a 50 μm gap and (C) the single port exit where the HTMSU tapers from 100 μm wide to 50 μm while the depth tapers from 150 to 80 μm over a 2.5 mm region that ends 2.5 mm from the Pt electrodes; (D) micrograph taken at 5× magnification showing the sinusoidal cell capture channels; and (E) 3D projection of the topology of the HTMSU obtained at 2.5 μm resolution using noncontact optical profilometry (arrows indicate the Pt electrode conduits).
Antibody Immobilization
Antibody immobilization was performed using a two-step process. Initially, the UV-modified HTMSU device, following thermal assembly, was loaded with a solution containing 4.0 mg/mL of 1-ethyl-3-[3-dimethylaminopro-pyl] carbodiimide hydrochloride (EDC), 6.0 mg/mL of N-hydrox-ysuccinimide (NHS) in 150 mM 2-(4-morpholino)-ethane sulfonic acid at pH = 6 (MES, Fisher Biotech, Fair Lawn, NJ), and buffered saline (Sigma-Aldrich, St. Louis, MO) for 1.0 h to form the succinimidyl ester intermediate. The EDC/NHS solution was then hydrodynamically replaced with a 1.0 mg/mL monoclonal anti-EpCAM antibody (R&D Systems Inc., Minneapolis, MN) solution contained in 150 mM PBS at pH = 7.4 (Sigma-Aldrich, St Louis, MO) and allowed to react for 4 h after which the device was rinsed with a solution of PBS (pH = 7.4) to remove any nonspecifically bound anti-EpCAM antibodies. For a complete description of this procedure, see the Supporting Information.
Apparatus
A PHD2000 syringe pump (Harvard Apparatus, Holliston, MA) was used to hydrodynamically process samples using the HTMSU. In order to interface the HTMSU to the pump, a luer lock syringe (Hamilton, Reno, NV) was fitted with a luer-to-capillary adapter (InnovaQuartz, Phoenix, AZ). The syringe pump was programmed to generate the appropriate volume flow rate to evaluate CTC cell capture efficiency and postcapture cell retention. The linear velocities were calculated from the cross-sectional area of the respective HTMSU capture channels and the programmed volume flow rate. The flow rates were validated by tracking, via optical microscopy, suspended cells over a fixed 80 μm region.
During evaluative experiments, the HTMSU was fixed to a programmable motorized stage of an Axiovert 200 M (Carl Zeiss, Thornwood, NY) microscope, which could monitor the transport of cells in the HTMSU using either fluorescence or brightfield imaging. Videos of cell transport were captured at 30 frames per second (fps) using a monochrome CCD (JAI CV252, San Jose, CA). For fluorescence, excitation was accomplished using a Xe arc lamp and dye-specific filter sets appropriate for the fluorescent dye used for labeling the MCF-7 cells (Carl Zeiss, Thornwood, NY). Each filter cube contained a dichroic mirror, an emission filter, and an excitation filter.
Integrated Conductivity Sensor
The conductivity electrodes consisted of Pt wires (~75 μm diameter) placed into guide channels that were embossed into the fluidic network and positioned orthogonal to the fluidic output channel (see Figure 1). A Pt wire was inserted into these guide channels prior to thermal assembly of the cover plate to the substrate. Once the wire was positioned, the substrate/wire assembly was placed between glass plates and clamped together and, finally, heated to slightly higher than the glass transition temperature of PMMA, which embedded the wire into the guide channels and spanned the entire depth of the output channel. To break through the wire to form the electrode pair, a high precision micromilling machine (KERN MMP 2522, KERN Micro- and Feinwerktechnik GmbH & Co.KG; Germany) with a 50 μm bit was used (see Figure 1B). Following machining of the Pt wire and UV activation, the cover plate was aligned to the embossed substrate via alignment marks and clamped together between two glass plates and subjected to thermal fusion bonding.
The conductivity sensor was assembled in-house (for details see Supporting Information). Conductivity measurements were conducted in TRIS-glycine buffer containing 0.25% (w/w) trypsin, 0.18 mM TRIS, 47 mM glycine, and 0.05% (v/v) Tween-20, referred to as the CTC releasing buffer. This CTC releasing buffer was carefully selected in large part for its relatively low conductivity (~50 μS/cm, pH 7.2) and the addition of trypsin to remove the bound CTCs from the capture channel surface for enumeration via single-cell conductometric counting.
Cell Suspensions
Citrated whole rabbit blood was purchased from Colorado Serum Company (Denver, CO). Upon collection, the blood was combined with 10% (w/w) sodium citrate to prevent coagulation. The MCF-7 cells (breast cancer cell line), growth media, phosphate buffered saline, trypsin, and fetal bovine serum was purchased from American type Culture Collection (Manassas, VA). Adherent MCF-7 cells were cultured to 80% confluence in Dulbecco’s modified Eagle’s Medium supplemented with high glucose containing 1.5 g L−1 sodium bicarbonate (NaHCO3), 15 mM HEPES buffer, and 10% fetal bovine serum (FBS). A 0.25% trypsin solution was prepared in 150 mM PBS and used to harvest the MCF-7 cells.
MCF-7 cells were stained with a fluorescein derivative, PKH67, containing a lipophilic membrane linker for fluorescence visualization experiments (Sigma-Aldrich, St. Louis, Mo). A modified protocol for cell staining was implemented whereby the dye concentration was increased 2-fold resulting in more evenly distributed fluorescent labels over the cell’s membrane. Cell counts for seeding experiments into whole blood were determined by counting three aliquots of cells in succession using a hemacytometer. The cell count accuracy was within 10%.
Results and Discussion
Model CTC System
The MCF-7 cell line, used as a model for CTC selection and enumeration via the integrated HTMSU, is a breast cancer cell line that possesses an overexpressed membrane antigen termed the epithelial cell adhesion molecule, EpCAM.30–33 MCF-7 cells are typically 15–30 μm in diameter (mean = 24 μm) and have been closely associated with micrometastatic breast cancer.34 EpCAM occurs at a frequency of 5.1 × 105 molecules per cell.35 Monoclonal antibodies for EpCAM are available, which can be used to selectively speciate these cells from mixed populations.
A number of experimental and device operational parameters were investigated to optimize the performance of the HTMSU: (1) Throughput - linear flow velocity, pressure drops, processing time; (2) recovery - capture channel geometry (shape and width), and cell flow dynamics; (3) purity - designing surfaces to minimize nonspecific adsorption and provide high selectivity for the target cells.
Pressure Drop
Because the goal of the HTMSU was to process whole blood directly in a high throughput format requiring high volumetric flow rates to reduce processing time, we employed capture channels with high aspect ratio (aspect ratio = channel height/channel width). For a capture channel width and height similar to the cell dimensions (aspect ratio = 1), capture of a cell within this channel would produce a large pressure drop due to obstruction and could possibly damage the captured cell making it unavailable for enumeration. Therefore, we adopted a capture channel geometry with an aspect ratio of 4.3; for a 35 μm channel width, the depth was 150 μm, which could easily be replicated using hot embossing. From eq S6 (see Supporting Information) and a blood viscosity of 4.8 cP (hematocrit = 0.4), the pressure drop for a channel 35 μm × 35 μm (L = 3.5 cm) was calculated to be 7.4 × 103 Pa, while a channel that possessed dimensions of 35 μm × 150 μm had a pressure drop of 2.9 × 103 Pa, a reduction of greater than 60%.
Flow Dynamics
It has been observed that laminar flow in a tube (capillary) having dimensions greater than 15% that of the cells being transported through it results in a marginal zone near the tube wall due to migration of the cells toward the central axis of the tube leaving a cell-free layer adjacent to the wall that can be as thick as 4 μm.35 Because the capture elements (antibodies) are poised on the channel wall, this focusing effect can reduce the number of encounters between the CTCs and their recognition elements. To investigate this phenomenon, we imaged CTCs stained with a membrane-specific fluorescein derivative as they were transported through an unmodified (no mAB) 35 μm wide microchannel that had a straight or sinusoidal configuration (see Figure 1). The results of these experiments are given in Figure 2; shown in the panels are the effects of channel shape and cell translational velocity (U) on the cells’ radial position within the microchannel. Evident in the linear channels (Figure 2A and 2B) is a cell-free zone, similar to what has been observed in straight capillaries,36 whose thickness increased with increases in U. However, for the sinusoidally shaped capture channels, CTC flow dynamics were quite different. First, there was no apparent marginal zone along one edge of the microchannel wall, and second, the cell radial distribution seemed to be unaffected by changes in U. The result would be an increase in the antigen/antibody encounter rate as the cells moved through the capture beds at the relatively high linear velocities required to process large input volumes in reasonable amounts of time using sinusoidal-shaped channels. We believe that the cells migrate to the outside of the curved channel due to two factors: (1) a cross-stream velocity component due to the reversal of the direction of curvature and (2) centrifugal force acting on the cells.
Figure 2.
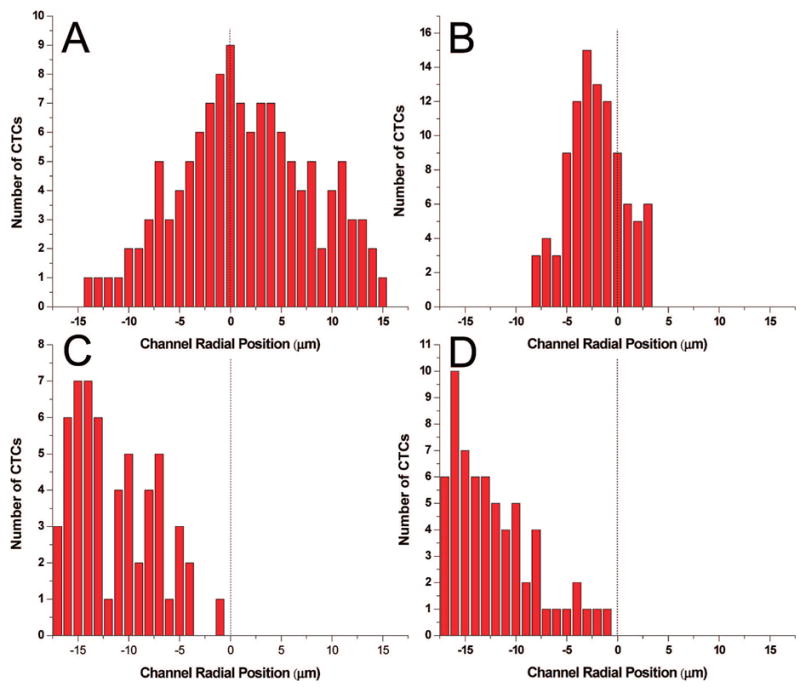
Histograms of the radial position of CTCs (centroid) in microchannels with Poiseulle flow at linear velocities (U) of 1.0 and 10 mm s−1 in both straight (A, B) and sinusoidal-configured (C, D) channels. The dashed line represents the microchannel’s central axis. (A and B) The radial position of several CTCs histogrammed from micrographs of the straight microchannel with linear flow rates of 1 and 10 mm s−1. (C and D) The radial position of several CTCs traversing 1/4 of a period of the sinusoidal microchannels with suspension linear velocities of 1 and 10 mm s−1 are shown. The cells were imaged using fluorescence microscopy with the cells stained using a fluorescein lipophilic membrane dye, PKH67.
To understand the fluid dynamics in terms of turbulence or Poiseulle flow for straight versus sinusoidally shaped micro-channels that may account for the results shown in Figure 2, we calculated the Reynolds and Dean numbers for the 35 μm curved channels (see Supporting Information, eqs S7–S12). The Dean number is a dimensionless quantity used for curved channels and accounts for the radius of curvature and the hydraulic diameter of the channel (see eq S9 in the Supporting Information).37 For high Reynolds numbers, Dean vortices form and create zones of recirculation. For our channel geometries, the Dean number was calculated to be ~1.1 for a translational velocity of 10 mm s−1 (see Figures S3 and S4 in the Supporting Information), well below the threshold number of 35.92, which has been identified as the critical value for the onset of flow instability.37
Effects of Channel Width and Linear Velocity on Capture Efficiency
In order to maximize the recovery of CTCs from whole blood, the dimensions and layout architecture of the capture channels and the linear flow velocity in which to operate the HTMSU were assessed. Experimental determination of CTC capture efficiency using cell selection channel widths of 20, 35, and 50 μm (straight channels) were evaluated as well as a 35 μm wide capture channel adopting a sinusoidal pattern. The results of these studies are depicted in Figure 3A as plots of CTC capture efficiency (%) versus linear velocity, U (mm s−1).
Figure 3.
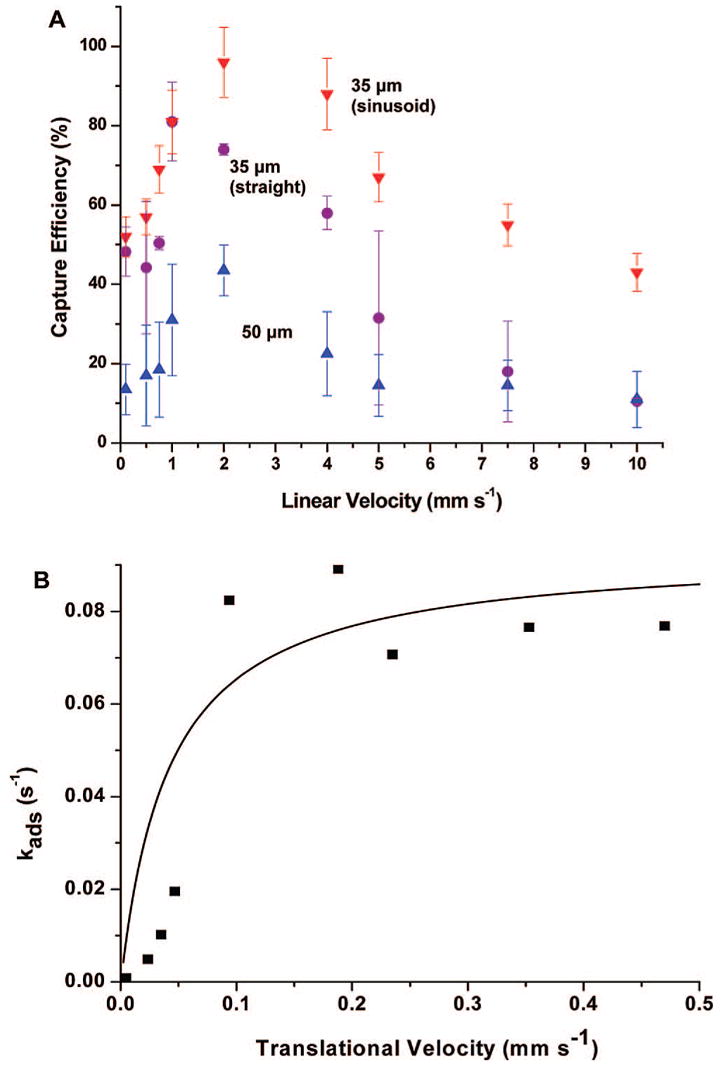
Data showing the capture efficiency of CTCs in spiked whole blood samples as a function of the cells’ translational velocity using 35 (red down triangles, sinusoid; purple circles, straight) and 50 μm (blue up triangles) wide microchannels. (A) The microfluidic device consisted of a single channel with the appropriate width and a depth of 150 μm. Following processing of the input buffer containing the MCF-7 cells, the number of captured cells was determined via brightfield microscopy by interrogating the entire length of the capturing channel. (B) The capture efficiency data as a function of the CTC translational velocity were replotted using eq S16 (see Supporting Information) with this data fit to Kf × Cx × Nr using the fitting parameter, kin (EpCAM/anti-EpCAM forward rate constant).
The 20 μm wide channel yielded 100% capture efficiency at all flow rates investigated. Because the CTCs were often larger than this channel, the cell membranes were in constant proximity to the antibody-coated microchannel walls. Also, most CTCs when observed via fluorescence microscopy did not enter the capture channel because their size was larger than the channel width. For those cells that were able to enter the selection bed, capture occurred within the first 1–2 mm of the 3.5 cm long channel. Due to these effects, the 20 μm device demonstrated consistent failure resulting from microchannel blockage. At higher linear flow velocities these blockages also led to compromising head pressures that resulted in device failure due to cover plate detachment from the embossed substrate. Therefore, adopting a channel width less than the cell average diameter was deemed inappropriate due to device failure and cell selection partly arising from size exclusion effects, which could lead to selection of a large number of leukocytes potentially complicating the enumeration process.
Inspection of the data in Figure 3A for capture channels with widths greater than the average CTC cell diameter showed that the capture efficiency increased with increasing linear velocity for all channel widths and shapes until a critical value was reached (~2 mm s−1) at which point the capture efficiency decreased. For all capture channel widths and shapes when operated at the same linear velocity, improved capture efficiencies were obtained for narrower channels with the highest efficiency demonstrated for the sinusoidal-shaped 35 μm wide channel (97%). This latter observation was consistent with the focusing effects noted for cells moving in a small channel experiencing Poiseulle flow (see Figure 2). Note that in practice as the velocity (1–10 mm s−1) is increased in the sinusoidal microchannels the CTCs deviate more from the microchannel centroid potentially resulting in a higher probability of cell/wall interactions.
To understand the capture efficiency dependence on linear velocity, we evaluated our data according to the Chang and Hammer model for cell adhesion between a surface-tethered antibody and a moving antigen.38 This model describes a two-state process:
| (1) |
| (2) |
where the first step (eq 1) accounts for transport of the solution antigen to the surface-bound antibody, which describes the encounter rate (ko), and the second step (eq 2) gives the probability (P) that an association event will occur during the time the antigen is in close proximity to the tethered antibody. The encounter rate, ko, increases with increasing linear flow velocity while the probability of a reaction, P, decreases (see Figure S5 in the Supporting Information). Analytical expressions for ko and P are provided in the Supporting Information based on the Chang/Hammer model.38 Below the cell translational velocity of 2 mm s−1, the capture efficiency is determined primarily by ko, but at U > 2 mm s−1, the capture rate is dominated by P. Plotted in Figure 3B is the cell adhesion rate, kads (see eq S16 in the Supporting Information), versus the cells’ translational velocity, which shows that kads asymptotically approaches a limiting value at translational velocities larger than 2 mm s−1. The data plotted in Figure 3B were then fit to the velocity-dependent intrinsic adhesion rate constant, kf, calculated from the product of ko and P (see eq S13 in the Supporting Information) with one adjustable parameter, kin, which represents the forward rate constant between EpCAM and its anti-EpCAM antibody. Literature values for the MCF-7 cell line and the EpCAM/anti-EpCAM complex were used to calculate P (see eq S15 in the Supporting Information as well as Table S2). The best fit was achieved for kin = 1 × 107 s−1, which is nearly 2 orders of magnitude larger than that reported in the literature for the EpCAM/anti-EpCAM forward rate constant (0.99 × 105 s−1).39 This disparity was most likely due to cooperativity effects resulting from the high expression level of EpCAM in this cell line.40 In addition, the nondeformable nature of cell adhesion assumed in the Chang/Hammer model was not observed in the current experiments (see Figure S6 in the Supporting Information).
Because the Chang/Hammer model is a local model that addresses only the interaction of the cell (antigen) and antibodies on the wall at a microscopic level, the external flow is accounted for by the inclusion of the mean velocity. Consequently, there is no change to Chang and Hammer’s model due to the sinusoidal channel geometries, but in its application, the local flow velocity will change due to the change in channel geometry.
A device with a single 35 μm × 150 μm channel operating at the optimal linear flow rate determined above to maximize the capture efficiency would exhaustively process 1 mL of input in 9.5 × 105 s (26 h). Simple increases in the linear velocity would not improve the sampling throughput without sacrificing capture efficiency, so the device was designed to contain multiple capture channels of similar dimensions fanning from a common input. For example, with 51 capture channels, the processing time was reduced to 1.9 × 103 s (31 min). The common output for all of the capture channels allowed for enumeration of cells selected from each channel (190 nL volume) in one step.
Shear Effects on Captured Cells
It was necessary to evaluate shear forces induced by the fluid traveling through the channels during cell selection and device washing prior to cell counting used to quantitatively enumerate the number of captured cells. Flow-induced shear could either detach the captured cells from the antibody-decorated walls or damage the cell prior to counting.
The total adhesion force (FA) between the cell and the antibody-decorated PMMA wall was evaluated using the model suggested by Bell.41 FA is derived from the product of the contact area, antibody surface density, and the adhesion force for a single EpCAM/anti-EpCAM bond, fc (see eq S18 in the Supporting Information). The value determined for fc was 6.7 × 10−6 dynes (see eq S20 in the Supporting Information) with a contact area of 16.6 μm2 for MCF-7 cells assuming the cells upon adhesion to the surface are nondeformable (see eq S19 in the Supporting Information). Using these values and an anti-EpCAM antibody PMMA surface density of ~2.3 × 1011 cm−2 produced a total adhesion force of 0.26 dynes.42 For cells that are flattened and elongated upon adhesion (see Figure S6 in the Supporting Information), the contact area was determined to be 456 μm2 and FA = 7.0 dynes.43 When the shear force (FS) generated by the laminar flow is equal to or greater than FA, the cell can be removed from the surface. The velocity dependent shear force, FS, was determined from Stokes’ law:
| (3) |
where r is the cell radius (12 μm for MCF-7 cells), η is the solution viscosity (4.8 cP for whole blood with a hematocrit level of 0.4), and νc is the critical linear velocity that can induce cell detachment. Rearrangement of eq 3 produced a value of 2.4 × 102 cm s−1 for νc assuming nondeformable contact. Insertion of the contact area and FA for the flattened and elongated captured cells yielded a value for νc of 6.4 × 103 cm s−1. Both of these values are significantly greater than the linear velocities used in the present experiments for optimizing the capture efficiency. Several captured cells were observed continuously during experiments in which linear velocities up to 10.0 cm s−1 were implemented and no cell damage or disruption of cell/wall adhesion was detected.
Detachment of Intact Cells for Quantitative Enumeration
Due to the strong adhesion force between the captured cells and the antibody-containing capture surface, thermodynamic release from the surface to allow for cell enumeration using the conductivity sensor was deemed inappropriate. The use of the proteolytic enzyme, trypsin, for the release of captured cells without causing cell disassembly, which would complicate single cell counting, was investigated. Figure 4 shows time-lapse micrographs of a surface-captured cell that was subjected to trypsin processing. Detachment was facilitated by proteolytic digestion in less than a 10 min incubation time. Rigorous evaluation of the trypsin release procedure indicated no cell damage was induced by this process.
Figure 4.

Time lapse micrographs of a captured MCF-7 cell: (A) Prior to exposure to the CTC releasing buffer and (B) exposed to the CTC releasing buffer for ~10 min. (C) The cell appears to be released from the PMMA surface, and finally, (D) the cell is swept away when the flow is initiated.
Conductivity Sensor for Cell Enumeration
Once the cells were released from the capture surface, they could be swept through the integrated Pt electrodes at the device output. The Pt electrodes were separated by 50 μm producing a cell constant, K (defined as the ratio of electrode gap to the electrode area), of ~0.01 μm−1. The cell constant was scaled to specifically detect CTCs due to their larger size with respect to leukocytes or erythrocytes. As shown in Figure 5A, the conductivity sensor with K = 0.01 μm−1 did not transduce the presence of leukocytes or erythrocytes due to their smaller size and their characteristic electrical properties. This is particularly attractive, because any nonspecific adsorption of leukocytes or erythrocytes during cell selection will not produce false positive signals providing an additional level of system specificity for the enumeration of CTCs.
Figure 5.
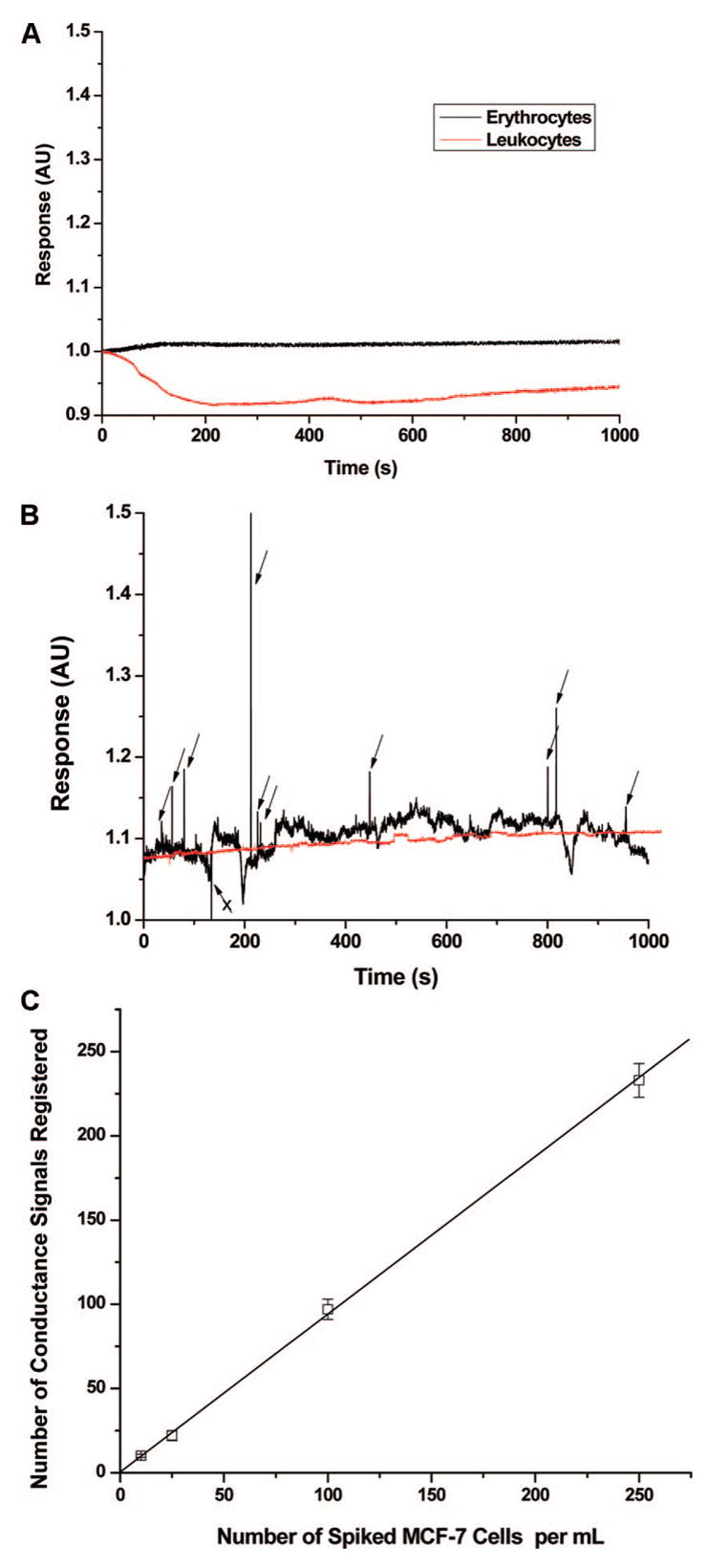
Conductance responses (in arbitrary units, AU) and calibration plots for CTCs are shown. (A) Conductometric response for suspensions of leukocytes and erythrocytes (cell density = 150 cells/μL) in TRIS-Glycine buffer transported through the integrated conductivity sensor at a volume flow rate of 0.05 μL/min. (B) Conductometric response of a 1.0 mL aliquot of whole blood spiked with 10 ± 1 CTCs or 0 CTCs and processed in the HTMSU at 2.0 mm/s. The isolated CTCs were released from the PMMA surface using the CTC releasing buffer and transported through the conductivity sensor at a volumetric flow rate of 0.05 μL/min. Peak identification was based on a signal-to-noise threshold of 3:1, which was determined by the peak height of the apparent response and the average peak-to-peak variation in the conductance of the CTC releasing buffer. The arrows designate those peaks scored as CTCs based on the aforementioned criteria. The arrow marked with an “X” possessed a conductivity response lower than the background buffer and as such was not scored as a CTC. Of the 10 ± 1 cells seeded into whole blood for this sample, 8 cells were scored above the 3σ threshold level. Also shown in this plot is a sample of whole blood containing no MCF-7 cells that was processed with the HTMSU device (red line). (C) Calibration plot for the number of CTCs seeded (10–250 cells mL−1) into whole blood versus the conductance responses registered using the conductivity sensor following the processing steps delineated in Figure 5B (m = 0.945, r2 = 0.9988), which shows the false positive rate. The data presented in Figure 5B were subjected to a three-point Savitsky–Golay smoothing.
Following antibody-mediated capture of CTCs seeded into whole blood using the HTMSU, the cells were released from the surface and hydrodynamically driven through the contact conductivity sensor.44 The chemical composition of CTCs make them ideal candidates for detection using conductivity due to their unique electrical properties compared to erythrocytes or leukocytes. For example, CTCs in general possess very low membrane potential and low impedance resulting from the intracellular migration of sodium ions (Na+) in compensation for the depleted potassium ion concentration (K+) of the intracellular fluid. Also, alteration in the cancer cell’s membrane content with respect to the types of glycoproteins and antigens they express result in an increase in the number of negatively charged sialic acid molecules that cap the extracellular domains of many integral membrane proteins.45
We next analyzed 1 mL of whole blood seeded with 10 ± 1 CTCs using the HTMSU, released the enriched cells, and quantitatively counted them with the conductivity sensor (see Figure 5B). Based on a signal-to-noise threshold of 3 (99.7% confidence level), there were 8 peaks in the conductance trace shown in Figure 5B that were assigned to CTCs of the 10 cells that were seeded into this sample. As can be seen from Figure 5B, only positive conductance spikes were scored as CTCs, while negative spikes were not scored as CTCs. The CTC scoring system was verified through brightfield microscopy, which indicated that CTCs produced positive signals relative to the background conductance generated by the CTC releasing buffer and the negative signals were produced from particulates. In addition, careful microscopic inspection indicated that the positive conductance spikes were indeed due to single cells. The disparity in the peak response for single CTCs was due to differences in cell morphology and composition partly resulting from any one of five mitosis phases of this cell line upon harvest.46
We also evaluated a blood sample that was not spiked with CTCs, the results of which are also plotted in Figure 5B. Careful inspection of this data indicated that there were no conductance signals that exceeded the 3σ criteria, indicating a false positive rate of 0.
For proper enumeration of the recovered CTCs using the HTMSU, the readout phase of the assay required a sampling efficiency near 100% so that every cell released from the capture channels could be detected with sufficient signal-to-noise to be scored as a CTC. To accomplish this and keep the conductivity cell constant (K) at a sufficient level to provide high signal-to-noise for the CTCs, the HTMSU was designed with a multilevel architecture (see Figure 1E). The 150 μm deep capture channels were tapered to a depth of ~80 μm at the conductivity sensor to match the Pt electrode diameter to ensure a sampling efficiency near 100%.
To verify the recovery and detection efficiency of the system, the seed number of CTCs in whole blood versus the number of cells enumerated over a broad physiologically relevant range (10–250 CTCs mL−1) was evaluated (see Figure 5C). The best fit line to these data indicated a slope of this calibration curve of 0.945 with an intercept near 0 (r2 = 0.9988), indicating near 100% recovery and detection of the CTCs seeded into these samples using the HTMSU, an indication of a very low false negative rate.
Conclusion
A novel polymer-based HTMSU was designed, fabricated, and tested for selecting, with high efficiency and specificity, CTCs directly from whole blood using surface-immobilized monoclonal antibodies targeted for membrane proteins unique to this class of cells. Due to the simple and low-cost production of these devices using microreplication technologies as well as their automated operation with no sample preprocessing required, the HTMSU can be envisioned as an important diagnostic tool for monitoring CTC levels in a variety of adenoma-based cancers for disease detection, cancer staging, or evaluating the effectiveness of therapeutic intervention. A compelling attribute of this microfluidic device is the ability to specifically detect the CTCs using an integrated conductivity sensor. The sensing platform did not respond to potential interferences, which could give rise to false-positive signals and was simple to implement both operationally (no cell staining as required for fluorescence) and instrumentally (no optical microscope or flow cytometer was required).
The HTMSU was designed to process large input volumes to search for rare events using molecular recognition via integral membrane proteins specific for the target. High throughput processing was realized by using a number of high-aspect ratio microchannels configured in parallel, providing the ability to process 1 mL of input in ~30 min. If the channel depth was increased to 250–an aspect ratio of 7.14 for 35 μm wide channels–and the number of parallel channels doubled, the sampling time for this same volume input could be reduced to 2.7 min. These changes could provide the ability to select extremely rare events requiring the need for processing larger input volumes (>10 mL).
The HTMSU is flexible in function as well with the ability to change the molecular recognition element to target other rare cells. For example, E. coli O157:H7 is a bacterial species typically found in a variety of fresh water bodies with EPA acceptable colony forming unit levels of <20 in wastewater and <200 in lakes and rivers per 100 mL. Using monoclonal antibodies specific for this bacterium and scaling the capture channel widths and conductivity sensor appropriate for these cell types, the reported device can be envisioned for field deployment to provide timely results for assisting in bacterial containment.
Supplemental Materials
Acknowledgments
We would like to thank the National Institutes of Health (National Cancer Institute, 1R33-CA09924601) for supporting this work. We also thank Dr. Robert Truax from the Louisiana State University School of Veterinary Medicine and Dr. Proyag Datta from the Louisiana State University Center for Advanced Micro-Devices for pathobiological services and replicating the microfluidic devices, respectively.
Footnotes
Supporting Information Available: Details of the HTMSU manufacturing process, conductivity sensor electronics, modeling of cell capture data, and calculation of adhesion strength. Figures S1–S6. Equations S1–S20. Tables S1–S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Leaf C. Fortune. 2004:76–94. [PubMed] [Google Scholar]
- 2.Paget S. Lancet. 1889;1:571. [Google Scholar]
- 3.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. Lancet Oncol. 2002;3:53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 4.Loberg RD, Fridman Y, Pienta BA, Keller ET, McCauley LK, Taichman RS, Pienta KJ. Neoplasia. 2004;6:302–309. doi: 10.1593/neo.03484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braun S, Marth C. New Engl J Med. 2004;351:824–6. doi: 10.1056/NEJMe048163. [DOI] [PubMed] [Google Scholar]
- 6.Singletary SE, Connolly JL. Ca–Cancer J Clin. 2006;56:37–47. doi: 10.3322/canjclin.56.1.37. [DOI] [PubMed] [Google Scholar]
- 7.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen L, Hayes DF. New Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 8.Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, Doyle GV, Matera J, Allard WJ, Miller MC, Fritsche HA, Hortobagyi GN, Terstappen L. J Clin Oncol. 2005;23:1420–1430. doi: 10.1200/JCO.2005.08.140. [DOI] [PubMed] [Google Scholar]
- 9.Cristofanilli M, Reuben JM, Fritsche HA, Hayes DF, Budd TG, Ellis MJ, Stopeck A, Miller CM, Matera J, Allard JW, Terstappen L. Breast Cancer Res Treat. 2004;88:S225–S225. [Google Scholar]
- 10.Molnar B, Ladanyi A, Tanko L, Sreter L, Tulassay Z. Clin Cancer Res. 2001;7:4080–4085. [PubMed] [Google Scholar]
- 11.Wood DP, Banerjee M. J Clin Oncol. 1997;15:3451–3457. doi: 10.1200/JCO.1997.15.12.3451. [DOI] [PubMed] [Google Scholar]
- 12.Uemura H. Hinyokika Kiyo. 1999;45:571–575. [PubMed] [Google Scholar]
- 13.Soria JC, Morat L, Durdux C, Housset M, Cortez A, Blaise R, Sabatier L. J Urol. 2002;167:352–356. [PubMed] [Google Scholar]
- 14.Kurusu Y, Yamashita Y, Ogawa M. Surgery. 1999;126:820–826. [PubMed] [Google Scholar]
- 15.Muller V, Pantel K. Breast Cancer Res Treat. 2004;6:258–61. doi: 10.1186/bcr942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pachmann K, Clement JH, Schneider CP, Willen B, Camara O, Pachmann U, Hoeffken K. Clin Chem Lab Med. 2005;43:617–627. doi: 10.1515/CCLM.2005.107. [DOI] [PubMed] [Google Scholar]
- 17.Pachmann K, Heiss P, Demel U, Tilz G. Clin Chem Lab Med. 2001;39:811–817. doi: 10.1515/CCLM.2001.134. [DOI] [PubMed] [Google Scholar]
- 18.Parareda A, Gallego S, Roma J, Llort A, Sabado C, Gros L, De Toledo Josep S. Oncol Rep. 2005;14:1021–7. [PubMed] [Google Scholar]
- 19.Chein RY, Yang YC, Lin YS. Electrophoresis. 2006;27:640–649. doi: 10.1002/elps.200500314. [DOI] [PubMed] [Google Scholar]
- 20.Furdui VI, Harrison DJ. Lab Chip. 2004;4:614–618. doi: 10.1039/b409366f. [DOI] [PubMed] [Google Scholar]
- 21.Grodzinski P, Yang J, Liu RH, Ward MD. Biomed Microdevices. 2003;5:303–310. [Google Scholar]
- 22.Balic M, Dandachi N, Hofmann G, Samonigg H, Loibner H, Obwaller A, van der Kooi A, Tibbe Arjan GJ, Doyle Gerald V, Terstappen Leon WMM, Bauernhofer T. Cytometry, Part B. 2005;68:25–30. doi: 10.1002/cyto.b.20065. [DOI] [PubMed] [Google Scholar]
- 23.Gross HJ, Verwer B, Houck D, Hoffman RA, Recktenwald D. Proc Natl Acad Sci USA. 1995;92:537–541. doi: 10.1073/pnas.92.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schutze K, Capron F, Franco D, Pazzagli M, Vekemans M, Lacour B, Brechot C, Paterlini-Brechot P. Am J Pathol. 2000;156:57–63. doi: 10.1016/S0002-9440(10)64706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Huang Y, Wang XB, Becker FF, Gascoyne PRC. Anal Chem. 1999;71:911–918. doi: 10.1021/ac981250p. [DOI] [PubMed] [Google Scholar]
- 26.Zabaglo L, Ormerod MG, Parton M, Ring A, Smith IE, Dowsett M. Cytometry, Part A. 2003;55A:102–108. doi: 10.1002/cyto.a.10071. [DOI] [PubMed] [Google Scholar]
- 27.Ghossein RA, Osman I, Bhattacharya S, Ferrara J, Fazzari M, CordonCardo C, Scher HI. Diagn Mol Pathol. 1999;8:59–65. doi: 10.1097/00019606-199906000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarley RL, Vaidya B, Wei S, Smith AF, Patel AB, Feng J, Murphy MC, Soper SA. J Am Chem Soc. 2005;127:842–843. doi: 10.1021/ja0454135. [DOI] [PubMed] [Google Scholar]
- 30.Alaiya AA, Franzén B, Auer G, Linder S. Electrophoresis. 2000;21:1210–1217. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1210::AID-ELPS1210>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 31.Crisan D, Ruark DS, Decker DA, Drevon AM, Dicarlo RG. Mol Diagn. 2000;5:33–8. doi: 10.1007/BF03262020. [DOI] [PubMed] [Google Scholar]
- 32.Simon B, Podolsky DK, Moldenhauer G, Isselbacher KJ, Gattonicelli S, Brand SJ. Proc Natl Acad Sci USA. 1990;87:2755–2759. doi: 10.1073/pnas.87.7.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szala S, Froehlich M, Scollon M, Kasai Y, Steplewski Z, Koprowski H, Linnenbach AJ. Proc Natl Acad Sci USA. 1990;87:3542–3546. doi: 10.1073/pnas.87.9.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao CG, Chianese D, Doyle GV, Miller MC, Russell T, Sanders RA, Terstappen LWMM. Int J Oncol. 2005;27:49–57. [PubMed] [Google Scholar]
- 35.Fung YC. Biomechanics: Mechanical Properties of Living Tissues. Springer-Verlag; New York: 1993. [Google Scholar]
- 36.Zheng JJ, Yeung ES. Anal Chem. 2002;74:4536–4547. doi: 10.1021/ac0257344. [DOI] [PubMed] [Google Scholar]
- 37.Ligrani PM, Niver RD. Phys Fluids. 1998;31:3605–3617. [Google Scholar]
- 38.Chang KC, Hammer DA. Biophys J. 1999;76:1280–1292. doi: 10.1016/S0006-3495(99)77291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willuda J, Honegger A, Waibel R, Schubiger PA, Stahel R, Zangemeister-Wittke U, Pluckthun A. Cancer Res. 1999;59:5758–5767. [PubMed] [Google Scholar]
- 40.Guthrie JW, Hamula CLA, Zhang HQ, Le XC. Methods. 2006;38:324–330. doi: 10.1016/j.ymeth.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Bell GI. Science (Washington, DC, United States) 1978;200:618–27. [Google Scholar]
- 42.Wei S, Soper SA, McCarley RL. Analyst. submitted for publication. [Google Scholar]
- 43.Feng J, Soper SA, McCarley RL, Murphy MC. Lasers in Surgery XIV Proceedings of the SPIE: San Jose, CA. 2004;5312:278–293. [Google Scholar]
- 44.Galloway M, Stryjewski W, Henry A, Ford SM, Llopis S, McCarley RL, Soper SA. Anal Chem. 2002;74:2407–2415. doi: 10.1021/ac011058e. [DOI] [PubMed] [Google Scholar]
- 45.Hakomori S. Immun Allergy Clin N Amer. 1990;10:781–802. [Google Scholar]
- 46.Sherr CJ. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.