

Abstract
The main objective of this study was to test the hypothesis that in vivo post-translational modifications in proteins, induced by the endogenously-generated reactive oxygen and nitrogen molecules, can alter protein function and thereby have an effect on metabolic pathways during the aging process. Succinyl-CoA:3-ketoacid coenzyme A transferase (SCOT), the mitochondrial enzyme involved in the breakdown of ketone bodies in the extrahepatic tissues, was identified in rat heart to undergo age-associated increase in a novel, nitro-hydroxy, addition to tryptophan 372, located in close proximity (∼10 Å) of the enzyme active site. Between 4 and 24 months of age, the molar content of nitration was more than doubled while specific enzyme activity increased significantly. The amount of SCOT protein, however, remained unchanged. In vitro treatment of heart mitochondrial soluble proteins with relatively low concentrations of peroxynitrite enhanced the nitration as well as specific activity of SCOT. Results of this study identify tryptophan to be a specific target of nitration in vivo, for the first time. We hypothesize that increases in tryptophan nitration of SCOT and catalytic activity constitute a plausible mechanism for the age-related metabolic shift towards enhanced ketone body consumption as an alternative source of energy supply in the heart.
The nature of the mechanisms causing age-associated alterations in various physiological functions is presently unclear. One hypothesis postulates that the progressive accumulation of macromolecular damage, induced by free radicals, is a primary causal factor underlying senescent changes (1, 2). Protein modifications are thought to be particularly relevant in the aging process because structurally altered proteins may lose function, undergo preferential degradation and/or elicit an immune response (3-6).
One specific type of post-translational protein modification involves the addition of a nitro group to the carbon of the aromatic ring of tyrosine by nitric oxide-derived reactive intermediates (7-10). In vitro exposure of proteins to the nitrating agent, peroxynitrite, has often been shown to cause an addition of a nitro group to tyrosine and to a lesser extent to tryptophan residues (11-13). Indeed, accumulation of tyrosine nitrated proteins has been observed in association with several pathological conditions (14, 15) as well as during the normal aging process (16-18). In contrast, in vivo nitration of tryptophan residues has not been reported thus far.
Notwithstanding, the main issue relevant to the elucidation of the relationship between protein nitration and the underlying causes of senescence is: what are the functional consequences of protein nitration? Although several different proteins have been reported to undergo nitration, with few exceptions (19, 20), the in vivo functional consequences of nitration remain to be demonstrated. Such information is deemed to be critical, because a post-translational structural modification in a protein is not an a priori indication of an alteration in activity (21). For instance, in vitro protein nitration has been found either to be associated with decreases (22-24), increases (25, 26) or no change in catalytic activity (27).
In this context, studies were conducted on soluble proteins from rat heart mitochondria at 4, 13, 19, and 24 months of age in order to identify the nitrated proteins and to determine whether putative nitration affected protein function. The rationale for selecting mitochondrial proteins was that they are thought to be particularly susceptible to nitration due to the presence of a putative mitochondrial nitric oxide synthase isoform (28, 29). Nitric oxide has also been shown to depress mitochondrial respiratory activity (30). Here, we report the novel finding that tryptophan 372, located in close vicinity of the enzyme active site of succinyl-CoA:3-ketoacid coenzyme A transferase (SCOT), is a specific in vivo target of nitration/oxidation, and that the amount of SCOT nitration and activity increase with age. SCOT catalyzes the conversion of the main ketone body, acetoacetate into acetoacetyl-CoA, which is subsequently metabolized via citric acid cycle for energy production. Ketone bodies (acetoacetate and 3-hydroxybutyrate) are primarily produced in the liver for utilization in the extrahepatic tissues, with heart and skeletal muscle being main users (31). Production of ketone bodies in the liver apparently doubles between 3 and 30 months of age in mice (32). Thus elevation in SCOT nitration and activity reported in this study are hypothesized to be reflective of an adaptive metabolic shift towards increased ketone body production and utilization during the aging process.
MATERIALS AND METHODS
Reagents
Unless stated otherwise, all chemicals were obtained from Sigma Chemical Co. (St. Louis, MO). Sources of other materials were: anti-nitrotyrosine mouse IgG2bk monoclonal antibody (clone 1A6) and peroxynitrite, Upstate Biotechnology (Lake Placid, NY); peroxidase conjugated goat anti-mouse and anti-rabbit IgG (H+L), Pierce (Rockford, IL); rabbit polyclonal antibody, produced against synthetic peptide KGPRFEKRIERLTTRDSP coupled to KLH protein, BioSource International (Camarillo, CA); percoll and chromatofocussing reagents, Amersham Corp.; sequencing grade modified trypsin, Promega (Madison, WI); complete protease inhibitor cocktail, Boehringer; broad range of prestained molecular weight markers (myosin, β-galactosidase, BSA, ovalbumin, carbonic anhydrase, soybean trypsin inhibitor, lysozyme and aprotinin, with polypeptide molecular masses of 209, 124, 80, 49.1, 34.8, 28.9, 20.6 and 7.1 kDa, respectively), Bio-Rad; 5-nitro-tryptophan, WAKO Pure Chemical Industries (Richmond, VA); 4-nitro-tryptophan, Dr. Russell King (Potato Reseach Center, Canada). The IgG fraction from rabbit immune serum was purified by ammonium sulfate precipitation and ion-exchange chromatography (33).
Animals
Male Fisher rats of different ages were obtained from the National Institute on Aging (Bethesda, MD) and fed ad libitum (AL). For specified purposes, hearts and kidneys from 100 young rats (2-month-old) were obtained from Pel-Freez Biologicals (Rogers, AK), and shipped overnight in ice-cold antioxidant buffer (50 mM potassium phosphate buffer, pH 7.4, 2 mM EDTA and 0.1 mM butylated hydroxytoluene) and processed immediately upon arrival. Neither the storage of the heart tissue in the antioxidant buffer at 4°C for up to 48 h nor the presence or absence of butylated hydroxytoluene had a discernible effect on the level of SCOT nitration.
Isolation of mitochondria and preparation of soluble proteins
In each experiment, tissues (liver, kidney, brain, heart, testis, lung, spleen, hind limb skeletal muscle) pooled from two rats were placed in ice-cold antioxidant buffer and homogenized in 10 vol (w/v) of the indicated tissue-specific isolation buffer : (i) liver - 0.25 M sucrose, 3 mM EDTA, 10 mM Tris, pH 7.4; (ii) kidney - 0.22 M mannitol, 70 mM sucrose, 10 mM EGTA, 2 mM Hepes, pH 7.4; (iii) heart - 0.3 M sucrose, 0.03 M nicotinamide, 20 mM EDTA, pH 7.4; (iv) skeletal muscle - buffer 1, containing 0.12 M KCl, 2 mM MgCl2, 1 mM EGTA, 0.5 mg/ml BSA, 20 mM Hepes, pH 7.4, and buffer 2, containing 0.3 M sucrose, 0.1 mM EGTA, 2 mM Hepes, pH 7.4; and (v) brain, testis, lung and spleen - 0.32 M sucrose, 1 mM EDTA, 10 mM Tris, pH 7.4. Each homogenization buffer was supplemented with freshly prepared 0.2 mM phenylmethylsulfonyl fluoride (PMSF) and complete protease inhibitor cocktail, at the concentration suggested by the manufacturer (Boehringer). Low and high speed differential centrifugations for mitochondrial isolation were, respectively, 1000 g for 10 min and 17,500 g for 10 min for liver; 600 g for 10 min and 8,500 g for 10 min for kidney; 700 g for 10 min and 10,000 g for 10 min for heart; 1300 g for 3 min and 21,200 g for 10 min for brain, testis, lung and spleen; 600 g for 12 min and 17,000 g for 12 min in buffer 1, and 1200 g for 12 min and 12,000 g for 12 min in buffer 2 for skeletal muscle. Mitochondria were isolated from all tissues within 1 to 2 h after tissue dissection, except the brain, which required a longer isolation time (up to 3 h) due to Percoll gradient centrifugation (34). Mitochondria from testis, lung and spleen were isolated by a procedure similar to that for the brain, except for the omission of Percoll gradient centrifugation. Mitochondrial pellets were resuspended in appropriate volumes of the respective tissue homogenization buffers, to achieve a concentration of 5-10 mg/ml protein, and stored in small aliquots at -80°C. Soluble mitochondrial proteins, located in the matrix and intermembrane space compartments, were obtained as follows: mitochondria (∼5 mg/ml protein) were placed on ice, sonicated for 30 sec and centrifuged at 100,000 g for 60 min to separate the soluble and the membraneous proteins. Pellets were resuspended in mitochondrial isolation buffer, followed by sonication and ultracentrifugation, as described above. Supernatants from both ultracentrifugation steps were combined and stored in aliquots at -80°C.
SDS Gel Electrophoresis and Western blot analysis
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out according to the method of Laemmli (35), with modifications suggested by Shagger and Jagow (36). Proteins were denatured by boiling for 5 min in sample buffer (60 mM Tris, containing 2% (w/v) SDS, 100 mM dithiothreitol, 10% (v/v) glycerin, and 0.1% (w/v) bromphenol blue) and subjected to electrophoresis on 1.0-mm gels, consisting of 4% stacking and 10% separating gels, using a Bio-Rad Miniprotean III gel apparatus. The electrophoretic separation was carried out at a constant current of 30 V for the stacking and 100 V for separating gels. Proteins were transferred to PVDF membrane (Bio-Rad) at 4°C in a buffer containing 16 mM Tris base, 120 mM glycine and 10% (v/v) methanol, at a constant current of 120 V for 60 min. For immunodetection, PVDF membranes were incubated with blocking solution containing 5% dry milk and 0.1% Tween 20 in TBS (10 mM Tris/HCl buffer, pH 7.5, and 150 mM NaCl) for 45 min at 37°C. Membranes were then washed 3 times, 5 min each, in TBS and incubated at 37°C with anti-nitrotyrosine or anti-SCOT antibodies diluted 1:2000 and 1:1000, respectively, for 1 h. After rinsing three times, 10 min each, with TBS, PVDF membranes were incubated with secondary antibody (goat anti-mouse at a dilution of 1:20,000 or anti-rabbit at a dilution of 1:100,000 IgG, coupled to horseradish peroxidase (Pierce)), for 1 h. After washing, the membranes were developed using the chemoluminescence detection kit (ECL-Plus) from Amersham Corp. Images of the immunoblots were digitized by using a flatbed scanner (Epson 2450). Analysis of densitometric data was performed using the public domain NIH Image program (developed at the US National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image). Contents of SCOT protein and nitration in different samples of mitochondrial matrix were calculated by comparisons with standard curves fitting increasing amounts of purified SCOT (10 to 40 ng) and mitochondrial matrix extracts from 4-month-old rat (2.5 to 15 μg). The relation between amounts of protein and band densities was found to be exponential, rather than linear, within a limited dynamic range of amounts of protein (indicated above) and was also dependent on the duration of film exposure during the chemoluminescence reaction.
Identification of nitrated protein
3NT-immunoreactive protein, shown in Fig. 1B, was purified as follows: mitochondrial soluble proteins (100 mg) isolated from heart tissue of 100 young rats were dialyzed against 25 mM Imidazol buffer containing 0.2 mM PMSF, pH 7.85, for 2 h, and then applied onto a chromatofocussing column (6×100 mm), equilibrated with the same buffer, and eluted with 100 ml of Polybuffer 74 (dilution 1:10), pH 3.9. The flow rate was 6 ml/h and fractions of 2 ml each were collected. pH was measured in every 5th fraction. Fractions containing the majority of the 58 kDa 3NT-immunopositive protein were concentrated to 200 μl, using Centricon 30 concentrators. Gel filtration was performed using a Shimadzu Class VP HPLC system and BioSep-SEC-S 3000 gel permeation column (5μm, 7.5×300 mm) obtained from Phenomenex (Torrance, CA) at room temperature. The column was equilibrated with 25 mM Tris buffer, pH 7.4, containing 75 mM NaCl, at a flow rate of 0.5 ml/min. Concentrated sample (50 μl) containing nitrotyrosine positive protein from the chromatofocussing step was then injected onto the column, and absorbance (200-600 nm) was monitored with a diode-array UV detector. Fractions of the eluate from the gel filtration column (0.5 ml) were collected between 12 and 20 min, and proteins were subjected to SDS-PAGE and analyzed by immunoblotting. This purification procedure yielded ∼200 μg of electrophoretically pure (>85%) SCOT protein.
FIGURE 1.

Western blot analysis of nitrated proteins in the soluble fraction of mitochondria from various tissues of the rat. Aliquots, containing 10 μg per lane of the supernatant of sonicated mitochondria, centrifuged at 100,000 g (soluble mitochondrial proteins), were separated by SDS-PAGE. A Coomassie-stained gel is shown in panel A. Panel B shows the distribution of mitochondrial enzymes enoyl-CoA hydratase (32 kDa) and lipoamide dehydrogenase (59 kDa) detected by Western blot using the respective polyclonal antibodies. Immunoblot analysis was performed with anti-nitrotyrosine monoclonal antibody (C) or with polyclonal anti-SCOT antibody (D). The arrow on the right indicates the position of the 58 kDa protein. M refers to the mixture of proteins, containing BSA (67 kDa) and prestained molecular weight markers, myosin, β-galactosidase, BSA, ovalbumin, carbonic anhydrase, soybean trypsin inhibitor, lysozyme and aprotinin, with polypeptide molecular masses of 210, 120, 80, 49, 35, 29, 20 and 7 kDa, respectively. Lane contents were as follows: 1, 3NT-BSA, 2-9, samples from heart, kidney, brain, skeletal muscle, lung, spleen, testis and liver of 4-month-old rat, respectively.
N-terminal sequencing
Purified SCOT (10 μg), separated by SDS-PAGE, was electroblotted onto PVDF membrane and subjected to N-terminal Edman degradation at the Microchemical Core Facility Laboratory of the University of Southern California. Sequence was ascertained by using the BLAST network service at the National Center for Biotechnology Information (NCBI). Numbering of amino acid residues in the text corresponds to the amino acid sequence of mature rat SCOT (NCBI accession number NP_001012221).
Mass spectrometry
Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were acquired on AXIMA CFR (Shimadzu, MD) operated in the positive ion linear mode, using α-cyano-4-hydroxycinnamic acid (10 mg/ml in 70% acetonitrile). Spectra represent the average of 100 laser shots. External mass calibration was provided by the [M+H]+ ions of methionine, tryptophan, 5-hydroxytryptophan and 5-nitrotryptophan. One milligram of dried sample of amino acids was dissolved in 100 of 0.1% trifluoroacetic acid (TFA). 1 μl of sample solution was mixed with 1 μl of matrix solution and deposited on the stainless-steel sample holder and air-dried. Three independent MALDI measurements were made for each sample to evaluate the reproducibility. In-solution and in-gel tryptic digestion of purified full length, C-terminal and N-terminal fragments of SCOT, analysis of tryptic peptide sequence tags by tandem mass spectrometry, and protein identification were performed as described in detail previously (21). Briefly, proteins from solution and/or from Coomassie stained gel pieces were reductively alkylated and digested with sequencing grade trypsin (Promega) overnight at 37°C. Tryptic digest products were extracted, dried and resuspended in 10 μl of 60% (w/v) acetic acid. Chromatographic separation of the tryptic peptides was achieved using a ThermoFinnigan Surveyor MS-pump in conjunction with a BioBasic-18 reverse phase capillary column (100×0.18 mm, ThermoFinnigan). Mass analysis was performed using a ThermoFinnigan LCQ Deca XP Plus ion trap mass spectrometer equipped with a nanospray ion source employing a 4.5 cm needle, using data-dependent acquisition mode. Protein identification was carried out with the MS/MS search software Mascot (Matrix Science), with confirmatory or complementary analyses by TurboSequest (Bioworks Browser 3.2, build 41, from ThermoFinnigan and Sonar MS/MS from Genomics Solutions). Alkylated modification (carbamidomethyl) was designated as fixed; in contrast, oxidation of methionine, histidine, tryptophan and/or nitration of tyrosine, tryptophan, were variable modifications in the Mascot search. NCBI rat genome database server complemented with the NCBI nonredundant protein database was used for the search.
SCOT Activity Assay
Enzyme activity was determined spectrophotometrically by monitoring acetoacetyl-CoA formation (37). The assay mixture typically consisted of 50 mM Tris/HCl, 10 mM MgCl2, 5 mM iodoacetamide, 0.1 mM succinyl-CoA, 50 mM acetoacetate, pH 8.5. Mitochondrial soluble proteins (30 μg) were added to the 1 ml cuvette and, after initiation of the reaction by acetoacetate, the change in absorbance was followed at 313 nm for 1-2 min using a Beckman DU-640 spectrophotometer. A linear relationship between SCOT activity and the amount of soluble protein from heart (10 to 50 μg protein range) mitochondria was established in preliminary experiments. Enzyme assays were carried out in duplicate and the average absorbance was used for calculations. Specific activity of SCOT was expressed as mol/min/mol enzyme (min-1). The molar content of SCOT in samples of the soluble fractions of rat heart mitochondria were estimated in Western blots using anti-SCOT antibody and purified SCOT as the standard, and expressed as pmol SCOT per mg mitochondrial soluble protein.
In vitro nitration of BSA
Fatty acid-free bovine serum albumin (BSA) (Sigma) was nitrated with peroxynitrite for use as a positive control for the Western blot analysis. Due to instability of BSA in the presence of degraded peroxynitrite and the occurrence of crosslinking reactions, nitrated protein was further purified by high performance gel permeation chromatography. Briefly, a 25 mg/ml solution of BSA was incubated with 1, 0.1 and 0.01 mM peroxynitrite for 15 min on ice in 25 mM sodium phosphate buffer, pH 7.4, containing 75 mM NaCl. Nitrated BSA (50 μl, 1.25 mg protein) was injected onto the gel filtration column (BioSep-SEC-S 3000), and eluted with buffer (25 mM sodium phosphate, pH 7.4, 75 mM NaCl) at a flow rate of 0.5 ml/min. The bulk of a peak with a retention time of 17 min, containing the nitrated BSA monomer, was collected, stored in small aliquots at -80°C, and used in SDS-PAGE and Western blot analysis. A negative control of BSA (non-nitrated) was prepared by a similar procedure in the absence of the peroxynitrite. Using amino acid analysis by HPLC/ECD, the molar content of 3NT in the nitrated BSA controls was determined to be 100 ± 12, 3.5 ± 0.2 and 0.115 ± 0.02 mmol/mol BSA (nitration with 1, 0.1 and 0.01 mM peroxynitrite, respectively), as described below.
Analysis of nitrated amino acids
Measurements of 3NT and nitro-hydroxytryptophan in nitrated BSA standards and purified full length SCOT, C-terminal and N-terminal fragments of SCOT were carried out by amino acid analysis of proteins in solution and/or on PVDF membranes after enzymatic hydrolysis, similar as described (38). Briefly, 10 to 100 μg of protein in solution was precipitated with 5% (w/v) meta-phosphoric acid, protein pellets were washed once with ice cold acetonitrile and dissolved in 100 μl of 0.1 M sodium acetate buffer, pH 4.7. Alternatively, proteins were separated by SDS-PAGE, electroblotted onto PVDF membrane, and Coomassie stained bands containing 1-10 μg of protein were cut and placed in 0.5 ml plastic tubes containing 100 μl of 0.1 M sodium acetate buffer, pH 4.7. Enzymatic hydrolysis of proteins was carried out in the presence of 5% (w/w) pronase from Staphylococcus griseus (Boehringer) at 50°C overnight. Hydrolysis was quenched by addition of 100 μl 10% (w/v) meta-phosphoric acid, samples were centrifuged at 18,000 g for 20 min, and supernatants were transferred to autosample micro vials for injection on HPLC. Amino acids (tyrosine, 3NT, tryptophan, 5-nitro-tryptophan, 4-nitrotryptophan, 5hydroxy-tryptophan, kynurenine) were separated by HPLC, fitted with a Shimadzu Class VP solvent delivery system using a reverse phase C18 Gemini column (4.6×150 mm, 5μm, Phenomenex). The mobile phase for isocratic elution consisted of 25 mM monobasic sodium phosphate, 12.5% methanol, pH 2.7, adjusted with 85% phosphoric acid. The flow rate was 1 ml/min. Under these conditions, the separation was completed in 30 min; 5-nitro-tryptophan was the last eluted peak, with a retention time of approximately 27 min. For the analysis of tyrosine and 3NT alone, the methanol was omitted from the solvent, which resulted in elution of 3NT as the last peak, with a retention time of approximately 25 min. Calibration standards of amino acids were prepared in 5% meta-phosphoric acid. Amino acids were detected with a model 5600 CoulArray electrochemical detector (ESA, Chelmsford, MA), equipped with a four-channel analytical cell, using potentials of +600, +700, +800 and +900 mV. The limit of electrochemical detection was 300 fmol (3-nitrotyrosine, 4- and 5-nitrotryptophan) and 200 fmol (tyrosine, tryptophan, 5-hydroxytryptophan and kynurenine), with the signal to noise ratio of 4:1. Each sample was injected twice, and the average of the peak areas was used for quantification. For the purpose of the UV spectrum analysis of eluting peaks, the SPD-M10AVP Diode-Array detector (Shimadzu) was connected upstream of the electrochemical detector, and UV spectra (200 to 800 nm wavelength range) of eluting peaks were acquired every 5 seconds during HPLC separations.
Synthesis of nitrated 5-hydroxy-tryptophan
5-Hydroxynitrotryptophan was produced by the reaction between 5-hydroxytryptophan and tetranitromethane. The procedure was carried out on ice in the dark (amber glass vial): 10mg solid 5-hydroxytryptophan was dissolved in 50μl of tetranintromethane and the solution was overlaid with 450 μl of 70% acetonitrile. The reaction components were gently stirred. It should be noted that the solution in the vial separated into two phases due to the insolubility of tetranintromethane (bottom) in acetonitrile (top). With time the dark red tetranitromethane phase migrated into the upper acetonitrile-containing phase. Progress of the reaction was monitored by examination 50 μl aliquot (100-fold dilution of upper phase of reaction mixture in 5% meta-phosphoric acid) by HPLC, as described above. The highest yield (∼70%) of 5-hydroxynitrotryptophan was obtained after 6 h of reaction. Further incubation resulted in the appearance of additional minor peaks with retention times of more than 30 min, probably due to the occurrence of multiple nitration reactions on the aromatic ring of tryptophan. In contrast, if the reaction was carried out at pH 8.0, using 50mM phosphate buffer, the formation of 5-hydroxy-nitrotryptophan was accompanied by relatively high amounts of multiple nitration by-products, and diminished stability of the target compound (rapid decay of 5-hydroxynitrotryptophan was detected within several minutes in phosphate buffer in the pH range from 7.0 to 8.0). Therefore, the absence of the buffer was deemed to be desirable for obtaining high yield and purity as well as prolonged stability (no significant decomposition of 5-hydroxynitrotryptophan was observed after storage at 4°C for up to 5 days if the reaction was acidified with 0.1% trifluoroacetic and/or 5% meta-phosphoric acid).
Analysis of data
Statistical analysis of data was performed using paired and unpaired t-test analysis, including Bonferroni correction (P < 0.001 was considered to be significant). The results are means ± SD.
RESULTS
Identification of SCOT as a specific target of nitration in the soluble fraction of mitochondria from various tissues
In an initial experiment, the supernatant fractions, resulting from 100,000 g centrifugation of sonicated mitochondria from heart, kidney, brain, upper hind limb skeletal muscles, lung, spleen, testis and liver of 4-month-old rats, were separated by SDS-PAGE and the gels were stained with Coomassie Blue or probed with anti-3NT antibody. Except in testis and liver, which showed no positive reaction for 3NT, all the other tissues showed only a single major immunopositive band, with a molecular weight of 58 kDa (Figure 1). The immunodensity of the 58 kDa band varied up to ∼10-fold in various tissues, with the rank order: heart > kidney > skeletal muscle > brain > lung > spleen. A comparison between 4- and 24-month-old rats indicated that the intensity of the 3NT-positive band increased with age in the heart and brain, remained unchanged in the skeletal muscle, and decreased in the kidney, lung and spleen (data not shown). To evaluate the reliability of the isolation procedure and the purity of mitochondrial preparations, we performed Western blots with polyclonal antibodies against two mitochondrial enzymes involved in oxidative decarboxylation and β-oxidation of fatty acids, namely lipoamide dehydrogenase (a component of pyruvate dehydrogenase complex) and enoyl-CoA hydratase, respectively. Results indicated the almost uniform presence of immunoreactive bands in our mitochondrial preparations (Figure 1B). The variations noted in the intensity of these two bands were deemed to be due to differences in the expression and/or the degree of purity of mitochondria. The specificity of the anti-3NT antibody was established by employing a series of controls (Figure 2A): e.g., experimentally nitrated BSA showed a positive immunoreaction, while non-nitrated BSA did not. Immunostaining of Western blots did not occur under the following conditions: i) in the absence of the primary antibody, ii) after reduction by dithionite, and iii) after pre-incubation of the primary antibody with free 3-NT. In contrast, pre-incubation of the primary antibody with free 5-nitro-tryptophan and/or 5-hydroxytryptophan did not abolish immunoreaction. In addition, using BSA controls, containing 100, 3.5 and 0.115 mmol 3NT per mol of protein, we determined the quantitative limit for the detectability of nitrated proteins in Western blot by 3NT antibody (Figure 2B). Nitrated BSA control was undetectable if the nitrotyrosine content was less than 115 μmol/mol protein. The effect of the reducing agent DTT, present in sample buffer, on SCOT nitration during SDS-PAGE analysis was investigated in order to evaluate potential loss of nitration due to reduction of nitro to amino group. The results showed that the absence or presence of DTT at concentrations of 1, 10, 100 mM in sample buffer had no effect in the levels of SCOT nitration during immunoblot analysis and no additional immunoreactive bands were detectable (see supporting information, Figure 2).
FIGURE 2.

Western blot experiments testing the specificity of the immunoreaction of the 3NT monoclonal antibody (Panel A). BSA (0.5 μg, lane 1), nitrotyrosine-containing BSA (0.5 μg, lane 2) and soluble proteins from heart mitochondria (5 μg, lane 3) were separated by SDS-PAGE and transferred to a PVDF membrane. Immunoblot analysis was carried out under the following conditions: in the presence (+) or absence (-) of 3NT monoclonal antibody; in the presence of free 3-nitrotyrosine; after reduction of the nitrotyrosine to aminotyrosine by dithionite; and in the presence of free 5-nitrotryptophan and/or 5-hydroxytryptophan. Panel B shows the determination of the amounts needed for the detection of nitrotyrosine-containing BSA in immunoblot (left), and in HPLC analysis using electrochemical detection (right).
BSA samples (0.5 μg and 1 μg per lane), containing indicated amounts of 3NT were separated by SDS-PAGE, transferred to a PVDF membrane and probed with 3NT-antibody.
The traces of HPLC chromatograms after enzymatic hydrolysis of 100 μg BSA, corresponding to indicated contents of 3NT, show the presence of the peak for 3NT at retention time of 25 min. Methanol was omitted from the solvent (see Materials and Methods section for details). Intensities of the signal from the electrochemical detector are indicated in vertical bars.
For identification, the immunopositive 58 kDa protein was purified from the soluble fraction of heart mitochondria by a combination of chromatofocussing (separation according to the isoelectric point) and high-performance gel filtration (Figure 3). The isolated 58 kDa nitrated protein exhibited an identical isoelectric point of 6.9-7.1, and a native molecular weight of 120 kDa (due to dimerization). The N-terminal sequence, VKFYTDPVKAVEGI corresponded to the first 14 amino acid residues of mature rat SCOT protein.
FIGURE 3.

Purification and identification of the 58 kDa nitrated protein from heart mitochondria as succinyl-CoA:3-ketoacid coenzyme A transferase (SCOT).
Soluble proteins from heart mitochondria (100 mg) were separated by chromatofocussing, as described in the Materials and Methods section. Panel A shows a protein elution chromatogram at 280 nm (solid line) and the corresponding pH gradient (dotted line). Proteins were separated by SDS-PAGE and the gels were stained with Coomassie Blue or immunostained with anti-3NT antibody (B). M refers to prestained molecular weight markers, as described in the legend of Fig. 1. Lane contents were as follows: C. 3NT-BSA (0.5 μg); S. soluble proteins isolated from heart mitochondria (10 μg); lanes 5 - 61 correspond to aliquots of fractions from chromatofocussing column eluate (25 μl). Panel C shows analysis of SCOT at various stages of purification. Gels were stained with Coomassie Blue or immunostained with anti-3NT or anti-SCOT antibody. Lane contents: M, prestained molecular weight markers (Fig. 1); 1, BSA (0.5 μg); 2, 3NT-BSA (0.5 μg); 3, whole heart mitochondria (5 μg); 4, soluble proteins isolated from heart mitochondria (5 μg); 5, chromatofocussing eluate (2.5 μg); and 6, gel filtration eluate (1.5 μg).
Further confirmation, that this protein was indeed SCOT, was provided by its positive reaction with a polyclonal antibody produced against the synthetic peptide KGPRFEKRIERLTTRDSP (the most exposed region of SCOT, as determined by hydrophobicity plot) coupled to KLH protein (Figure 1D). The rank order of the intensity of the immunoreaction of the band with this antibody was essentially identical to that observed with the monoclonal anti-3NT antibody. The ratio of the optical density of 3NT immunostaining to that of SCOT protein was essentially constant in all the tissues with detectable bands, averaging 0.74 ± 0.26, n=6 (in 4-month-old rat), thereby indicating that the degree of SCOT nitration was essentially similar in different tissues.
MS/MS analysis of SCOT preparations resulted in a positive match of 8 tryptic peptides to SCOT database sequences, which corresponded to 32% sequence coverage (Table I.). SCOT co-purified with several other mitochondrial proteins, which were identified as aconitase, creatine kinase, and hydroxyacyl-CoA dehydrogenase. These proteins were co-purified in the chromatofocussing step during the SCOT isolation procedure: aconitase (86 kDa), creatine kinase (43 kDa) and SCOT (58 kDa) were eluted from the column in fractions 5-15, 11-15 and 13-25, respectively (see Figure 3). None of the tryptic peptides, matched for indicated proteins, contained any detectable 3NT in the MS/MS analysis. Surprisingly, in contrast to previous reports indicating that aconitase and creatine kinase, among other mitochondrial proteins, are also nitrated in rat heart (17), our results indicated the lack of nitration in immunoblots as well as the absence of nitrotyrosine containing peptides in MS/MS analysis. However, such lack of nitration can be due to relatively low amounts of the 3NT-containing peptides and/or incomplete protein sequence coverage in MS/MS analysis.
Table 1.
Results of mass spectrometric analysis of proteins identified in enriched SCOT isolates from heart and kidney mitochondria
| Protein | NCBI accession number | Molecular Mass (kDa) | Peptides matched§ | Sequence coverage (%) |
|---|---|---|---|---|
| 3-oxoacid CoA transferase (SCOT)* | NP_001012221.1 | 57.5 | 8 (147) | 32 |
| Aconitase 2 | NP_077374.2 | 86.1 | 12 (21) | 19 |
| L-3-hydroxyacyl-Coenzyme A dehydrogenase | NP_476534.1 | 34.5 | 4 (6) | 16 |
| Protein expressed in non-metastatic cells 2 | NP_114021.2 | 17.4 | 3 (3) | 22 |
| Creatine kinase | NP_036662.1 | 43.2 | 1 (1) | 3 |
| Hemoglobin alpha 1 chain | NP_037228.1 | 15.5 | 1 (1) | 8 |
Numbers of unique peptides found, and their abundance (in parenthesis).
Data based on combined results of MS/MS analysis of “in-solution” and “in-gel” tryptic digests of SCOT.
Identification of the site of nitration
Mature rat SCOT protein contains 11 tyrosine and 3 tryptophan residues (NCBI accession number NP_001012221). Localization of the nitrated residue was facilitated by a known feature of the SCOT catalytic mechanism, namely the formation of a covalent adduct between the active site glutamyl residue (Glu 303) and acetoacetyl-CoA, with the subsequent formation of an oxyproline intermediate and cleavage of the α-peptide bond (39). The SCOT protein was thus cleaved at the Glu 303 residue into N-terminal and C-terminal fragments in the presence, but not in the absence, of acetoacetyl-CoA (Figure 4). Briefly, purified SCOT was incubated in the presence or absence of 1 mM acetoacetylCoA for 5 min at room temperature, heated at 70°C for 60 min and then subjected to SDS-PAGE. Proteins were electrotransferred to PVDF membrane, and the membrane was probed with anti 3NT monoclonal and anti-SCOT polyclonal antibodies, or the gel was stained with Coomassie Blue (Figure 4A), which revealed the presence of two fragments with molecular weights of 37 and 21 kDa. The 37 kDa N-terminal fragment showed a positive immunoreaction with anti-SCOT antibody (which recognizes the amino terminal sequence of SCOT), but not with the anti-3NT antibody. In contrast, the 21 kDa C-terminal fragment reacted positively only with the anti-3NT antibody.
FIGURE 4.

Identification of the site of nitration in SCOT.
Panel A: M refers to prestained molecular weight markers, as described in the legend to Fig. 1; 0.5 μg 3NT-BSA (lane 1) was the positive control. Purified SCOT (10 μg) from heart mitochondria (lane 2) was incubated in the absence (lane 3) or presence (lane 4) of 1 mM acetoacetyl-CoA at room temperature for 5 min and heated at 70°C for 60 min, following which the samples were subjected to SDS-PAGE. Gels were stained with Coomassie Blue or electrotransferred to a PVDF membrane and probed with anti-3-nitrotyrosine antibody or anti-SCOT antibody. Panel B presents a schematic representation of the mature SCOT protein, containing 11 tyrosine and 3 tryptophan residues (indicated by Y and W, respectively). Substrate-induced auto-cleavage of SCOT occurs at the active site glutamate residue (G303) and produces a 37 kDa N-terminal fragment and a 21 kDa C-terminal fragment. The positions of the fragments are indicated in panel A by the arrows. The shaded box in panel B indicates the location of the KGPRFEKRIERLTTRDSP peptide, which was recognized by the anti-SCOT polyclonal antibody. The N-terminal fragment of SCOT was recognized by the anti-SCOT antibody and not by the 3NT antibody. The C-terminal fragment was recognized by the anti-3NT antibody.
Amino acid analysis of the electroblotted SCOT bands (up to 100 μg pure protein) revealed the presence of an unknown peak (designated as X) with the retention time of 21 min in both the full length and C-terminal fragment, but not in the N-terminal fragment of SCOT protein (Figure 5). In contrast, no detectable peaks (detection limit 300 fmol on column) were observed at 7 min (retention time of 3NT) or 25 and 27 min (retention time of 4-nitrotryptophan and 5-nitro-tryptophan, respectively). Furthermore, no additional peaks were detected (detection limit 200 fmol) i) at retention times corresponding to hydroxy- and/or dihydroxytryptophan (5 and 8 min, respectively), and ii) if the enzyme was purified from the heart of 20-month-old rat. The unknown amino acid, present in peak X, displayed a UV spectrum similar to 3NT and 5-nitro-tryptophan, a broad absorption peak maximum at 360 nm, which is characteristic of nitrated aromatic products (6, 7). Mass spectrometric analysis (MALDI-TOF) of the fraction, collected during the HPLC separation between 20 and 22 min (Figure 5, trace c), indicated the presence of several peaks with a mass/charge value of 266.34, 250.3, 228 and 212 (Figure 6). The protonated molecules of hydroxynitrotryptophan are responsible for ions at m/z 266.34, which was consistent with a +61 modification of tryptophan, corresponding to nitro-(46-1=45 mass) and hydroxy-(17-1=16 mass) additions. Photodecomposition of the protonated molecular ion of nitroxydroxytryptophan corresponded to ions that have lost single oxygen atom [MH-O]+ (peak at m/z 250.3). Such characteristic photodecomposition reaction of aromatic amino acids and peptides, containing nitrobenzyl moieties, under laser light during MALDI analysis, is well established (40) and is often viewed as a “fingerprint” of the protein nitration (41). The low intensity of the ion peak corresponding to hydroxynitrotryptophan (m/z=266.34) is originated from the concentration effect, which has been shown in MALDI-TOF analysis of picomolar amounts of nytrobenzyl compounds, indicating on the prevalence of the photodecomposition ions due to the loss of oxygen (40, 41). Using picomolar amounts of the standard (5-nitrotryptophan) in MALDI-TOF analysis, we observed a similar shift of peak intensity in spectra: higher intensity of ion peaks corresponding to the photodecomposition product of 5-nitrotryptophan ([MH-O]+ ions with m/z 234.7) compared to 5-nitrotryptophan ([MH]+ ions with m/z 250.7) (data not shown). The conversion of nitro-hydroxy adduct of tryptophan to its amino-analog, by reduction with dithionite, was confirmed by the presence of ions at m/z 236.38, corresponding to hydroxyaminotryptophan, concomitant with the disappearance of the peaks with m/z 266.34 and 250.3, corresponding to hydroxynitrotryptophan (Figure 6B ). The low intensity of the ion peak corresponding to hydroxyaminotryptophan (m/z=236.38) is due to the excess of dithionite, most intense peak (m/z 173.92), used for the reduction reaction.
FIGURE 5.
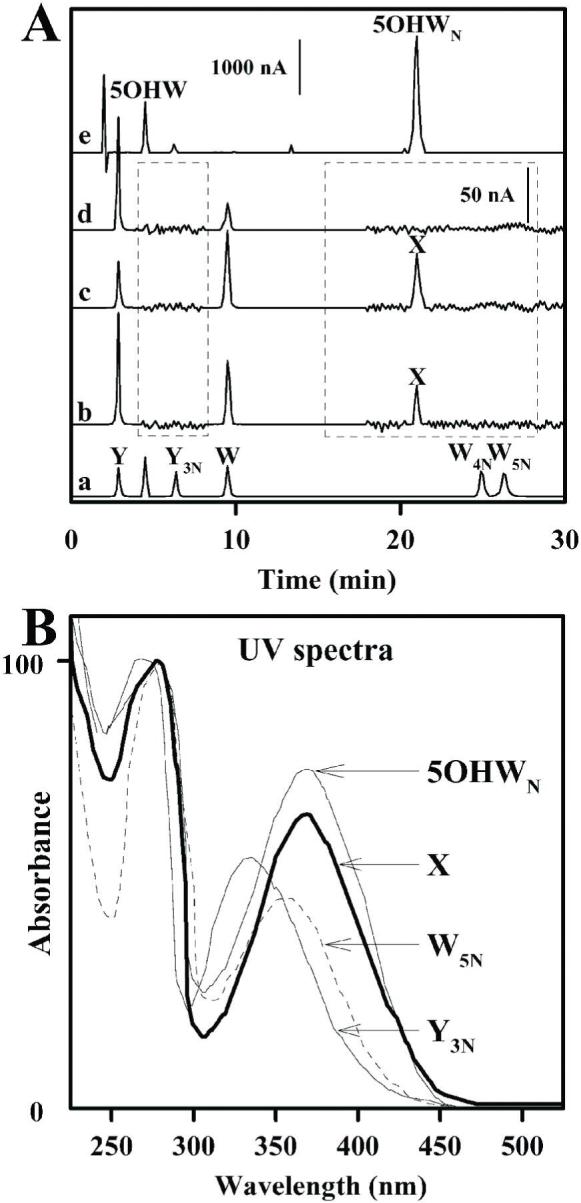
Identification of the modified amino acid in SCOT.
Panel A shows the HPLC chromatograms of amino acids after enzymatic hydrolysis of full length SCOT protein (trace b), C-terminal fragment (trace c) and N-terminal fragment (trace d). Trace a shows calibration standards of tyrosine, 3-nitrotyrosine, tryptophan, 4- and 5-nitrotryptophan (indicated as Y, Y3N, W, W4N and W5N, respectively). Trace e shows separation of aliquot of 6h reaction between 5-hydroxytryptophan (peak labeled as 5OHW, retention time 4.9 min) and tetranitromethane. Peak with retention time of 21 min is labeled as 5OHWN (5-hydroxynitrotryptophan). Intensities of the signal from the electrochemical detector are indicated in vertical bars: 1000 nA for the chromatograms outside and 50 nA for those inside the dotted boxes, with retention times from 4 to 8 and 18 to 30 min. X indicates a peak with a retention time of 21 min, present in the chromatograms of samples of full length and C-terminal fragment of SCOT. Panel B shows UV spectra of 3-nitrotyrosine (Y3N), 5-nitrotryptophan (W5N), synthetic 5-hydroxynitrotryptophan (5OHWN) and modified amino acid from nitrated SCOT (X).
FIGURE 6.

MALDI-TOF analysis of modified tryptophan residue isolated from nitrated SCOT synthetic nitrated 5-hydroxytryptophan, and other related tryptophan derivatives.
A, mass spectrum of peak X (collected during HPLC separation of pronase digest of the C-terminal fragment of SCOT and shown in Figure 4, trace c). The mass/charge value of 266.34 for the amino acid in peak X corresponds to tryptophan containing +61 Da (i.e. nitro- and hydroxy-) addition. B, mass spectrum of peak X treated with sodium dithionite. The ion peak with m/z value of 236.38 corresponds to amino-hydroxytryptophan, 173.92 to dithionite. C, mass spectrum of an aliquot taken from the reaction between 5-hydroxytryptophan and tetranitromethane, after 6 h of incubation (see Figure 4, trace e). The ion peak with m/z of 266.35 corresponds to 5-hydroxynitrotryptophan, 250.38 and 234.4 - to ions of photodecomposition products derived from 5-hydroxynitrotryptophan due to the loss of single and double oxygen, respectively. The ion peaks with m/z of 221.5 and 204.5 corresponds to ions of remaining un-reacted 5-hydroxytryptophan and photodecomposition product derived from 5-hydroxytryptophan due to the loss of water molecule, respectively. D, mass spectrum of 5-nitrotryptophan standard (m/z=250.7). The ion peak with m/z of 234.7 corresponds to ions of photodecomposition product derived from 5-nitrotryptophan due to the loss of single oxygen. E, mass spectrum of 5-hydroxytryptophan standard (m/z= 221.6). The ion peak with m/z of 204.6 corresponds to ions of photodecomposition product derived from 5-hydroxytryptophan due to the loss hydroxyl group. Ion peaks with m/z values of 192, 212 and 228 are originated from the matrix (α-cyano-4-hydroxycinnamic acid).
5-Hydroxynitrotryptophan was synthesized in order to directly compare its properties to those of amino acid X, isolated from the C-teminal fragment of SCOT and identified as hydroxynitrotryptophan. Several different approaches were employed for the synthesis of hydroxynitrotryptophan, including i) reaction between tryptophan and peroxynitrite, ii) oxidation of 5-nitrotryptophan, and iii) nitration of 5-hydroxytryptophan. All of these three reactions resulted in the appearance of multiple products; however the most efficient and appropriate method for the synthesis of the hydroxynitrotryptophan was deemed to be the reaction between 5-hydroxytryptophan and tetranitromethane, as described in detail in the Materials and Methods section. Figure 5 (trace e) shows an HPLC chromatogram of the aliquot taken 6 h after the reaction between 5-hydroxytryptophan and tetranitromethane. A major product (retention time 21 min) was formed by the nitration of 5-hydroxytryptophan (retention time 5 min). This synthetic amino acid displayed UV-spectrum with a peak at 360 nm, which is characteristic of the addition of nitro group. MALDI-TOF analysis of the reaction mixture (Figure 6C) revealed the presence of several major peaks with a mass/charge value of 266.35, 250.38, 234.4 and 204.5, in addition to 221.5 (protonated 5-hydroxytryptophan). The protonated molecules of 5-hydroxynitrotryptophan were responsible for the ions at m/z 266.35, which is consistent with a +45 modification of 5-hydroxytryptophan, corresponding to nitro-addition (mass: 46-1=45). Photodecomposition of the protonated molecular ion of nitroxydroxytryptophan corresponded to the ions that have lost single oxygen atom [MH-O]+ (peak at m/z 250.38) and double oxygen atoms [MH-O-O]+ (peak at m/z 234.4). Photodecomposition of the protonated molecular ion of 5-hydroxytryptophan corresponded to the ions that have lost hydroxyl group [MH-OH]+ (peak at m/z 204.5). Similar phenomenon, involving loss of hydroxyl group during MALDI-TOF analysis of tryptophan standard due to photodecomposition reaction under the laser light, has been described recently (42). MALDI-TOF analysis of the standards (Figure 6D, 6E), 5-nitrotryptophan and 5-hydroxytryptophan, was in agreement with observed ion peaks corresponding to hydroxy and nitro derivatives of tryptophan and their specific photodecomposition products.
It should be pointed out that potential artificial introduction of the hydroxy group into the phenyl ring of nitro-tryptophan in SCOT, similar to the frequently reported oxidation of methionine and tryptophan residues in proteins during in-gel sample processing and analysis by MS/MS (40, 41), was ruled out because identical results of amino acid and MS/MS analysis of SCOT were obtained in independent experiments, i.e. using “in-gel” and “in-solution” procedures.
Quantitative analysis of nitro-hydroxy-tryptophan in SCOT, purified from heart mitochondria of young rats, was carried out using HPLC measurements of enzymatic digests, using 5-nitro-tryptophan as a calibration standard. The molar content of this modification was determined to be 36 ± 3.1 mmol/mol SCOT protein (n=4), based on the 58 kDa molecular weight. Further quantification of the nitration of SCOT was carried out in Western blot analysis, by comparisons of the band densities in mitochondrial samples to those of SCOT purified from young rats.
MS/MS analysis of full length and C-terminal SCOT protein bands (Figure 7) revealed a series of tryptic peptides, including YGDLANWMIPGK, corresponding to amino acid residues 366 - 377, to be either unmodified (precursor ion 683.22 with a cross-correlation score XC= 2.39) or modified by +16 (oxidation) at methionine 373 (precursor ion 691.31 with a cross-correlation score XC= 4.35) and +61 (oxidation and nitration) at tryptophan 372 (precursor ion 713.3 with a cross-correlation score XC= 3.52). The MS/MS spectrum, shown in Figure 7A, indicated the presence of 9 y and 9 b out of the potential 11 ions. The most intense ion, y6 with observed mass of 792.3 (the theoretical mass for this peptide is 792.37), indicated the mass addition of +61. The presence of the ion peak b7 (m/z 880.5) also indicated the +61 addition. Such modification by 61 Da can be accounted for by a nitration (+45) and oxidation (+16) of the tryptophan 372 residue or the nitration of tryptophan 372 and oxidation of methionine 373 residues. However, the presence of the ion peaks y5 (m/z=545.6) and b8 (m/z=1011.37) indicated the absence of the oxidation of the methionine 373 in this peptide. Thus, residue 372 in the SCOT sequence was identified as hydroxynitrotryptophan. The MS/MS spectrum of unmodified peptide showed 6 y and 6 b out of potentially 11 ion peaks (Figure 7B), which indicated that neither methionine and/or tryptophan were oxidized nor the tryptophan was nitrated. The MS/MS spectra (Figure 7C) showed the presence of a series of ion peaks (y4, y5, y6 and corresponding b6, b7, b8) indicating the oxidation of methionine only. It is worth reporting that using MS/MS analysis, we never observed the indicated SCOT peptide exhibiting only nitro addition to tryptophan 372 residue. In addition, all three MS/MS spectra in Figure 7 indicated the unique fragmentation pattern of this specific peptide, that is, the relative dominance of the y3 and b9, but not of y4 or y5 ion peaks. Such specific fragmentation pattern may occur due to the presence of the proline residue (dominant peaks y3 and b9), which can cause suppression and/or disappearance of some of the following fragments (including y4 and y5) as was shown previously in similar proline-containing peptides (45).
FIGURE 7.

Identification of tryptophan 372 as the site of nitration in SCOT by tandem mass spectrometry.
MS/MS spectra for SCOT tryptic peptide Y366GDLANWMIPGK377, containing hydroxynitrotryptophan (A), unmodified peptide (B) and oxidized methionine (C). The ion annotation is based upon results presented in Sequest’s “display ion view” window, corroborated by the dta file. M# indicates oxidized methionine and W* a hydroxynitrotryptophan.
Effect of age on SCOT nitration, protein amount and catalytic activity in heart mitochondria
A comparison of 4-, 13-, 19- and 24-month-old rats indicated that the specific activity of SCOT in heart mitochondria increased by 30% (P<0.0001, n=9) between 4 and 24 months of age (Figure 8). Furthermore, this gain in activity was accompanied by an increase of nitro-hydroxytryptophan content from 36 to 80 mmol/mol SCOT (Figure 8). During the duration of aging, the average amount of SCOT protein remained unchanged (56.3 ± 4.9 pmol SCOT/mg matrix protein).
FIGURE 8.

Age-associated increase in 3NT content and specific activity of SCOT in rat heart. Panel A: Immunoblot analysis of heart mitochondrial proteins from rats of various ages, demonstrating increased anti-3NT and constant anti-SCOT reactivity at older ages. Panel B: Specific activity of SCOT, content of the nitro-hydroxytryptophan and SCOT protein amounts are presented as percentages of the amounts in the 4-month-old rats (5.15 ± 0.15 × 103 min-1, 36 ± 2.2 mmol/mol SCOT and 56.3 ± 4.9 pmol/mg matrix proteins, respectively). All values represent mean ± standard deviation of n=6 for immunoblots and n=9 for activity assays; * indicates significant difference vs 4-month control (P<0.001). Activity in 13-month-old rats was not determined.
Effect of nitration on SCOT activity
SCOT activity, measured in samples of the soluble fraction of mitochondria, represents the sum of the activities of non-modified SCOT and nitrated SCOT. For instance, in heart mitochondria, the specific activity of the enzyme was increased from 5.15 to 6.7 × 103 min-1 (4- vs. 24-month old rat), whereas the nitro-hydroxytryptophan content was elevated from 36 to 80 mmol/mol SCOT (Figure 8). Modification of one tryptophan residue implies that 2.03 (0.036 × 56.3) and 4.5 (0.080 × 56.3) pmol of the enzyme (56.3 pmol SCOT per milligram of matrix protein) were modified in the young and the old rats, respectively. Since there was no alteration in the total content of SCOT and no evidence of other posttranslational modifications of this protein, the gain of catalytic activity with age can be attributed to the changes in the amounts of unmodified and nitrated SCOT, as follows:
-
Catalysis in young rats:
A [54.27] + Amod [2.03] = 5.15 × 103 [56.3]
-
Catalysis in old rats:
A [52.07] + Amod [4.5] = 6.7 × 103 [56.3]
A - specific activity of unmodified SCOT; Amod - specific activity of nitrated SCOT; numbers in brackets indicate amounts of enzyme in pmol SCOT per mg of matrix protein.
The specific activity of nitrated SCOT (Amod) was calculated to be 38.5 ± 3.5 × 103 min-1, in contrast to A = 3.9 ± 0.3 × 103 min-1 of unmodified enzyme, suggesting that nitration causes a ∼10-fold increase in specific activity.
Effect of in vitro peroxynitrite-induced nitration on catalytic activity of SCOT
Soluble proteins from heart mitochondria of 2-month-old rats were incubated with 0 to 500 μM peroxynitrite. SCOT activity was 24% higher (P<0.0001) at 25 μM peroxynitrite, compared to the control (Figure 9), but at concentrations higher than 50 μM, there was a linear decrease in activity (data not shown). This observation was attributed to the simultaneous oxidation effects of peroxynitrite on a variety of amino acid residues (cysteine, methionine, tyrosine, histidine among others) in addition to nitration of aromatic amino acids (46). Due to the multiplicity and complexity of such reactions, further characterization of the effects of high concentrations of peroxynitrite on SCOT was not pursued.
FIGURE 9.

In vitro nitration and modulation of SCOT activity by peroxynitrite. Mitochondrial soluble proteins from heart and kidney mitochondria of 2-month-old rats were incubated with 25 μM peroxynitrite for 15 min. Specific activity (left) is expressed in min-1 and nitration content (right) in mmol/mol SCOT protein.
Analysis of SCOT treated with 25 μM peroxynitrite revealed an increase of nitration from 36 to 66 mmol/mol SCOT. Modification of one tryptophan residue per protein molecule would suggest that 2.03 (0.036 × 56.3) and 3.75 (0.066 × 56.3) pmol of the enzyme was modified in control and by peroxynitrite treatment, respectively. An increase of catalytic activity (from 4.91 to 6.07 × 103 min-1) upon peroxynitrite treatment can be attributed to the changes in the relative amounts of unmodified and nitrated SCOT as follows:
-
Catalysis in the absence of peroxynitrite:
A[54.27] + Amod [2.03] = 4.91 × 103 [56.3]
-
Catalysis in the presence of peroxynitrite:
A[52.55] + Amod[3.75] = 6.07 × 103 [56.3]
A - specific activity of unmodified SCOT; Amod - specific activity of nitrated SCOT; numbers in brackets indicate amount of enzyme in pmol SCOT per mg of matrix protein.
Therefore, the catalytic activity of nitrated SCOT (Amod = 41.3 ± 2.2 × 103 min-1) was calculated to be ∼12 times higher than that of the unmodified enzyme (A = 3.5 ± 0.4 × 103 min-1), this is in good agreement with the in vivo values obtained for the respective activities of SCOT in the rat heart during aging.
DISCUSSION
Results of this study indicate that the mitochondrial protein SCOT, a rate-limiting enzyme in ketone body utilization, is an in vivo target of nitration in most tissues of the rat, including heart, kidney, skeletal muscle, brain, lung, and spleen, but not the liver and testis, where it has been shown to be virtually absent (37, 47). Whereas the nitration of SCOT was originally noted in the kidney and heart of endotoxin- and/or streptozotocin-treated diabetic rats (48, 49), results of the present study demonstrate that nitration is not restricted to the heart and kidney or to a diabetic and/or inflammatory condition, but occurs also in other tissues expressing this enzyme, and even in young healthy rats.
Using mass spectrometry (MALDI-TOF, tandem MS/MS) and amino acid analysis, we have identified and characterized the specific post-translational modification of SCOT, namely simultaneous nitration and oxidation of tryptophan 372. In addition, no hydroxy- or nitro adducts of tryptophan or 3NT were detected within the limits of sensitivity of the methodology used here. It should be emphasized that the amino acid analysis with coulochem electrochemical detection would allow detection of nitrated or hydroxylated derivatives of tyrosine and tryptophan above 200-300 fmol, which would correspond to nitration content of >50 μmol/mol SCOT (single amino acid modification). Therefore, the presence of nitrative-oxidative modifications of SCOT (nitrotyrosine, nitrotryptophan, hydroxytryptophan) at levels below the limits of quantification by HPLC/electrochemical detection, can not be ruled out. However, the hypothetical presence of nitrated tyrosines at such extremely low levels will be undetectable by immunoblotting, as indicated by our study with BSA, containing 115 μmol nitrotyrosine per mol of protein. In contrast, methionine oxidation, observed for non-modified tryptic peptides, was quite likely introduced during “in-gel” sample handling for MS/MS analysis (43). While both in vitro and in vivo nitration of tyrosine residues has been documented extensively in a variety of proteins (9-20, 22-27), the present demonstration of tryptophan nitration/oxidation in SCOT is the first evidence supporting the existence of this particular mechanism targeting a specific aromatic amino acid (tryptophan 372), among 11 tyrosine and 3 tryptophan residues in SCOT.
Additional direct evidence, confirming the identity of the hydroxy-nitro modification of tryptophan residue in SCOT protein, can be obtained by comparison with hydroxynitrotryptophan. However this particular compound is not available commercially and to our knowledge there are no reports describing its synthesis. Consequently, we successfully synthesized 5-hydroxynitrotryptophan by reacting tetranitromethane and 5-hydroxytryptophan. The modified tryptophan residue isolated from SCOT, and the synthetic 5-hydroxynitrotryptophan, had identical retention times in HPLC analysis, similar UV-spectra that are characteristic of nitrated species, as well as analogous mass spectral data in MALDI-TOF analysis, indicating the presence of hydroxynitrotryptophan and its distinctive photodecomposition products.
A noteworthy implication of the present study is that immunoreaction of 3NT antibody (clone 1A6 from Upstate Biotechnology, which has been used in a majority of relevant studies on protein nitration, see references 11, 18, 20, 24, 48, 49) occurs not only with proteins containing 3NT but also with at least one protein containing nitro-hydroxytryptophan. This is probably due to the structural similarities between the phenyl ring of tyrosine and tryptophan carrying nitro- and hydroxy-groups (Figure 10). In contrast to free 3NT, nitro-tryptophan and/or hydroxy-tryptophan alone did not diminish the immunoreactivity of SCOT with the anti-3NT antibody in Western blot analysis. These results are in agreement with those of Ikeda et al. (13), who, using 3NT antibody, reported the absence of immunointeraction between 3NT antibody and nitrotryptophan-containing human Cu,Zn-SOD. It is plausible that the simultaneous presence of both modifications (nitro- and hydroxy-) on the phenyl ring of tryptophan is required for the immunoreaction to occur (Figure 10). However, due to the limited stability of the synthetic 5-hydroxynitrotryptophan in solutions compatible with Western blot, the immuno-competition experiments with 3NT-antibody could not be performed. Such instability of the 5-hydroxynitrotryptophan in solution could be due to the presence of the reactive by-products of the tetranitromethane and/or the continuation of the reaction towards multiple nitrations on the aromatic and indole rings of tryptophan. Further studies on purification and characterization of the synthetic 5-hydroxynitrotryptophan will be required to improve the stability of this compound.
FIGURE 10.

Structural similarity between the phenyl rings of 5-hydroxy-6-nitrotryptophan (left) and 3-nitrotyrosine (right).
Since the amount of SCOT protein remained stable and the degree of SCOT nitration increased with age, we hypothesized that the enhanced enzyme activity may be due to the accrual of nitrated SCOT. Indeed our estimations of specific activities of unmodified and nitrated SCOT accord with the observed amounts of SCOT protein, amounts of nitration, and enzyme activity of SCOT during aging in rat heart. In vitro nitration of glutathione S-transferase by peroxynitrite has also been demonstrated to cause an increase in activity (24, 25). Results of this study indicate that such an activating mechanism may also exist for SCOT in vivo in the heart. Thus our hypothesis that low degree of nitration tends to increase SCOT catalytic activity is based on correlation between SCOT nitration, protein amount and catalytic activity in heart mitochondria during in vivo aging. Notwithstanding, to address this issue more directly, it would be necessary to compare the activity of non-nitrated and nitrated SCOT. However, this was rendered unfeasible by the fact that SCOT is nitrated in most tissues of rat at all ages and separation of nitrated from non-nitrated SCOT by immunoprecipitation can not be achieved due to the lack of reactivity between the native enzyme and the 3NT antibody (data not shown). This later finding is in fact in agreement with the study by Kanski et al. (17), which did not detect SCOT as nitration target using immunoprecipitation of cardiac proteins with 3NT-antibody.
The observed age-related alterations in nitration and protein amount of SCOT in heart mitochondria raise the question about the possible causes underlying this phenomenon. The increase in the amount of modified SCOT in aged animals could be due to the increase in the turnover of non-modified enzyme and/or accumulation (i.e. decreased degradation) of modified SCOT. Our data indicate that the total amount of SCOT remains the same in heart mitochondria during aging, whereas the amount of modified SCOT nearly doubles and the non-modified SCOT declines in old rats. Such differential changes may reflect the age-related shifts in turnover rates for non-modified and modified SCOT, in favor the latter. Presently, limited information is available about synthesis/degradation of SCOT. The half-life of SCOT degradation (46-50 h), reported in neuronal cells (50), was comparable to that of the total cellular proteins (28 h). It seems unlikely that degradation rates of SCOT in heart mitochondria decrease with age since our results indicated that SCOT protein amount was similar at all ages. Thus the accumulation of nitrated SCOT in the heart of old rats can be due to the increased rate of enzyme modification, decreased turnover of nitrated enzyme and/or a combination of both of these processes. However, at present it is not possible to rule out either of these two possibilities. Moreover, there are conflicting reports about whether nitrated proteins are marked for increased or decreased degradation (51).
In contrast to the present and previous findings (44, 45) that only a single mitochondrial soluble protein exhibited nitration, Kanski et al. (17) using 2D gel electrophoresis, Western analysis and immunoprecipitation with 3NT-antibody identified 13 putatively nitrated proteins in the heart of aged rats, including aconitase, creatine kinase, enoyl-CoA hydratase and 3-oxoacyl-CoA thiolase, all of which are located in the mitochondrial matrix. However, they did not detect SCOT nitration, and mass spectrometric confirmation of actual nitrated peptides was not presented. Similarly the same authors, using Western analysis and matching anti-3NT antibody, did not report nitration of any of the mitochondrial proteins in rat skeletal muscle (16) listed in ref. 16. Moreover, in their most recent study (18), they used exactly the same anti-3NT antibody as in our study, but none of the previously reported mitochondrial proteins (16, 17) were confirmed to be nitrated using Western blot and tandem mass spectrometric analysis. Results of the present study, based on the immunoblot and mass spectrometric analysis of enriched mitochondrial proteins (aconitase and creatine kinase) showed the absence of nitration. However, due to the partial sequence coverage of aconitase (19%) and creatine kinase (3%) in MS/MS analysis and/or low amounts of 3NT-containing peptides, it is possible that such peptides were not detected. To investigate the source of the discrepancy between our respective results, using conditions and antibody dilutions identical to those of (17), we detected SCOT and some additional immunoreactive bands as well as considerable background immunoreaction, although such bands lacked specificity towards anti-3NT antibody, as indicated by the various controls described in Figure 2. It is also possible that if the nitration content in other mitochondrial proteins is much lower than in SCOT, their immunoreaction with anti-3NT antibody will be weaker or undetectable.
An obvious question raised by this study is why nitration and oxidation of Trp 372 affects SCOT activity. Structural studies indicate that the active site of SCOT, which consists of several amino acid residues (Asn 51, Tyr 76, Gln 99, Glu 241, Arg 242, Asn 279, Lys 327, Lys 380, Gly 383, Gly 384, Ala 385), is located in close proximity to Glu 303, a key catalytic residue (52, 53). The Trp 372 residue is positioned on the tip of the flexible loop, which is accessible to solvent and located within ∼10 Å from Glu 303 on its opposite side (Figure 10). Activated Glu 303 forms a thioester with succinyl-CoA, which is bound in an extended conformation, such that the distance between the sulphur atom and 3′-phosphate on the ribose ring is 20 Å (53). Accordingly, it is quite conceivable that Trp 372 may interact with the tail of succinyl-CoA directly or indirectly (through water molecules), and that nitration of Trp 372 may further increase the hydrophobicity of the environment of the substrate binding site (Tyr 76, Ala 385), possibly favoring an accelerated binding of the substrate and/or ejection of the product during the catalytic cycle, thereby increasing the rate of SCOT catalysis. It is also plausible that Trp 372 modification, i.e. addition of bulky nitro and hydroxyl groups, may influence the orientation and/or binding of the succinyl-CoA in the active site pocket, resulting in changes of the SCOT conformation, stability, folding or subunit interaction. Such proximity of Trp 372 to the active site residues of the enzyme further supports the hypothesis that the increase in nitration of Trp 372 with age is correlated with enhanced SCOT activity.
Although the nature of the chemical species nitrating/hydroxylating tryptophan 372 in vivo is still uncertain, peroxynitrite involvement is considered plausible in mitochondria, because of the presence of an isoform of nitric oxide synthase and high rate of O2-. generation in this organelle (9, 27, 28). In vitro incubation of mitochondrial proteins with 25 μM peroxynitrite indicated that there was a significant gain in SCOT activity and nitration. Quantitative comparisons of the activity and nitration content of the enzyme, treated with peroxynitrite, revealed that the specific activity of modified SCOT was ∼12-fold higher than that of the unmodified enzyme. This finding provides additional support for our hypothesis that nitration has an activating effect on catalytic function of SCOT. However, additional in vitro experiments using pure SCOT and different nitrating agents (peroxynitrite, tetranitromethane among the others) will be needed to test our hypothesis about the link between the nitrative-oxidative modification of tryptophan 372 and the enhanced enzyme activity of SCOT.
Among the variety of factors which may contribute to the decline of cellular functions in the heart during senescence, mitochondrial dysfunction and metabolic remodeling of energy-producing pathways are thought to play a key role (55, 56). Indeed decreases in the activities of the electron transport chain (57), aconitase and ATP synthase (21), among others, have been reported to occur in the heart during aging. Such disturbances may collectively contribute to deterioration in mitochondrial function and produce a state of energy deprivation in the senescent heart; in contrast, the increase in SCOT activity can be viewed as an adaptive mechanism to compensate for an age-related energetic imbalance.
Conclusions
We have identified a novel in vivo posttranslational protein modification - nitrohydroxy addition to the tryptophan residue 372 in SCOT, a key enzyme in ketolysis. Identification of this modification was established by mass spectrometry and by direct comparison with de novo synthesized 5-hydroxynitrotryptophan. A correlation between an increase in activity and content of hydroxynitrotryptophan of SCOT was detected in vivo as well as in vitro. As there is an apparent increase in the production of ketone bodies during aging (32) and the heart is one of the primary user of ketone bodies for energy, SCOT nitration and an increase in its activity during aging may be reflective of an adaptive shift in energy metabolism in the heart.
Supplementary Material
FIGURE 11.

Ribbon diagram of a monomer of SCOT (A) showing the proposed enzyme active site (B) and structure of the substrate succinyl-CoA (C). Positions of Trp 372 and key residues lining the succinyl-CoA binding cavity (Asn 51, Tyr 76, Gln 99, Glu 241, Arg 242, Asn 279, Lys 327, Lys 380, Gly 383 and Gly 384) are indicated. Figure was drawn using the program VMD (53), using PDB data file 1ooy of the crystal structure of pig heart SCOT (47). Numbering of amino acid residues corresponds to the sequence of mature rat SCOT. For the purpose of clarity, only the side chains of the amino acid residues are shown (B); whereas the positions of the α-carbon atoms and sulphur atom of the substrate (C) are depicted as balls. Bars: 5Å.
ACKNOWLEDGEMENTS
We thank Melissa Ferguson, Kathleen Rice and Barbara Sohal for their technical assistance. Justin Jones participated in some preliminary studies. MS/MS analysis and protein identifications were performed at the Proteomics Core Facility at the University of Southern California, School of Pharmacy, and the authors thank Rodrigo Aguilera for technical support. This work is dedicated to the memory of Dr. Andrei Nedospasov (Russian Academy of Sciences) for stimulating discussions on “tryptophan paradoxes”.
The abbreviations used are
- SCOT
succinyl-CoA:3-ketoacid coenzyme A transferase
- 3NT
3-nitro tyrosine
- MS/MS
tandem mass spectrometry
Footnotes
This research was supported by the grant RO1 AG 13563 from the National Institute on Aging - National Institutes of Health.
REFERENCES
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 4.Stadtman ER, Levine RL. Protein oxidation. Ann. NY Acad. Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 5.Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 9.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamakura F, Ikeda K. Modification of tryptophan and tryptophan residues in proteins by reactive nitrogen species. Nitric Oxide. 2006;14:152–161. doi: 10.1016/j.niox.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Herold S, Shivashankar K, Mehl M. Myoglobin scavenges peroxynitrite without being significantly nitrated. Biochemistry. 2002;41:13460–13472. doi: 10.1021/bi026046h. [DOI] [PubMed] [Google Scholar]
- 12.Yamakura F, Matsumoto T, Ikeda K, Taka H, Fujimura T, Murayama K, Watanabe E, Tamaki M, Imai T, Takamori K. Nitrated and oxidized products of a single tryptophan residue in human Cu,Zn-superoxide dismutase treated with either peroxynitrite-carbon dioxide or myeloperoxidase-hydrogen peroxide-nitrite. J Biochem. 2005;138:57–69. doi: 10.1093/jb/mvi095. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda K, Yukihiro Hiraoka B, Iwai H, Matsumoto T, Mineki R, Taka H, Takamori K, Ogawa H, Yamakura F. Detection of 6-nitrotryptophan in proteins by Western blot analysis and its application for peroxynitrite-treated PC12 cells. Nitric Oxide. 2007;16:18–28. doi: 10.1016/j.niox.2006.04.263. [DOI] [PubMed] [Google Scholar]
- 14.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch. Biochem. Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 15.Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol. Rev. 2002;54:619–634. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- 16.Kanski J, Alterman MA, Schöneich C. Proteomic identification of age-dependent protein nitration in rat skeletal muscle. Free Radic. Biol. Med. 2003;35:1229–1239. doi: 10.1016/s0891-5849(03)00500-8. [DOI] [PubMed] [Google Scholar]
- 17.Kanski J, Behring A, Pelling J, Schöneich C. Proteomic identification of 3-nitrotyrosine-containing rat cardiac proteins: effects of biological aging. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H371–381. doi: 10.1152/ajpheart.01030.2003. [DOI] [PubMed] [Google Scholar]
- 18.Kanski J, Hong SJ, Schöneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J. Biol. Chem. 2005;280:24261–24266. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- 19.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schöneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem. J. 1999;340:657–669. [PMC free article] [PubMed] [Google Scholar]
- 20.Sharov VS, Galeva NA, Kanski J, Williams TD, Schöneich C. Age-associated tyrosine nitration of rat skeletal muscle glycogen phosphorylase b: characterization by HPLC-nanoelectrospray-tandem mass spectrometry. Exp Gerontol. 2006;41:407–16. doi: 10.1016/j.exger.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Yarian CS, Rebrin I, Sohal RS. Aconitase and ATP synthase are targets of malondialdehyde modification and undergo an age-related decrease in activity in mouse heart mitochondria. Biochem. Biophys. Res. Commun. 2005;330:151–156. doi: 10.1016/j.bbrc.2005.02.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Proc. Natl. Acad. Sci. U.S.A. 1998;95:7659–7663. doi: 10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- 24.Boulos C, Jiang H, Balazy M. Diffusion of peroxynitrite into the human platelet inhibits cyclooxygenase via nitration of tyrosine residues. J. Pharmac. Exp. Therap. 2000;293:222–229. [PubMed] [Google Scholar]
- 25.Ji Y, Bennett BM. Activation of microsomal glutathione s-transferase by peroxynitrite. Mol. Pharmacol. 2003;63:136–146. doi: 10.1124/mol.63.1.136. [DOI] [PubMed] [Google Scholar]
- 26.Ji Y, Neverova I, Van Eyk JE, Bennett BM. Nitration of tyrosine 92 mediates the activation of rat microsomal glutathione s-transferase by peroxynitrite. J. Biol. Chem. 2006;281:1986–1991. doi: 10.1074/jbc.M509480200. [DOI] [PubMed] [Google Scholar]
- 27.Soulere L, Claparols C, Perie J, Hoffmann P. Peroxynitrite-induced nitration of tyrosine-34 does not inhibit Escherichia coli iron superoxide dismutase. Biochem. J. 2001;360:563–567. doi: 10.1042/0264-6021:3600563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem. 2002;277:38079–38086. doi: 10.1074/jbc.M205256200. [DOI] [PubMed] [Google Scholar]
- 29.Lacza Z, Snipes JA, Zhang J, Horvath EM, Figueroa JP, Szabo C, Busija DW. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free Radic. Biol. Med. 2003;35:1217–1228. doi: 10.1016/s0891-5849(03)00510-0. [DOI] [PubMed] [Google Scholar]
- 30.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 31.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980;60:143–187. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 32.Hagopian K, Ramsey JJ, Weindruch R. Influence of age and caloric restriction on liver glycolytic enzyme activities and metabolite concentrations in mice. Exp Gerontol. 2003;38:253–266. doi: 10.1016/s0531-5565(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 33.Dunbar BS, Schwoebel ED. Preparation of polyclonal antibodies. Methods Enzymol. 1990;182:663–670. doi: 10.1016/0076-6879(90)82051-3. [DOI] [PubMed] [Google Scholar]
- 34.Sims NR. Mitochondrial Isolation from Brain: Strategy, Techniques, and Criteria for Purity. In: Lash LH, Jones DP, editors. Mitochondrial dysfunction. Academic Press; San Diego, CA: 1993. pp. 29–41. [Google Scholar]
- 35.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 37.Williamson DH, Bates MW, Page MA, Krebs HA. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem. J. 1971;121:41–47. doi: 10.1042/bj1210041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shigenaga MK, Lee HH, Blount BC, Christen S, Shigeno ET, Yip H, Ames BN. Inflammation and NO(X)-induced nitration: assay for 3-nitrotyrosine by HPLC with electrochemical detection. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3211–3216. doi: 10.1073/pnas.94.7.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howard JB, Zieske L, Clarkson J, Rathe L. Mechanism-based fragmentation of coenzyme A transferase. Comparison of alpha 2-macroglobulin and coenzyme A transferase thiol ester reactions. J. Biol. Chem. 1986;261:60–65. [PubMed] [Google Scholar]
- 40.Nielsen KR, Pennington MW. Mass spectral analysis of peptides containing nitrobenzyl moieties. Lett. Pept. Sci. 1995;2:301–305. [Google Scholar]
- 41.Sarver A, Scheffler NK, Shetlar MD, Gibson BW. Analysis of peptides and proteins containing nitrotyrosine by matrix-assisted laser desorption/ionization mass spectrometry. J. Am. Soc. Mass. Spectrom. 2001;12:439–448. doi: 10.1016/S1044-0305(01)00213-6. [DOI] [PubMed] [Google Scholar]
- 42.Smaniotto A, Comai S, Bertazzo A, Costa CV, Allegri G, Seraglia R, Traldi P. A mass spectrometric investigation on the possible role of tryptophan and 7-hydroxytryptophan in melanogenesis. J. Mass Spectrom. 2006;41:921–930. doi: 10.1002/jms.1051. [DOI] [PubMed] [Google Scholar]
- 43.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu. Rev. Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 44.Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J. Biol. Chem. 2003;278:19587–19590. doi: 10.1074/jbc.C300135200. [DOI] [PubMed] [Google Scholar]
- 45.Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 46.Alvarez B, Radi R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids. 2003;25:295–311. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- 47.Fukao T, Song XQ, Mitchell GA, Yamaguchi S, Sukegawa K, Orii T, Kondo N. Enzymes of ketone body utilization in human tissues: protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and and cytosolic acetoacetyl-CoA thiolases. Pediatr. Res. 1997;42:498–502. doi: 10.1203/00006450-199710000-00013. [DOI] [PubMed] [Google Scholar]
- 48.Marcondes S, Turko IV, Murad F. Nitration of succinyl-CoA:3-oxoacid CoA-transferase in rats after endotoxin administration. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7146–7151. doi: 10.1073/pnas.141222598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H2289–2294. doi: 10.1152/ajpheart.2001.281.6.H2289. [DOI] [PubMed] [Google Scholar]
- 50.Haney PM, Bolinger L, Raefsky C, Patel MS. Turnover of succinyl-CoA:3-oxoacid CoA-transferase in glioma and neuroblastoma cells. Specific influence of acetoacetate in neuroblastoma cells. Biochem. J. 1984;224:67–74. doi: 10.1042/bj2240067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Souza JM, Choi I, Chen Q, Weisse M, Daikhin E, Yudkoff M, Obin M, Ara J, Horwitz J, Ischiropoulos H. Proteolytic degradation of tyrosine nitrated proteins. Arch. Biochem Biophys. 2000;380:360–366. doi: 10.1006/abbi.2000.1940. [DOI] [PubMed] [Google Scholar]
- 52.Coros AM, Swenson L, Wolodko WT, Frazer ME. Structure of the CoA transferase from pig heart to 1.7 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1717–1725. doi: 10.1107/S0907444904017974. [DOI] [PubMed] [Google Scholar]
- 53.Bateman K, Brownie ER, Wolodko WT, Fraser ME. Structure of the mammalian CoA transferase from pig heart. Biochemistry. 2002;41:14455–14462. doi: 10.1021/bi020568f. [DOI] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Sample J, Cleland JGF, Seymour AML. Metabolic remodeling in the aging heart. J. Mol. Cell Cardiol. 2006;40:56–63. doi: 10.1016/j.yjmcc.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 56.Csiszar A, Labinskyy N, Orosz Z, Ungvari Z. Altered mitochondrial energy metabolism may play a role in vascular aging. Med. Hypotheses. 2006;67:904–908. doi: 10.1016/j.mehy.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 57.Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, Hoppel CL. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J. Mol. Cell. Cardiol. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.