

Abstract
The oxidation products obtained from the reaction of peroxynitrite (ONOO−) with dG include - among others - 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodG), 2,2-diamino-4[(2-deoxy-β-D-erythro-pentafuranosyl)amino]-5(2H)-oxazolone (oxazolone), spiroiminodihydantoin and N1-(β-D-erythro-pentofuranosyl)-5-guanidinohydantoin (guanidinohydantoin). In the present work, the formation of these products from the treatment of calf thymus DNA with varying amounts of ONOO− was studied quantitatively in vitro. 13C-, 15N-labeled standards were synthesized for the nucleosides of interest, and calf thymus DNA was reacted with ONOO− and digested enzymatically down to the nucleoside level. Specific modifications in the DNA were measured by HPLC separation followed by electrospray ionization tandem mass spectrometric analysis in the selected reaction-monitoring mode. Artifacts of the above four oxidation products, arising from oxidation of dG and/or 8-oxodG during DNA digestion and subsequent work-up, were evaluated with 7-15N-dG and/or stable-isotope labeled 8-oxodG as internal standards. Levels of artifactual 8-oxodG were about 5/106 nucleosides. The artifacts of spiroiminodihydantoin and guanidinohydantoin, arising from 8-oxodG, were 3.7% and 0.6% of the measured 8-oxodG values, respectively. No artifacts of oxazolone were detected. 8-OxodG and oxazolone were formed dose-dependently in DNA treated with ONOO−, while the levels of spiroiminodihydantoin and guanidinohydantoin increased significantly at low ONOO− doses, and then dropped off at higher ONOO− doses. The complexity of these dose-response relationships is likely due to the dual role of peroxynitrite as both an oxidant and a nucleophile in competition with water.
Keywords: 8-oxodG, oxazolone, spiroiminodihydantoin, guanidinohydantoin, peroxynitrite, calf thymus DNA
Introduction
Peroxynitrite (ONOO−) is a powerful oxidizing agent formed by the diffusion-controlled reaction of nitric oxide with superoxide (1,2). Inflammatory cells such as neutrophils and macrophages, which are capable of simultaneously producing nitric oxide and superoxide, can therefore generate ONOO− in vivo. In chronic inflammatory states, these cells can overproduce ONOO− over prolonged periods of time, and excess ONOO− formation has been implicated in a number of pathological conditions such as rheumatoid arthritis and neurodegenerative diseases (2–4) as well as in increased cancer risk (5). In addition to reacting with a number of cellular substrates, ONOO− reacts with DNA, resulting in both sugar (6–8) and nucleobase damage. Of the four DNA bases, guanine, with the lowest oxidation potential, is the most reactive (9–11). Major oxidation products arising from the reaction of dG with ONOO− include 8-oxodG (12–14) oxazolone, and its precursor, imidazolone (15,16) (Scheme 1). In addition, 8-oxodG is at least 1000 times more reactive toward ONOO− than is dG (17), resulting in the formation of secondary oxidation products such as oxazolone (18), spiroiminodihydantoin (13), and guanidinohydantoin(19) (Scheme 1). These four DNA oxidation products are all pro-mutagenic. 8-OxodG is a well-known DNA oxidation product, and has been shown to cause G → T transversions in both prokaryotic and eukaryotic cells (20–22). The mutagenic potential of oxazolone following DNA synthesis in vitro suggests that the formation of oxazolone in DNA mainly leads to G → T and to some extent G → C transversions (23). The replication of oxazolone-containing DNA in E. coli, also predominantly produces G → T transversions (24) and oxazolone has also been shown to constitute a blocking lesion during DNA synthesis (23) in vitro. The frequencies of the G → T transversions for oxazolone were found to be notably high (95 %) in comparison to the low mutation frequency (7%) of 8-oxodG (24). Both diastereoisomers of spiroiminodihydantoin have been shown to be strong inhibitors of DNA polymerase extension in E.coli cells, causing a mixture of G → T and G → C transversions (24). The mutation frequency for spiroiminodihydantoin is at least an order of magnitude higher than that for 8-oxodG (25). Guanidinohydantoin is also a highly mutagenic lesion and causes exclusive G → C transversions (25). To assess the formation profile of these highly mutagenic lesions in vitro, the current work focused on the qualitative as well as quantitative formation of 8-oxodG, oxazolone, guanidinohydantoin, and spiroiminodihydantoin in calf thymus DNA treated with ONOO− (bolus), and we now report quantitation, by isotope-dilution-HPLC-tandem mass spectrometry, of these modifications.
Scheme 1.
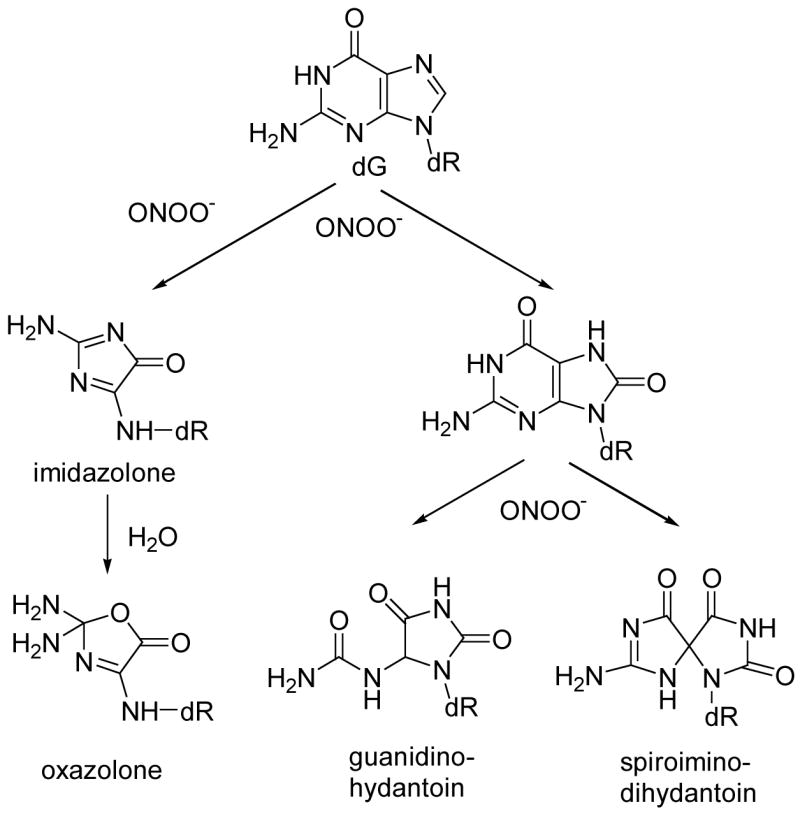
Products of oxidation of dG by peroxynitrite
Materials and methods
The uniformly 13C, 15N labeled dGTP (13C and 15N > 98%) was obtained from Cambridge Isotope Laboratories (Andover, MA). The syntheses of 1,2,7-15N3-8-oxodG and 3,7,8-15N3-N1-(-D-erythro-pentofuranosyl)-5-guanidinohydantoin have been reported (26). NAP-10 DNA desalting columns were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). Nuclease P1 and white-potato acid phosphatase were obtained from Calbiochem (San Diego, CA). Snake-venom phosphodiesterase (type I), bovine spleen phosphodiesterase (type II), alkaline phosphatase and calf thymus DNA were from Sigma (St. Louis, MO). Stock solutions of ONOO− were prepared from ozone and sodium azide as described earlier (27) and stored at −80 °C until needed. 7-15N-dG was a generous gift from Dr. T. M. Harris of Vanderbilt University. Microcon YM-10 centrifugal columns were purchased from Millipore (Bedford, MA). All other chemicals, reagents and solvents were purchased from Sigma-Aldrich, EM Science and other chemical distributors. UV/Vis measurements were made on an HP8452 diode-array spectrophotometer (Agilent Technologies, Palo Alto, CA). HPLC analyses were carried out on an Agilent 1100 Series HPLC system with binary pumps, a degasser, an auto-injectorand a diode array detector. Quantitative LC-MS/MS analyses were conducted on an Agilent 1100 HPLC pump system interfaced with an Sciex API 3000 tandem quadrupole mass spectrometer (Applied Biosystems, Framingham, MA). ESI-MS experiments for recoveries, isotopic purities, and structure confirmations were done on an Agilent (Palo Alto, CA) MSD-TOF.
Synthesis of labeled standards
1. 13C, 15N labeled dG
The uniformly 13C, 15N labeled dGTP (0.5 mg) was dissolved in water and incubated with 1 unit of alkaline phosphatase (in a 10 × alkaline phosphatase buffer) at 37 °C for 2 h. The enzyme was removed by centrifugation on a Microcon YM-10 column. Conversion of the labeled dGTP to the corresponding labeled dG was complete. The labeled dG was purified by HPLC using a 250 × 2.1 mm, 5 μm Supelcosil LC-18-DB column (Supelco, Bellafonte, PA). The mobile phase consisted of 50 mM ammonium acetate (A) and acetonitrile (B) with a gradient of 0–32% B over 40 min at a flow rate of 0.25 mL/min (dG retention time 14.0 min).
2. 13C, 15N labeled 8-oxodG
13C, 15N labeled 8-oxodG was synthesized from the uniformly 13C, 15N labeled dG as described by Kasai and Nishimura (28). Labeled 8-oxodG was formed as a minor product and purified by HPLC using a 250 × 4.6 mm, 5 μm Columbus C18 column (Phenomenex, Torrance, CA). The mobile phase described above was delivered as a gradient of 0–20% B over 40 min at a flow rate of 1 mL/min (retention times: labeled dG, 18.0 min, labeled 8-oxodG, 19.6 min, see supporting information Figure 1; ESI-MS of labeled 8-oxodG: m/z 299.0 [M+H]+ and m/z 177.9 [BH+H]+, see supporting information Figure 2).
3. 13C, 15N labeled oxazolone
A mixture of the labeled dG (500 μM) and riboflavin (100 μM) in water was irradiated with visible light for 40 min (29). The reaction mixture was purified by HPLC using a 250 × 4.6 mm, 5 μm Synergi Hydro RP column (Phenomenex, Torrance, CA) with UV detection wavelengths at 230 nm and 320 nm. The mobile phase was water (A) and acetonitrile (B) with a gradient of 100 % A for the first 15 min and then a ramp to 30% B over 10 min at a flow rate of 1.0 mL/min (imidazolone retention time 12.7 min). The fraction eluting between 12 and 13 min was collected and incubated at 37 °C overnight to form oxazolone (see supporting information Figure 3). The resulting solution was freeze-dried to give labeled oxazolone (ESI-MS: m/z 259.1 [M+H+]+, m/z 214.0 [M+H+-CO2]+, 138.0 [BH+H+]+, 122.1 [M+H+-137]+, see supporting information Figure 4).
4. 13C, 15N labeled spiroiminodihydantoin
Irradiation of an aerated D2O solution of the uniformly 13C, 15N labeled dG (200 μM) and methylene blue (100 μM) with visible light for 4 h resulted in the formation of the corresponding spiroiminodihydantoin (13,30,31). The reaction mixture was purified by HPLC using a 50 × 2.0 mm, 5 μm, Luna amino column (Phenomenex, Torrance, CA). The mobile phase was 50 mM ammonium acetate (A) and acetonitrile (B) delivered isocratically (75% B and 25% A) at 0.2 mL/min (two diastereoisomers of spiroiminodihydantoin retention times at 20.0 and 23.0 min, see supporting information Figure 5; ESI-MS: m/z 315.1 [M+H+]+, m/z 194.0 [BH+H+]+, m/z 122.1 [M+H+-193]+, see supporting information Figure 6).
Molar extinction coefficients for spiroiminodihydantoin and oxazolone
3H-dG was used as the starting material for the syntheses of 3H-oxazolone and 3H-spiroiminodihyantoin according to procedures described above. The specific activity of dG was determined by scintillation counting (0.77 mCi/mmol), and purified 3H-oxazolone and 3H-spiroiminodihyantoin were used for determination of the molar extinction coefficients.
Reaction of Calf Thymus DNA with ONOO−
Concentrations of the ONOO− stock solutions were determined for serial dilutions with 0.1 N NaOH by measuring the absorbance at λ = 302 nm (ε = 1670 M−1 cm−1) (32). Calf thymus DNA (2230 μg) was dissolved in 1900 μL of potassium phosphate (150 mM) and sodium bicarbonate (25 mM) (pH 7.2). The guanine content in this solution was calculated to be ~ 1100 μM. The solution was divided into aliquots (100 μL) and each aliquot was diluted to 400 μL with potassium phosphate/sodium bicarbonate buffer (guanine content ~ 275 μM). Varying amounts of the ONOO− stock solution were added to the buffered DNA solution followed by rapid vortexing for 1 min. The samples were left at room temperature for 1 hour and then desalted by passing through NAP-10 columns. Recovery of the DNA was over 90% as determined by UV/Vis spectroscopy. In control experiments, calf thymus DNA treated with a deactivated ONOO− solution was assayed under the same conditions to determine the basal levels of the four nucleosides of interest. The treatment of calf thymus DNA with ONOO− at each dose was carried out in duplicate.
Enzymatic digestion of calf thymus DNA and HPLC purification of 8-oxodG, spiroiminodihydantoin, guanidinohydantoin and oxazolone
The protocols for enzymatic digestion of calf thymus DNA prior to analysis for 8-oxodG, spiroiminodihydantoin, and guanidinohydantoin, were similar except that the metal chelator desferrioxamine was added (0.1 mM) to the digestion buffer in the analysis of 8-oxodG in order to minimize artifactual formation of that analyte. Typically, calf thymus DNA (117 μg) was digested by incubation with 10 units of nuclease P1 in a mixture of 150 μL of H2O, 25 μL of NaOAc (1 M, pH 5.1), and 20 μL of ZnCl2 (2 mM). A known amount of 13C, 15N labeled 8-oxodG, spiroiminodihydantoin, or 3,7,8-15N3-5-guanidinohydantoin was added to each DNA sample before the digestion. The samples were incubated at 37 °C overnight followed by the addition of 25 μL of sodium carbonate buffer (1 M, pH 9.2), snake venom phosphodiesterase (0.02 units), and alkaline phosphatase (4 units). The resulting mixture was incubated for 7 h at 37 °C. Since oxazolone is base-labile, the enzymatic digestion of DNA for the analysis of this compound was carried out under acidic conditions. Typically, calf thymus DNA (117 μg) was digested by incubation with 10 units of nuclease P1 and 1 unit of white-potato acid phosphatase in a mixture of 150 μL of H2O, 65 μL of NaOAc (1 M, pH 5.1), and 50 μL of ZnCl2 (2 mM). A known amount of 13C, 15N labeled oxazolone was added to each DNA sample before the digestion. After incubation at 37 °C overnight, the enzymes in the digestion mixture were removed with Microcon YM-10 centrifugal columns. The filtrate was then concentrated by freeze-drying. DNA oxidation products typically require some prepurification and concentration in order to maximize sensitivity and minimize the likelihood of coeluting interferences. Purification of 8-oxodG was performed using a 50 × 2.1 mm, 5 μm Zorbax Eclipse XDB-C18 column (Agilent Technologies, Palo Alto, CA). The mobile phase was 50 mM ammonium acetate (A) and acetonitrile (B) with a gradient of 0–15% B over 40 min at a flow rate of 0.2 mL/min (8-oxodG retention time at 7.0 min; see supporting information Figure 7). Purification of spiroiminodihydantoin was performed using a 50 × 2.0 mm, 5 μm, Luna-amino column (Phenomenex, Torrance, CA). The mobile phase was 50 mM ammonium acetate (A) and acetonitrile (B) as an isocratic mixture of 75% B and 25% A at 0.2 mL/min (spiroiminodihydantoin retention time at 20.4 and 23.2 min, see supporting information Figure 8). Guanidinohydantoin was prepurified using a 4.6 × 250 mm, 5 μM Zorbax Eclipse XDB C18 column with 100% 0.1% TFA in water at a flow rate of 0.4 mL/min. The HPLC fractions containing guanidinohydantoin were concentrated and further purified on a Thermosil-Keystone 3.0 × 100 mm, 5 μM Hypercarb column (Phenomenex, Torrance, CA), with 0.1% TFA in water (A) and acetonitrile (B) delivered as a gradient of 0–9% B over 15 min at 0.4 mL/min. Oxazolone was purified on a Thermosil-Keystone Hypercarb column (100 × 2.1 mm, 3 μm) using water (A) and acetonitrile (B) as a gradient of 0–30% B over 45 min at a flow rate of 0.2 mL/min (oxazolone retention time at 14.8 min, see supporting information Figure 9). The HPLC fractions containing 8-oxodG, spiroiminodihydantoin, guanidinohydantoin or oxazolone were collected and concentrated by freeze-drying.
Artifacts of 8-oxodG formed by oxidation of dG during DNA digestion and work-up
Untreated calf thymus DNA (145.6 μg) was digested as described above in the presence of 75 nmol of 7-15N-dG and 62.4 pmol of 1,2,7-15N3-8-oxodG as internal standards along with desferrioxamine (0.1 mM). 8-OxodG was purified by HPLC on a 4.6 × 250 mm, 5 μm Zorbax XDB-C18 column. The mobile phase consisted of water (A) and acetonitrile (B); 0–7% B over 28 min at a flow of 0.8 mL/min. Fractions containing 8-oxodG (between 23.8 and 25.4 min) and dG/7-15N-dG (between 20.6 and 22 min) were collected and concentrated.
Artifacts of spiroiminodihydantoin, guanidinohydantoin, and oxazolone from oxidation of 8-OxodG during DNA digestion and work-up
Untreated calf thymus DNA (145.6 μg) was digested as described above in the presence of desferrioxamine (0.1 mM), and 7-15N-dG (75 nmol). In measuring the artifacts of spiroiminodihydantoin and oxazolone, 62.4 pmol of 1,2,7-15N3-8-oxodG and 126.5 pmol of the uniformly 13C, 15N labeled spiroiminodihydantoin or 62.4 pmol of 1,2,7-15 13C, 15N labeled oxazolone were N3-8-oxodG and 86.4 pmol of the uniformly added to DNA before the digestion. For guanidinohydantoin, 62.4 pmol of the uniformly 13C, 15N labeled 8-oxodG and 40.2 pmol of 3,7,8-15N3-5-guanidinohydantoin were added to DNA before the digestion. Purification of 8-oxodG and dG/7-15N-dG from the digestion mixture was accomplished as described above. The HPLC fraction between 0–6 min on the 4.6 × 250 mm, 5 μm Zorbax XDB C18 column was collected for spiroiminodihydantoin, guanidinohydantoin, or oxazolone and freeze-dried. This fraction was further purified on a 3.0 × 150 mm, 5 μm Thermo-Keystone Hypercarb column; ammonium acetate (10 mM, pH 7.0: A) and acetonitrile (B) delivered from 0–18.7% B over 28 min at 0.2 mL/min. Fractions containing spiroiminodihydantoin (retention times of 14 and 16 min), guanidinohydantoin (retention time at 18 min), and oxazolone (retention time of 21 min) were collected and freeze-dried.
LC-MS/MS analysis of 8-oxodG, spiroiminodihydantoin, oxazolone, and guanidinohydantoin
The dried samples containing 8-oxodG, oxazolone, spiroiminodihydantoin or guanidinohydantoin and their corresponding isotopomers were dissolved in appropriate solvents: 8-oxodG in 50 mM ammonium acetate, spiroiminodihydantoin and oxazolone in 3:7 and 3:2 acetonitrile/water mixtures, respectively, and guanidinohydantoin in water. An aliquot of each sample was analyzed by HPLC-ESI-MS/MS using an Agilent 1100 series HPLC system interfaced with an API-Sciex 3000 triple quadrupole mass spectrometer equipped with either a TurboIonSpray™ source or, for the analysis of guanidinohydantoin, a nanoelectrospray ionization source. Chromatographic isolation of 8-oxodG was achieved using a 50 × 2.1 mm, 5 μm C18 Zorbax Eclipse XDB-C18 column, using 50 mM ammonium acetate (A) and acetonitrile (B) delivered from 0 to 15% B over 40 min at 0.2 mL/min. Oxazolone and spirominodihydantoin were separated on a 4.6 × 75 mm, 5 μm Zorbax XDB-C18 column, with a mobile phase of 0.1% acetic acid in water (pH 4.5) (A) and acetonitrile (B) delivered isocratically (40% A and 60% B) at 0.3 mL/min. Guanidinohydantoin was isolated on a 15 cm PicoFritTM capillary column (360 μM OD x 75 μM ID) (New Objective, Woburn, MA) packed with Alltech C18 (5 μM). The mobile phase - 0.4% acetic acid in water (A) and 0.4% acetic acid in acetonitrile (B) - 97:3, respectively - was delivered at 90 nL/min. The mass spectrometer was operated in the positive ion mode, with all instrument paramenters optimized for maximum sensitivity. Samples were analyzed in the selected reaction-monitoring (SRM) mode, which monitors collision-induced dissociation of a precursor ion to an abundant characteristic product ion. The analyses monitored the transitions of m/z 284 → m/z 168, m/z 285 → m/z 169, and m/z 287 → m/z 171 (or m/z 299 → m/z 178) for 8-oxodG, 7-15N-8-oxodG, 1,2,7-15N3-8oxo-dG (or the uniformly 13C, 15N labeled 8-oxodG), respectively (see supporting information Figure 10). Transitions of m/z 247 → m/z 131, m/z 249 → m/z 133 and m/z 259 → m/z 138 were monitored for oxazolone, 2,2-15N2-oxazolone (from oxidation of 1,2,7-15N3-8-oxodG) and the uniformly13C,15N labeled oxazolone (see supporting information Figure 11). Transitions of m/z 300 → m/z 184, → m/z 303 → m/z 187, and m/z 315 → m/z 194 were monitored for spiroiminodihydantoin, 15 N3-spiroiminodihydantoin (from oxidation of 1,2,7-115N3-8-oxodG), and the uniformly 13C, 15N labeled spiroiminodihydantoin (see supporting information Figure 12 and 13). Transitions of m/z 274 → m/z 158, m/z 288 → m/z 167, and m/z 277 → m/z 161 were monitored for guanidinohydantoin, the uniformly 13C, 15N labeled guanidinohydantoin (from oxidation of the uniformly 13C, 15N labeled 8-oxodG) and 3,7,8-15N3-5-guanidinohydantoin (see supporting information Figure 14). Calibration curves for the labeled and unlabeled forms of each of the four DNA oxidation products were constructed by plotting the SRM signal ratios between the labeled and unlabeled forms against their corresponding concentration ratios. Quantitation of 8-oxodG, oxazolone, spiroiminodihydantoin, and guanidinohydantoin in each sample was achieved using the SRM signal ratio between analyte of interest and its isotope labeled internal standard and the response curve.
The limit of detection of 8-oxodG, oxazolone, spiroiminodihydantoin or guanidinohydantoin was measured by adding a known amount of the corresponding labeled nucleoside to DNA. Typically, 200 femtomol of the labeled nucleoside was added to 117 μg of untreated DNA (the level of the labeled nucleoside is 5.6 nucleosides/107 nucleosides). DNA was digested to the nucleoside level. The labeled nucleoside was purified by HPLC and analyzed by LC-MS/MS as described above. For each labeled nucleoside, the signal-to-noise ratio e was ~ 10. The limit of detection was ~ 1 nucleoside/107 nucleosides with a signal to noise ratio of 2. Assuming a response ratio of ~ 1 between signals of the unlabeled and the labeled nucleosides in the SRM mode, the limit of detection of each oxidation product (the unlabeled nucleoside) would also be ~1 nucleoside/107 nucleosides.
dG from DNA digestion
The amount of dG in the HPLC fraction containing 7-15N-dG and dG from DNA digestion was measured by nano-LC-ESI-MS/MS on a capillary C18 column at a flow rate of 120 nL/min. The mobile phase was an isocratic mixture of 0.4% acetic acid in water (A) and 0.4% acetic acid in acetonitrile (B) at 97% A and 3% B. SRM transitions of 268 152 and 269 153 were used to measure dG and 7-15N-dG, respectively. The amount of dG in each sample was calculated from the response curve obtained from the SRM signal ratios between dG and 7-15N-dG and their corresponding concentration ratios.
8-OxodG and spiroiminodihydantoin in DNA digested with different enzymes
Untreated calf thymus DNA was digested with enzymes in various combinations:nuclease P1 and white potato acid phosphatase, nuclease P1, phosphodiesterase I and alkaline phosphatase, phosphodiesterase I (5’-exo digestion) and alkaline phosphatase, phosphodiesterase II (3’-exo digestion) and alkaline phosphatase (33). The uniformly 13C, 15N labeled spiroiminodihydantoin and 1,2,7-15N-8-oxodG were added to DNA before the digestion. Spiroiminodihydantoin and 8-oxodG were purified and quantitated as described above.
Results
We studied the formation in vitro and quantitation of four important DNA damage products - 8-oxodG, oxazolone, spiroiminodihydantoin, and guanidinohydantoin - arising from treatment of calf thymus DNA with ONOO− via bolus addition. To ensure reliable quantitation, we synthesized 13C, 15N labeled standards for each of the four analytes (26). Calf thymus DNA was reacted with ONOO−, desalted, and digested down to the nucleoside level using a cocktail of enzymes. The presence of a particular oxidation product in the ONOO− treated DNA was determined by HPLC purification followed by LC-MS/MS analysis in SRM mode. The optimized HPLC-ESI-MS/MS conditions allowed detection at femtomole levels (on-column) for all four oxidation products.
Analysis of 8-oxodG
The uniformly 13C, 15N labeled 8-oxodG was synthesized with a 10% yield. Its identity was confirmed by its characteristic UV spectrum (λmax= 248 and 292 nm) and by ESI-MS, which gave a protonated molecule at m/z 299 ([M+H+]+). Two fragment ions were observed, at m/z 122 ([M+H+-178]) and m/z 178 ([BH+H]+), corresponding to the protonated 13C5-deoxyribose and 13C5, 15N5-8-oxoguanine(see supporting information Figure 2). The amount of 8-oxodG formed in calf thymus DNA treated with 0, 0.011, 0.022, 0.055, 0.11, 0.22, 0.55, 1.1, 2.2 and 4.4 μmol of ONOO− (corresponding to 0, 0.1, 0.2, 0.5, 1, 2, 5, 10, 20 and 40 equivalents of the guanine content in DNA) was determined by HPLC-MS/MS. To minimize formation of 8-oxodG from metal-catalyzed oxidation of dG during the DNA digestion and work-up, 0.1 mM of desferrioxamine, a metal chelator, was added to the digestion mixture (34). To evaluate its effectiveness, 75 nmol of 7-15N-dG (~ the same amount as dG obtained from complete digestion of 117 μg of DNA) was added to DNA before the digestion. The labeled dG served two purposes, i.e., 1.) as an internal standard for LC-MS/MS analysis of dG, and 2.) to calculate the amount of 8-oxodG that formed adventitiously from dG during the overall experiment, assuming that 7-15N-dG would be oxidized to the same extent as dG during DNA digestion and work-up. During the MS/MS analysis of 8-oxodG, SRM transitions of m/z 284 → m/z 168, m/z 285 → m/z 169, m/z 287 → m/z 171 (or m/z 299 → m/z 178) were used to quantitate 8-oxodG and 7-15N-8-oxodG with 1,2,7-15N3-8oxo-dG (or the uniformly 13C, 15N labeled 8-oxodG) as the internal standard (supporting information Figure 10). The presence of a significant amount of the m/z 285 → m/z 169 transition demonstrated that 7-15N-dG was oxidized to 7-15N-8-oxodG during the digestion and work-up despite the presence of desferrioxamine in the digestion mixture, suggesting in turn that artifactual oxidation of dG to 8-oxodG also occurred. The amount of dG obtained from DNA digestion was quantitated using LC-MS/MS with 7-15N-dG as the internal standard. SRM transitions of m/z 268 → m/z 152 and m/z 269 → m/z 153 were used to measure dG and 7-15N-dG, respectively. With the molar ratio between dG and 7-15N-dG and the amount of 7-15N-8-oxodG known, the artifactual 8-oxodG was calculated according to the equation: 8-oxodG artifacts = 7-15N-8-oxodG * molar ratio (dG/7-15N-dG); this amounted to approximately 5 8-oxodG/106 nucleosides. There is some uncertainty with respect to this value due to the contribution of the 13C isotope peak from the unlabeled 8-oxoG to the monoisotopic 12C signal from the labeled standard. Since this effect would be at most about 6% for a 1:1 mixture of the two compounds (1.1% 13C for each of the 5 carbons in the fragment ion), we did not apply a correction. The amount of authentic 8-oxodG in calf thymus DNA resulting from ONOO− treatment was calculated by subtracting the artifacts from the measured 8-oxodG values. The overall trend for the formation of 8-oxodG was dose-dependent except at 5 equivalents of ONOO−, at which the 8-oxodG level was lower than for either 2 equivalents or 10 equivalents of ONOO− (Figure 1).
Figure 1.

The amount of 8-oxodG formed in calf thymus DNA treated with 0, 0.011, 0.022, 0.055, 0.11, 0.22, 0.55, 1.1, 2.2 and 4.4 μmol of ONOO− was determined by HPLC-MS/MS. To minimize formation of 8-oxodG from metal-catalyzed oxidation of dG during the DNA digestion and work-up, 0.1 mM of desferrioxamine, a metal chelator, was added to the digestion mixture (34). Despite this precaution, artifactual 8-oxodG was observed, and the levels were estimated as described earlier. The amount of authentic 8-oxodG in calf thymus DNA resulting from ONOO− treatment was calculated by subtracting the artifacts from the measured 8-oxodG values. The data points represent duplicate analyses.
Analysis of oxazolone
The uniformly 13C, 15N labeled oxazolone and 3H-oxazolone were synthesized as described in materials and methods. Their identities were confirmed by their UV spectra (λ max=232 nm) and by comparing their HPLC retention times to that of the unlabeled oxazolone. ESI-MS of the uniformly 13C, 15N labeled oxazolone gave the [M+H+]+ ion at m/z 259 along with diagnostic fragment ions at m/z 214 ([M-CO2+H]+), m/z 138 (protonated 13C3, 15N4-oxazolone base) and m/z 122 (protonated 13C5-deoxyribose). The molar extinction coefficient for the oxazolone nucleoside was measured at 0.009 M−1 cm−1 (lambda; max=232 nm in water). Artifacts of oxazolone formed from oxidation of 8-oxodG during the DNA digestion and work-up were evaluated by adding 1,2,7-15N3-8oxodG to calf thymus DNA before the digestion and by monitoring the transitions of m/z 247 → m/z 131 (oxazolone), m/z 249 → m/z 133 (2,2-15N2-oxazolone), and m/z 259 → m/z 138 (the uniformly 13C, 15N labeled oxazolone) during the MS/MS analysis (supporting information Figure 11). Only the transition corresponding to the internal standard (m/z 259 → m/z 138) was observed, i.e., the background levels of oxazolone in calf thymus DNA and the artifactual formation of oxazolone from 8-oxodG during DNA digestion and work-up were both below the detection limit of 1 oxazolone/107 bases. The amount of oxazolone formed in calf thymus DNA treated with 0, 0.11, 0.55, 1.1, 2.2 and 4.4 μmol of ONOO− (corresponding to 0, 1, 5, 10, 20 and 40 equivalents of the guanine content in DNA) was determined using LC-MS/MS with the 13C, 15N labeled oxazolone as the internal standard. That oxazolone was not detected in untreated calf thymus DNA also suggests that oxidation of dG to oxazolone during the DNA digestion and work-up is relatively insignificant. Figure 2 illustrates the dose-dependent formation of oxazolone from calf thymus DNA treated with different amounts of ONOO−.
Figure 2.
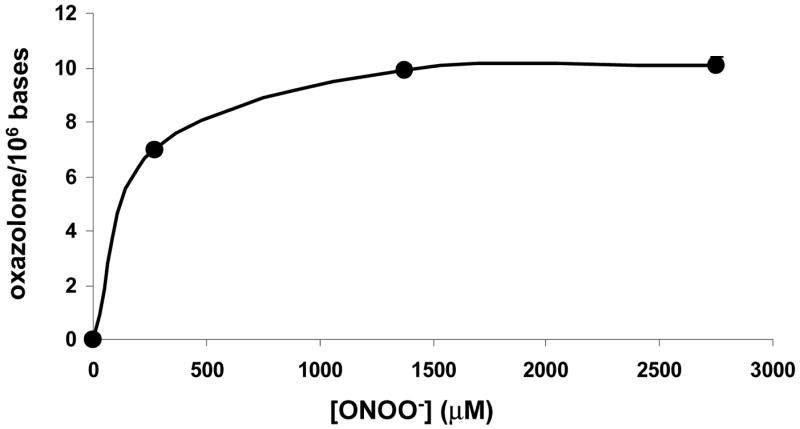
Formation of oxazolone in calf thymus DNA treated with 0, 275, 1375, and 2750 μM ONOO−. Analysis was by LC-MS/MS with the 13C, 15N labeled oxazolone as the internal standard. The data points represent duplicate analyses; the error bars fall within the data markers. Oxazolone was not detected in untreated calf thymus DNA, suggesting that oxidation of dG to oxazolone during the DNA digestion and work-up is relatively insignificant. The data points represent duplicate analyses.
Analysis of spiroiminodihydantoin
The 13C, 15N labeled spiroiminodihydantoin and 3H-spiroiminodihydantoin were synthesized as described in materials and methods. Identities of the labeled compounds were confirmed by UV spectra and HPLC retention times. ESI-MS of the 13C, 15N labeled spiroiminodihydantoin gave a protonated molecule at m/z 315 ([M+H+]+), along with characteristic fragment ions including m/z 194 (protonated 13C5, 15N5-spiroiminodihydantoin base) and m/z 122 (protonated 13C5-deoxyribose). The molar extinction coefficient for the spiroiminodihydantoin nucleoside was determined to be 0.0105 M−1cm−1 at λ max=230 nm, which is over two-fold higher than the reported value of 4.3 mM−1cm−1 (31). The amount of spiroiminodihydantoin formed in calf thymus DNA treated with 0, 0.011, 0.022, 0.055, 0.11 and 1.1 μmol of ONOO− (corresponding to 0, 0.1, 0.2, 0.5, 1 and 10 equivalents of the guanine content in DNA) was determined by LC-MS/MS with 128 pmol of the 13C, 15N labeled spiroiminodihydantoin as the internal standard. Artifacts of spiroiminodihydantoin formed from 8-oxodG during DNA digestion and work-up were evaluated by adding 1,2,7-15N3-8-oxodG and the uniformly 13C, 15N labeled spiroiminodihydantoin to DNA before the digestion and by subsequently monitoring SRM transitions of m/z 300 → m/z 184 (spiroiminodihydantoin), m/z 303 → m/z 187 (15N3-spiroiminodihydantoin from oxidation of 1,2,7-15N3-8-oxodG), and m/z 315 → m/z 194 (the uniformly 13C, 15N labeled spiroiminodihydantoin) (supporting information Figure 12). There were significant SRM signals of both spiroiminodihydantoin and 15N3-spiroiminodihydantoin. The amount of15N3-spiroiminodihydantoin formed during the digestion and work-up was 3.7 ± 0.3 mol% of 1,2,7-15N3-8-oxodG added to the digestion mixture. Assuming that 8-oxodG would be oxidized to the same extent as the isotopomer, the amount of spiroiminodihydantoin from artifactual oxidation of 8-oxodG was calculated as artifacts = 0.037 * 8-oxodG. The authentic spiroiminodihydantoin was calculated by subtracting the artifacts from the measured spiroiminodihydantoin values. Figure 3 depicts spiroiminodihydantoin formed from DNA treated with different amounts of ONOO−. Although it was formed in a dose-dependent manner at low ONOO− doses (ONOO− up to 0.2 equivalent of guanine content in DNA), its formation dropped significantly at higher ONOO− doses.
Figure 3.

Formation of spiroiminodihydantoin formed in calf thymus DNA treated with varying amounts of peroxynitrite. Quantitation was by LC-MS/MS with 13C, 15N labeled spiroiminodihydantoin as the internal standard. Artifactual formation was evaluated by adding 1,2,7-15N3-8-oxodG and uniformly 13C, 15N labeled spiroiminodihydantoin to DNA before the digestion and by subsequently monitoring SRM transitions of m/z 300 → m/z 184 (spiroiminodihydantoin), m/z 303 → m/z 187 (15N3-spiroiminodihydantoin from oxidation of 1,2,7-15N3-8-oxodG), and m/z 315 → m/z 194 (the uniformly 13C, 15N labeled spiroiminodihydantoin). The data points represent duplicate analyses.
Analysis of guanidinohydantoin
Synthesis of 3,7,8-15N3-5-guanidinohydantoin, starting with 1,7,NH2-15N3-2’-deoxyguanosine, is described in detail elsewhere (26). The amount of guanidinohydantoin formed in calf thymus DNA treated with 0, 0.011, 0.022, 0.055, 0.22 and 2.2 μmol of ONOO− (corresponding to 0, 0.1, 0.2, 0.5, 2 and 20 equivalents of the guanine content in DNA) was determined by LC-MS/MS with 40.5 pmol of 3,7,8-15N3-5-guanidinohydantoin as the internal standard. Artifactual formation of guanidinohydantoin was evaluated by adding the 13C, 15N labeled 8-oxodG and 3,7,8-15N3-5-guanidinohydantoin to DNA before the digestion and by subsequently monitoring SRM transitions of m/z 274 → m/z 158 (guanidinohydantoin), m/z 288 → m/z 167 (the uniformly 13C, 15N labeled guanidinohydantoin from oxidation of the uniformly 13C, 15N labeled 8-oxodG), and m/z 277 → m/z 161 (3,7,8-15N3-5-guanidinohydantoin) (supporting information Figure 14). Significant amounts of guanidinohydantoin and its 13C, 15N isotopomer were detected. The 13C, 15N labeled guanidinohydantoin formed during DNA digestion and work-up was 0.6 ± 0.06 mol% of the uniformly 13C, 15N labeled 8-oxodG added to DNA before the digestion. Based on the assumption that 8-oxodG would be oxidized to the same extent as the labeled 8-oxodG in the digestion mixture, the amount of artifactual guanidinohydantoin was calculated according to the equation: artifacts = 0.006 * 8-oxodG, and the authentic guanidinohydantoin was calculated by subtracting the artifacts from the measured guanidinohydantoin values. Figure 4 depicts guanidinohydantoin formed from DNA treated with different amounts ONOO−. As with spiroiminodihydantoin, formation of guanidinohydantoin was dose-dependent at low ONOO− doses (ONOO− up to 0.2 equivalent of total guanine in DNA) and its formation dropped significantly to the basal level at high ONOO− doses.
Figure 4.

The levels of guanidinohydantoin formed in calf thymus DNA treated with varying guanidinohydantoin as the internal standard. Artifactual formation of guanidinohydantoin was amounts of ONOO− were determined by LC-MS/MS with 40.5 pmol of 3,7,8-15N3-5-evaluated by adding the 13C, 15N labeled 8-oxodG and 3,7,8-15N3-5-guanidinohydantoin to DNA before the digestion and by subsequently monitoring SRM transitions of m/z 274 m/z 158 (guanidinohydantoin), m/z 288 m/z 167 (the uniformly 13C, 15N labeled guanidinohydantoin from oxidation of the uniformly 13C, 15N labeled 8-oxodG), and m/z 277 → m/z 161 (3,7,8-15N3-5-guanidinohydantoin). The data points represent duplicate analyses.
Yields of 8-oxodG and spiroiminodihydantoin from different digestion methods
Cadet, et al., reported that enzymatic hydrolysis of phosphodiester bonds between 4,8-dihydro-4-hydroxy-8-oxo-dG (later to be corrected as spiroiminodihydantoin (13) and normal 2’-deoxyribonucleosides in oligonucleotides was inhibited with both endonucleases and exonucleases (33). In order to evaluate the effects of various enzymes on the formation of 8-oxodG and spiroiminodihydantoin, untreated calf thymus DNA was digested with 1.) nuclease P1 and white potato acid phosphatase; 2.) nuclease P1, phosphodiesterase I and alkaline phosphatase; 3.) phosphodiesterase II (3’-exo digestion) and alkaline phosphatase; and 4.) phosphodiesterase I (5’-exo digestion) and alkaline phosphatase. The uniformly 13C, 15N labeled spiroiminodihydantoin and 1,2,7-15N-8-oxodG were added to the DNA before digestion. The results are summarized in Table 1. Surprisingly, the yields of spiroiminodihydantoin were similar among the various enzyme combinations. The yield of 8-oxodG from acidic digestion, however, was significantly lower than that from other enzymatic digestion methods.
Table 1.
Yields of Spiroiminodihydantoin and 8-OxodG by Various Enzymatic Digestion Methods
| Enzymes | DNA digestion yield | 8-oxodG /106 bases | Spiroiminodi-hydantoin/106 bases |
|---|---|---|---|
| NP1/acid phosphatase | 83 ± 1.5% | 4 ± 0.7 | 10 ± 0.4 |
| NP1/AP (12 h) | 87 ± 0.3% | 15 ± 1.8 | 9 ± 0.6 |
| NP1/AP (24 h) | 90 ± 1% | 15 ± 2.0 | 9 ± 0.7 |
| phosphodiesterase I (5’-exo)/AP | 71 ± 8% | 12 ± 1.7 | 8 ± 0.8 |
| phohphodiesterase II (3’-exo)/AP | 36 ± 4% | 14 ± 1.9 | 10.8 ± 1.2 |
AP = alkaline phosphatase, NP1= nuclease P1
Discussion
The reaction of ONOO− with dG yields a number of primary and secondary oxidation products including 8-oxodG, oxazolone, spiroiminodihydantoin and guanidinohydantoin. Indications that these four products may also be formed from oxidation of guanine base in DNA or oligonucleotides have emerged during the past decade. 8-OxodG is a ubiquitous DNA oxidation product both with isolated DNA and cellular DNA (35). The level of 8-oxodG has been reported to increase dose-dependently in calf thymus DNA treated with singlet O2 (36). Oxazolone and its precursor imidazolone have been observed as oxidation products of guanine or 8-oxoguanine in oligonucleotides and calf thymus DNA treated with Mn-TMPyP/KHSO5 (37), ONOO− (11, 18), photochemical oxidants (38–41), and γ-radiation (11). Spiroiminodihydantoin has been observed as the final oxidation product of guanine base (likely via 8-oxoguanine) in oligonucleotides by carbonate radical anions (42) and by chromium species (43). Guanidinohydantoin has been detected in DNA treated with peroxo-Cr(V) (44) or in oligonucleotides containing 8-oxoguanine (45). Cadet and co-workers reported that the levels of oxazolone and 8-oxodG increased and decreased, respectively, in calf thymus DNA treated with ONOO− (11). However, neither spiroiminodihydantoin nor guanidinohydantoin in calf thymus DNA treated with ONOO− has been reported. In this paper we describe the identification and quantitation, by LC-MS/MS, of 8-oxodG, oxazolone, spiroiminodihydantoin and guanidinohydantoin in calf thymus DNA following treatment with ONOO−. Detection of these oxidation products in vitro is a necessary first step toward profiling various ONOO− related lesions that may be formed in vivo. Oxazolone, guanidinohydantoin and spiroiminodihydantoin have been shown in various studies to be strong blocking and/or highly mutagenic lesions (24, 25). Future research may reveal that the increased cancer risks associated with excess ONOO− production can be directly linked to the formation of specific lesions in DNA.
LC-MS/MS was chosen to detect and quantitate these DNA oxidation products because it combines the power of on-line purification by liquid chromatography with the specificity and sensitivity of tandem mass spectrometry. The SRM mode was used because all of the four products share the characteristic fragmentation pattern of splitting of the N-glycosidic bond from the pseudo-molecular ions ([M+H+]+) of the analytes. The [M+H+]+ ions were selected in Q1, fragmented in Q2, and the protonated bases ([BH+H+]+) of the analytes were selected in Q3. This specific and sensitive assay allowed a detection limit of 1 lesion/107 bases for all four DNA oxidation products. All the stable-isotope labeled standards of the four DNA oxidation products were tested to assure that no false positives were introduced by the internal standards.
Artifactual formation of 8-oxodG during DNA digestion and work-up is common (46). It was reported that the presence of desferrioxamine during digestion would greatly reduce fenton-type oxidation of dG (47). Our results confirm that this reduction is not complete. Under our DNA digestion and work-up conditions, artifactual 8-oxodG was about 5/106 nucleosides. Artifacts of oxazolone, spiroiminodihydanotin and guanidinohydantoin could also emerge during the DNA digestion and work-up, from oxidation of dG or 8-oxodG or both. This possibility was assessed by LC/MS using the various SRM transitions described earlier. No 2,2-15N2-Oxazolone was detected, suggesting little or no artifactual formation of oxazolone. 15N3-spiroiminodihydantoin and the uniformly 13C, 15N labeled guanidinohydantoin, however, were detected at 3.7 mol% and 0.6 mol% of the corresponding labeled 8-oxodG, respectively. In fact, the formation of these two compounds from oxidation of 8-oxodG during the DNA digestion and work-up, with preferential formation of spiroiminodihydantoin at pH~7, was not surprising (31,48,49) and it is clear that care must be taken during their analysis to avoid false positives.
The dose-response studies of the formation of 8-oxodG, oxazolone, spiroiminodihydantoin and guanidinohydantoin in DNA treated with ONOO− provided better understanding of the mechanism of formation of these lesions in DNA. The basal level of 8-oxodG in calf thymus DNA determined by us was approximately 12 8-oxoguanine/106 bases (or about 50 8-oxoguanine/106 guanine bases), which is similar to literature values (11 8-oxoguanine/106 bases (36) or 61.6 8-oxoguanine/106 guanine bases (50). The amount of 8-oxodG increased up to 2 equivalents of ONOO− treatment and then began to decrease until 10 equivalents of ONOO− treatment, after which it increased again in a dose-dependent fashion. This behavior could be due to differences, at each ONOO− dose, in the competition between the accumulation of 8-oxoguanine from oxidation of guanine by ONOO− and further reaction of 8-oxoguanine with ONOO− to its secondary oxidation products.
It was somewhat surprising that the background level of oxazolone in calf thymus DNA was below the detection limit of 1 lesion/107 bases. Since it is a stable end-product from reaction of dG and ONOO−, its formation in a dose-dependent manner in DNA is expected, e.g., Douki and Cadet have also shown that oxazolone is produced from the reaction of calf thymus DNA with peroxynitrite (11).
Spiroiminodihydantoin and guanidinohydantoin, both considered as secondary oxidation products from the oxidation of 8-oxodG (13,19,31,49), were formed in DNA in a dose-dependent manner at low ONOO− doses with their levels increasing up to 0.2 equivalents of ONOO− treatment. Their formation, however, dropped significantly at high ONOO− doses (> 10 equivalents of ONOO− treatment). This could be explained as follows (Scheme 2): 8-oxodG undergoes two one-electron oxidations to a quinonoid intermediate (I). At low ONOO− doses, water (the predominant nucleophile) reacts with I to form II, which then undergoes decomposition to spiroiminodihydantoin or guanidinohydantoin. At higher ONOO− doses, however, ONOO− may also act as a nucleophile and react with I to afford III, which could then undergo decomposition to oxazolone, oxaluric acid or cyanuric acid (19). The background levels of spiroiminodihydantoin and guanidinohydantoin in calf thymus DNA were 12/ 106 bases and 4/106 bases, respectively, perhaps due to oxidation of pre-existing 8-oxoguanine in DNA.
Scheme 2.
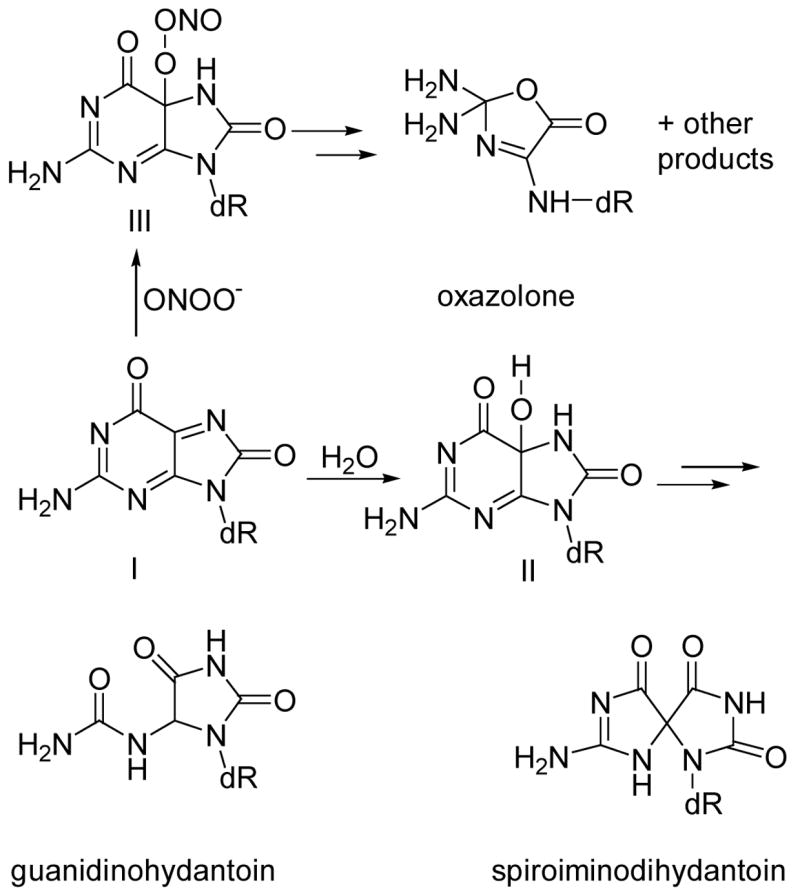
Secondary oxidation products of 8-oxodG
Conclusion
In conclusion, we have quantitated 8-oxodeoxyguanosine, oxazolone, guanidinohydantoin and spiroiminodihydantoin from peroxynitrite-treated calf thymus DNA. The formation of these compounds demonstrates that ONOO− inflicts complex oxidative damage to guanine bases in DNA in vitro and suggests that elevated concentrations of ONOO− may induce oxidative damage to DNA under inflammatory conditions in vivo. Efforts to detect these lesions in DNA from tissues of mouse models of inflammation are underway.
Supplementary Material
Representative LC/MS and LC/MS/MS chromatograms, mass spectra, UV spectra, and calculation methods.
Acknowledgments
Thanks to NIH (Grant No. CA26371 the MIT Center for Environmental Health Sciences (Grant No. ES002109) for financial support. We thank Dr. Julie Marr (Agilent) for her insight and technical assistance with the LC-MSD-TOF, and Agilent for graciously providing full access to the instrument.
Abbreviations
- ONOO−
peroxynitrite
- dG
2’-deoxyguanosine
- 8-oxodG
8-oxo-7, 8 dihydro-2’-deoxyguanosine
- oxazolone
2,2-diamino-4[(2-deoxy-β-D-erythro-pentafuranosyl)amino]-5(2H)-oxazolone
- imidazolone
2-amino-5-[2-deoxy-β-D-erythro-pentafuranosyl)amino]-4H-imidazol-4-one
- guanidinohydantoin
N1-(β-D-erythro-pentofuranosyl)-5-guanidinohydantoin
- dGTP
2’-deoxyguanosine triphosphate
- SRM
selected reaction monitoring
References
- 1.Beckman JS. Oxidative Damage and Tyrosine Nitration by Peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 2.Huie RE, Padmaja S. The reaction of NO with superoxide. Free Rad Res Comm. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 3.Szabo C, Zingarelli B, O'Connor M, Salzman AL. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaur H, Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation. Nitrotyrosine in serum synovial fluid from rheumatoid patients. FEBS Lett. 1994;350:9–12. doi: 10.1016/0014-5793(94)00722-5. [DOI] [PubMed] [Google Scholar]
- 5.Roediger WEW, Lawson MJ, Radcliffe BC. Nitrite from inflammatory cells-a cancer risk factor in ulcerative colitis? Dis Colon Rectum. 1990;33:1034–1036. doi: 10.1007/BF02139219. [DOI] [PubMed] [Google Scholar]
- 6.Salgo MG, Stone K, Squadrito GL, Battista JR, Pryor WA. Peroxynitrite causes DNA nicks in plasmid pBR322. Biochem Biophys Res Comm. 1995;210:1025–1030. doi: 10.1006/bbrc.1995.1759. [DOI] [PubMed] [Google Scholar]
- 7.Szabo C, Ohshima H. DNA damage induced by peroxynitrite: subsequent biological effects. Nitric Oxide: Biol Chem. 1997;1:373–385. doi: 10.1006/niox.1997.0143. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy LJ, Moore K, Jr, Caulfield JL, Tannenbaum SR, Dedon PC. Quantitation of 8-oxoguanine and strand breaks produced by four oxidizing agents. Chem Res Toxicol. 1997;10:386–392. doi: 10.1021/tx960102w. [DOI] [PubMed] [Google Scholar]
- 9.Burney S, Niles JC, Dedon PC, Tannenbaum SR. DNA damage in deoxynucleosides and oligonucleotides treated with peroxynitrite. Chem Res Toxicol. 1999;12:513–520. doi: 10.1021/tx980254m. [DOI] [PubMed] [Google Scholar]
- 10.Burney S, Caulfield JL, Wishnok JS, Tannenbaum SR. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res. 1999;424:37–49. doi: 10.1016/s0027-5107(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 11.Douki T, Cadet J. Peroxynitrite mediated oxidation of purine bases of nucleosides and isolated DNA. Free Radic Res. 1996;24:369–380. doi: 10.3109/10715769609088035. [DOI] [PubMed] [Google Scholar]
- 12.Niles JC, Burney S, Singh S, Wishnok JS, Tannenbaum SR. Peroxynitrite reaction products of 3',5'-di-O-acetyl-8-oxo-7,8-dihydro-2'-deoxyguanosine. Proc Natl Acad Sci U S A. 1999;96:11729–11734. doi: 10.1073/pnas.96.21.11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene blue. Org Lett. 2001;3:963–966. [PubMed] [Google Scholar]
- 14.Yermilov V, Yoshie Y, Rubio J, Ohshima H. Effects of carbon dioxide/bicarbonate on induction of single-strand breaks and formation of 8-nitroguanine, 8-oxoguanine, and base propenal mediated by peroxynitrite. FEBS Lett. 1996;399:67–70. doi: 10.1016/s0014-5793(96)01288-4. [DOI] [PubMed] [Google Scholar]
- 15.Cadet J, Berger M, Buchko GW, Joshi PC, Raoul S, Ravanat JL. 2,2-Diamino-4-[(3,5-di-O-acetyl-2-deoxy-beta-D-erythro- pentofuranosyl)amino]-5-(2H)-oxazolone: a novel and predominant radical oxidation product of 3',5'-Di-O-acetyl-2'-deoxyguanosine. J Am Chem Soc. 1994;116:7403–7404. [Google Scholar]
- 16.Raoul S, Berger M, Buchko GW, Joshi PC, Morin B, Weinfeld M, Cadet J. Novel oxidation products of deoxyguanosine:oxazolone and imidazolone nucleosides. J Chem, Soc Perkin Trans 2. 1996;2:371–381. [Google Scholar]
- 17.Uppu RM, Cueto R, Squadrito GL, Salgo MG, Pryor WA. Competitive reactions of peroxynitrite with 2'-deoxyguanosine and 7,8-dihydro-8-oxo-2'-deoxyguanosine (8-oxodG): Relevance to the formation of 8-oxodG in DNA exposed to peroxynitrite. Free Rad Biol Med. 1996;21:407–411. doi: 10.1016/0891-5849(96)00220-1. [DOI] [PubMed] [Google Scholar]
- 18.Tretyakova NY, Niles JC, Burney S, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligonucleotides containing 8-oxoguanine. Chem Res Toxicol. 1999;12:459–466. doi: 10.1021/tx980235c. [DOI] [PubMed] [Google Scholar]
- 19.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin and guanidinohydantoin are the dominant reaction products of 8-oxoguanosine oxidation at low fluxes of peroxynitrite: mechanistic studies with 18O. Chem Res Toxicol. 2004;17:1510–1519. doi: 10.1021/tx0400048. [DOI] [PubMed] [Google Scholar]
- 20.Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionization radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 21.Moriya M, Ou C, Bodepudi V, Johnson F, Takeshita M, Grollman AP. Site-specific mutagenesis using a gapped duplex vector: a study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat Res. 1991;254:281–288. doi: 10.1016/0921-8777(91)90067-y. [DOI] [PubMed] [Google Scholar]
- 22.Klein JC, Bleeker MJ, Lutgerink JT, Van BJ, Brugghe HF, Van DE, Van der Marcel GA, Van BJ, Westra JG, Berns AJ. Use of shuttle vectors to study the molecular processing of defined carcinogen-induced DNA damage: mutagenicity of single O4-ethylthymine adducts in HeLa cells. Nucleic Acids Res. 1990;18:4131–4137. doi: 10.1093/nar/18.14.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duarte V, Gasparutto D, Jaquinod M, Cadet J. In vitro DNA synthesis opposite oxazolone and repair of this DNA damage using modified oligonucleotides. Nucleic Acids Res. 2000;28:1555–1563. doi: 10.1093/nar/28.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson PT, Delaney JC, Gu F, Tannenbaum SR, Essigmann JM. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry. 2002;41:914–921. doi: 10.1021/bi0156355. [DOI] [PubMed] [Google Scholar]
- 25.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Wishnok JS, Tannenbaum SR. Synthesis of 3,7,8-15N3-(beta-D-erythropentafuranosyl)-5-guanidinohydantoin. J Label Cmpds Radiopharm. 2003;46:1269–1277. [Google Scholar]
- 27.Pryor WA, Cueto R, Jin X, Koppenol WH, Ngu-Schwemlein M, Squadrito GL, Uppu PL, Uppu RM. A practical method for preparing peroxynitrite solutions of low ionic strength and free of hydrogen peroxide. Free Rad Biol Med. 1995;18:75–83. doi: 10.1016/0891-5849(94)00105-s. [DOI] [PubMed] [Google Scholar]
- 28.Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984;12:2137–2145. doi: 10.1093/nar/12.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasparutto D, Ravanat JL, Gérot O, Cadet J. Characterization and chemical stability of photooxidized oligonucleotides that contain 2,2-diamino-4-[(2-deoxy-D-erythro-pentofuranosyl)amino]-5(2H)-oxazolone. J Am Chem Soc. 1998;120:10283–10286. [Google Scholar]
- 30.Ravanat J, Cadet J. Reaction of singlet oxygen with 2'-deoxyguanosine and DNA isolation and characterization of the main oxidation products. Chem Res Toxicol. 1995;8:379–388. doi: 10.1021/tx00045a009. [DOI] [PubMed] [Google Scholar]
- 31.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 32.Hughes MN, Nicklin HG. The chemistry of pernitrites. Part I Kinetics of decomposition of pernitrous acid. J Chem Soc. 1968:450–452. [Google Scholar]
- 33.Romieu A, Gasparutto D, Molko D, Ravanat JL, Cadet J. Synthesis of oligonucleotides containing the (4R) and (4S) diastereoisomers of 4,8-dihydro-4-hydroxy-8-oxo-2'-deoxyguanosine. Eur J Org Chem. 1999;1:49–56. [Google Scholar]
- 34.Ravanat JL, Douki T, Duez P, Gremaud E, Herbert K, Hofer T, Lasserre L, Saint-Pierre C, Favier A, Cadet J. Cellular background level of 8-oxo-7,8-dihydro-2'-deoxyguanosine: an isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis. 2002;23:1911–1918. doi: 10.1093/carcin/23.11.1911. [DOI] [PubMed] [Google Scholar]
- 35.Helbock HJ, Beckman KB, Ames BN. 8-Hydroxy-deoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzymol. 1999;300:156–166. doi: 10.1016/s0076-6879(99)00123-8. [DOI] [PubMed] [Google Scholar]
- 36.Ravanat JL, Saint-Pierre C, Di Mascio P, Martinez G, Medeiros MHG, Cadet J. Damage to isolated DNA mediated by singlet oxygen. Helv Chim Acta. 2001;84:3702–3709. [Google Scholar]
- 37.Vialas C, Claparos C, Pratviel G, Meunier B. Guanine oxidation in double-stranded DNA by Mn-TMPyP/KHSO5: 5,8-dihydroxy-7,8-dihydroguanine residue as a key precursor of imidazolone and parabanic acid derivatives. J Am Chem Soc. 2000;122:2157–2167. [Google Scholar]
- 38.Adam W, Grimm GN, Saha-Moeller CR, Dall'Acqua F, Miolo G, Vedaldi D. DNA damage by tert-butoxyl radicals generated in the photolysis of a water-soluble, DNA-binding peroxyester acting as a radical source. Chem Res Toxicol. 1998;11:1089–1097. doi: 10.1021/tx980089a. [DOI] [PubMed] [Google Scholar]
- 39.Adam W, Arnold MA, Grimm GN, Saha-Moeller CR, Dall'Acqua F, Miolo G, Vedaldi D. 4-tert-Butylperoxymethyl-9-methoxypsoralen as intercalating photochemical alkoxyl-radical source for oxidative DNA damage. Photochem Photobiol. 1998;68(4):511–518. [PubMed] [Google Scholar]
- 40.Adam W, Saha-Moeller CR, Schoenberger A. Photooxidation of 8-oxo-7,8-dihydro-2'-deoxyguanosine by thermally generated triplet-excited ketones from 3-(hydroxymethyl)-3,4,4-trimethyl-1,2-dioxetane and comparison with Type I and Type II photosensitizers. J Am Chem Soc. 1996;118:9233–9238. [Google Scholar]
- 41.Adam W, Andler S, Saha-Moeller CR, Schoenberger A. Inhibitory effect of ethyl oleate hydroperoxide and alcohol in photosensitized oxidative DNA damage. J Photochem Photobiol B: Biology. 1996;34:51–58. doi: 10.1016/1011-1344(95)07235-7. [DOI] [PubMed] [Google Scholar]
- 42.Joffe A, Geacintov NE, Shafirovich V. DNA lesions derived from the site selective oxidation of guanine by carbonate radical anions. Chem Res Toxicol. 2003;16:1528–1538. doi: 10.1021/tx034142t. [DOI] [PubMed] [Google Scholar]
- 43.Sugden KD, Campo CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem Res Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 44.Joudah L, Moghaddas S, Bose RN. DNA oxidation by peroxo-chromium(V) species: oxidation of guanosine to guanidinohydantoin. Chem Comm. 2002:1742–1743. doi: 10.1039/b205407h. [DOI] [PubMed] [Google Scholar]
- 45.Duarte V, Muller JG, Burrows CJ. Insertion of dGMP and dAMP during in vitro DNA synthesis opposite an oxidized form of 7,8-dihydro-8-oxoguanine. Nucleic Acids Res. 1999;27:496–502. doi: 10.1093/nar/27.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halliwell B. Why and how should we measure oxidative DNA damage in nutritional studies? How far have we come? Am J Clin Nutr. 2000;72:1082–1087. doi: 10.1093/ajcn/72.5.1082. [DOI] [PubMed] [Google Scholar]
- 47.Helbock HJ, Beckman KB, Shigenaga MK, Walter PB, Woodall AA, Yeo HC, Ames BN. DNA oxidation matters: The HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proc Natl Acad Sci U S A. 1998;95:288–293. doi: 10.1073/pnas.95.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo W, Muller JG, Burrows CJ. The pH-dependent role of superoxide in riboflavin-catalyzed photooxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 49.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 50.Collins A, Gedik C, Wood S, White A, Dubois J, Duez P, Rees JF, Legall R, Degand L, Loft S, Jensen A, Poulsen H, Weimann A, Jensen B, Cadet J, Douki T, Ravanat JL, Faure H, Tripier M, Morel I, Sergent O, Cillard P, Morin B, pe B, Phoa N, Hartwig A, Pelzer A, Dolara P, Casalini C, Guglielmi F, Luceri C, Kasai H, Kido R, Olinski R, Bialkowski K, Durackova Z, Hlincikova L, Korytar P, Dusinska M, Mislanova C, Vina J, Lloret A, Moller L, Hofer T, Gremaud E, Fay L, Stadler R, Eakins J, Pognan F, O'Brien J, Elliott R, Astley S, Bailley A, Herbert K, Chauhan D, Kelly F, Dunster C, Lunec J, Podmore I, Patel P, Johnson S, Evans M, White A, Tyrrell R, Gordon M, Wild C, Hardie L, Smith E. Inter-laboratory validation of procedure for measuring 8-oxo-7,8-dihydroguanine/8-oxo-7,8-dihydro-2'-deoxyguanosine in DNA. Free Rad Res. 2002;36:239–245. doi: 10.1080/10715760290019246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative LC/MS and LC/MS/MS chromatograms, mass spectra, UV spectra, and calculation methods.