

Abstract
Two dicopper(I) complexes containing tertiary N-methylated hexaaza ligands which impose different steric constrains to the Cu ions have been synthetized, and their reactivity towards O2 has been compared with a mononuclear related system, highlighting the importance of cooperative effects between the metal centers in O2 activation.
A number of proteins involved in dioxygen transport and activation contains a dinuclear Cu active site.1 The arguably best known are hemocyanin, catechol oxidase and tyrosinase, which react with O2 generating O2-bound species which have been spectroscopically and structurally characterized.1,2 Traditionally, the mimicking of these dicopper centers was performed either using mononuclear complexes that self-assemble when reacting with O2, or via dinucleating ligands designed with the purpose of favoring O2 binding by spatial pre-organization of the dimetallic site.3 Particularly remarkable are m-xylyl linked dinuclear Cu(I) complexes which react with O2 to form well-characterized (μ-η2: η2-peroxo)dicopper(II) species that in selected cases undergo intra-4 or intermolecular5 regioselective hydroxylation of an aromatic ring, thus mimicking tyrosinase activity.
Here, we report the reactivity with O2 of two related dinuclear copper(I) complexes supported by two hexaaza ligands (L16 and L2, scheme 1) based on a m-xylyl spacer, and compare it with the one recently reported for the mononuclear analogue [Cu(MeAN)]B(C6F5)4 (MeAN = N, N, N′, N′, N″-pentamethyldipropylenetriamine).7 The three complexes contain ligands that bind copper atoms within very similar coordination environments and give rise to electronically and structurally comparable metal sites. However, they exhibit rather unexpected differences in their reaction with O2 that may be understood on the basis of the relative ability of the ligand to promote a synergistic actuation of the two Cu ions along the O2 binding process.
Scheme 1.
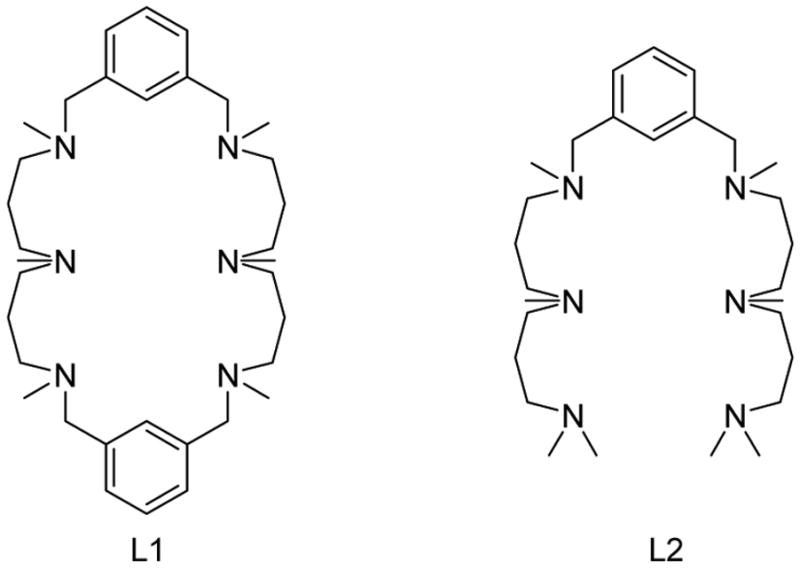
Schematic structure of ligands L1 and L2.
L1 and L2 were prepared following a three-step synthetic method involving dialdehyde-amine condensation, NaBH4 hydrogenation and HCHO/HCOOH permethylation (see Sup. Inf.). Dicopper complexes [Cu2(L1)](X)2 (1X2) and [Cu2(L2)](X)2 (2X2) (X = CF3SO3, SbF6 and BArF; BArF = [B{3,5-(CF3)2-C6H3}4]−) were prepared. 1(BArF)2 and 2(SbF6)2 were characterized by X-ray diffraction analysis.8
1(BArF)2 and 2(SbF6)2 contain discrete dinuclear cationic complexes (Figure 1). The coordination geometries and metrical parameters of the metal sites are not very different. Within each complex, the two Cu sites are pseudo or symmetrically related and the Cu···Cu distances are 7.040(3) and 7.019(2) Å, respectively. Each Cu ion contains an N3 coordination set and adopts a distorted trigonal planar geometry. The average Cu-N distance is 2.066(3) Å for 1(BArF)2 and 2.024(3) Å for 2(SbF6)2. The main difference between the two structures can be found in the N1-Cu1-N3 angle which is 156.52(10)º in 1(BArF)2 and 148.84(11)º (150.62(13)º for the pseudo-symmetrically related N4-Cu2-N6 angle) in 2(SbF6)2, which presumably reflects subtle ligand strains imposed by the more rigid macrocyclic L1 backbone. These structural parameters closely resemble the ones found in [Cu(MeAN)]B(C6F5)4,7 where Cu(I) adopts a tricoordinated geometry using a similar nitrogen-based donor-set ligand. The low coordination number and geometry attained in these complexes is particularly interesting, because it provides available coordination sites for interaction with external molecules such as O2 and indeed is the most common coordination structure found in Cu-dependent O2-processing proteins.1,2
Figure 1.
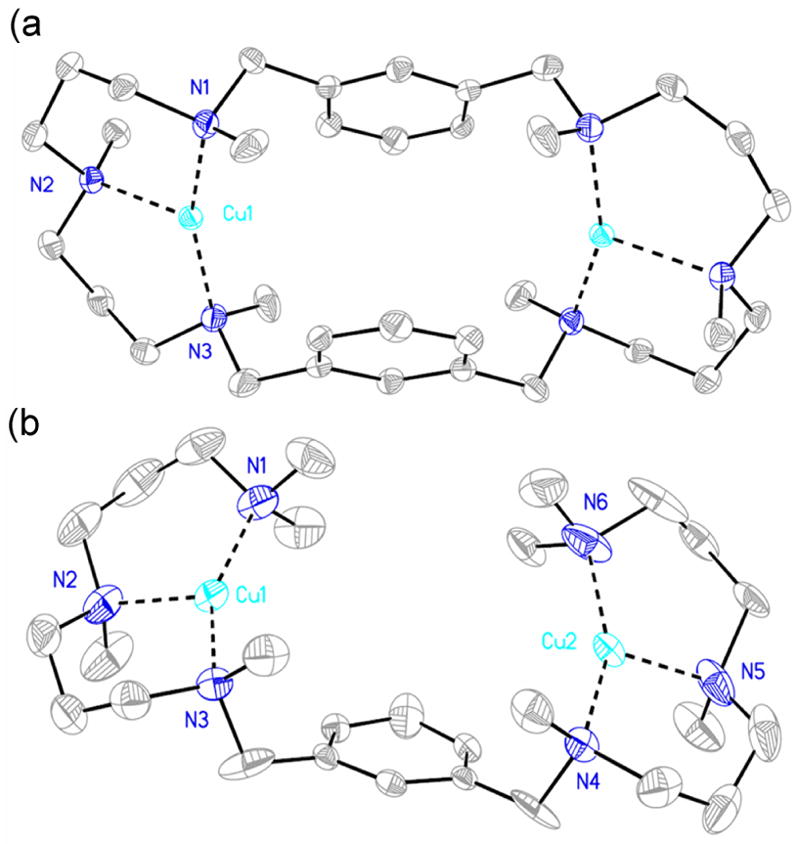
ORTEP diagrams for the cationic part of (a) 1(BArF)2; Cu-N1 2.007(3) Å, Cu-N2 2.192(2) Å, Cu-N3 1.999(3) Å, N3-Cu-N1 156.52(10)º, N3-Cu-N2 102.50(10)º, N1-Cu-N2 100.98(10)º and (b) 2(SbF6)2. Cu1-N1 1.990(3) Å, Cu1-N2 2.120(3) Å, Cu1-N3 1.986(3) Å, Cu2-N4 1.974(2) Å, Cu2-N5 2.100(3) Å, Cu2-N6 1.973(3) Å, N3-Cu1-N1 148.84(11)º, N3-Cu1-N2 105.08(10)º, N1-Cu1-N2 105.59(11)º, N6-Cu2-N4 150.62(13)º, N6-Cu1-N5 104.34(15)º, N4-Cu1-N5 104.95(12)º.
Multinuclear NMR studies performed in acetone-d6 for 1(BArF)2 and 2(BArF)2 indicated that the solid state structures are retained in solution. Pulse field gradient spin echo NMR (PGSE) measurements in acetone-d6 at −80 °C provide diffusion rates of 0.98±0.12*10−11 m2*s−1, and 0.89±0.11*10−10 m2*s−1 for 1 and 2, respectively, which indicates that both species have similar hydrodynamic radii, which, in turn, points out towards a monomeric nature of the species in solution.10
The electronic properties of the metal sites were studied by FT-IR analyses of the corresponding Cu(I)-CO adducts, which were generated in situ by bubbling CO through CH2Cl2 solutions.11 Remarkably, ν(CO) frequencies are 2083 and 2085 cm−1 in 1 and 2, respectively, which indicates that Cu(I) sites in both complexes are electronically comparable.12 We conclude that the Cu(I) ions in 1 and 2 possess electronically and structurally analogous properties.
In spite of the structural similarities between the complexes described, they show an unexpectedly different reactivity towards O2. Compound 1X (X = CF3SO3 and BArF) does not react with O2 in acetone at 198K, but irreversibly reacts with O2 at 273K to generate copper(II) species. However, no accumulation of any apparent intermediate was detected, and therefore we conclude that decomposition of any reaction intermediate is always faster than its formation. On the other hand, reaction of 2X ((X = CF3SO3, SbF6 and BArF) with O2 in acetone, CH2Cl2 or THF at low temperature show the relatively fast formation (within seconds) of a yellow species which was formulated as a bis-μ-oxo dicopper(III) species (3) on the basis of its UV-visible and resonance Raman spectra. The UV-vis spectrum of 3 in THF at −80 ºC exhibits two prominent bands at 308 nm (ε = 20000 M−1·cm−1) and at 413 nm (ε = 28000 M−1·cm−1). Resonance Raman experiments carried out in acetone using laser excitation at 413 nm reveal a characteristic Cu2O2 breathing vibration peak at 600 cm−1 that shows a - 23 cm−1 downshift when 18O2 is used (Figure 2). These are common spectral features for a Cu2III(μ-O)2 core which led us to formulate 3 as [Cu2III(μ-O)2L2]2+.13
Figure 2.
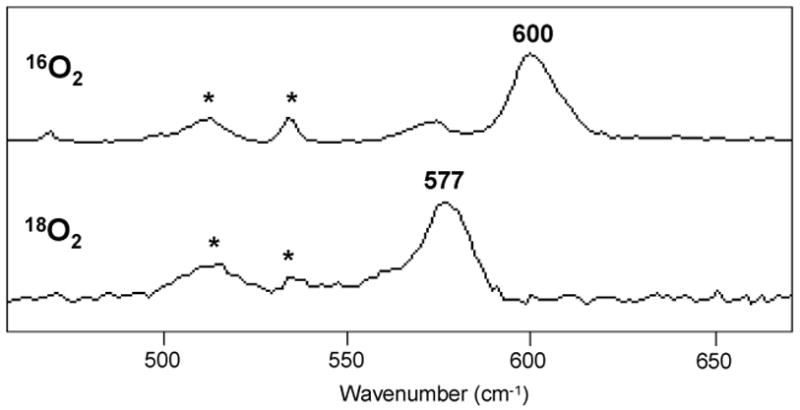
Resonance Raman spectra (λex = 413 nm) of 3 generated from 16O2 (top) and from 18O2 (bottom) in frozen acetone (77 K). Solvent peaks are marked with *
The diffusion rate for 3 measured in acetone-d6 at −80 ºC is 1.25±0.14*10−10 m2*s−1, which compares well with the values obtained for 1 and 2 and strongly supports the intramolecular nature of O2 binding. 3 constitutes then the first example of a Cu2III(μ-O)2 species formed within a dicopper complex containing a xylyl spacer,14 as to date only intramolecular Cu2II(μ-η2: η2-O2) intermediates have been characterized for systems using this type of linker.4,5 Besides, despite several examples of [Cu2III(μ-O)2L2] species, where L is a bidentate diamine, have already been reported,3 3 constitutes a rare example of such species supported by tridentate ligands.7,15,16 Although it is well established that Cu2III(μ-O)2 species are usually close in energy to their Cu2II(μ-η2: η-O2) isomers,3,15 the structure of 3 was found to be unperturbed by either the counterion (CF3SO3 or BArF) or the solvent (THF, acetone or CH2Cl2). DFT calculations at the B3LYP level (see Sup. Inf.) indicate that, for 3, the Cu2III(μ-O)2 structure is 35.5 kJ·mol−1 more stable than Cu2II(μ-η2: η2-O2), thus substantiating the sole observation of the bis-μ-oxo isomer. These results clearly contrast those reported by Karlin et. al. in the oxygenation of a related mononuclear [Cu(MeAN)]B(C6F5)4,7 where side-on Cu2II(μ-η2: η2-O2) species are formed.
Stopped-flow kinetic analysis of the oxygenation reactions indicates that 2 reacts reversibly with O2 to generate 3.17 At low temperatures (from −80 to −50 ºC), the equilibrium is shifted to the right, and the formation of 3 is essentially quantitative. Under these conditions, the reaction is first order in [O2] and first order in [2] (See Sup. Inf.):
Activation parameters for the oxygenation reactions are characterized by a rather low ΔH≠ = 9.5 ± 2 kJ·mol−1 and a large negative ΔS≠ = −175 ± 10 J·K−1·mol−1. The simple second-order rate law observed at low temperature is consistent with a stepwise reaction scheme (Scheme 2) similar to the oxygenation mechanisms of other dicopper(I) complexes.3b,17
Scheme 2.
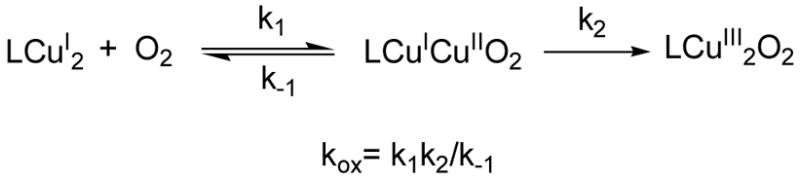
This mechanistic picture (Scheme 2) involves reversible reaction of O2 with 2 to generate a putative superoxo CuICuIIO2− species in a left-lying preequilibrium process, followed by intramolecular collapse into the final dinuclear [Cu2III(μ-O)2L2]2+ structure. The kinetic parameters and rate law associated with the formation of 3 resemble those determined for (μ-η2: η2-peroxo)dicopper(II) species bearing tridentate bis(pyridylethyl)amine ligands tethered by m-xylyl scaffolds.4 It is also interesting to point out that the oxygenation of the related mononuclear [Cu(MeAN)]+ complex exhibits a second order rate law in copper complex.7 Comparison between the kinetic parameters associated to the formation of 3 and [CuII2(μ-η2: η2-O2)(MeAN)2]+ indicates a higher ΔH≠ and a lower ΔS≠ for the former, probably associated with its more organized dinuclear structure.18 Direct interpretation of these parameters is hampered by the different rate law, and the multistep nature of the oxygenation reaction. Also unexpected is the dramatic difference in the O2 reactivity of 1 and 2. Given the comparable coordination sphere and electronic properties of the Cu(I) ions in both complexes, it is rather unlikely that the initial O2 binding to a single Cu(I) (k1·k−1−1) depends on the particular complex, and therefore the different O2 reactivity highlights the important role played by the second metal ion. The reaction in complex 1 is slower than in 2 presumably because there is an enthalpic barrier to surmount to bring the two Cu ions together, due to some strain from the ligand. In complex 2, the ligand is flexible, allowing copper sites to approach close enough to promote their synergistic actuation in O2 binding/reduction. Instead, the rather rigid nature of the macrocyclic ligand L1 imposes a higher barrier to this process, shutting down the reaction. The stability of 3, which allows its spectroscopic characterization, in comparison with the lack of stability of any reaction intermediate formed along the 1 + O2 pathway may also be explained on the basis of the different structural strains imposed by the ligands.
In summary, comparison of the O2 chemistry associated to [Cu(MeAN)]+, 1 and 2 constitutes a remarkable example of the importance of the cooperative actuation of two metal centers in the activation of O2 and highlights the challenge in designing suitable dinuclear scaffolds for modeling O2 processing proteins containing a dimetallic active site.19
Supplementary Material
Full details for the preparation and characterization of 1-2, kinetic data and details on the computational calculations. CIF files and details of the crystallographic characterization of 1-2.
Acknowledgments
Financial support from MEC of Spain through projects CTQ2005-08797-C02-01 and BQU2003-02884, from NIH (GM-38767 to LQ) and from NSF (Chem 0111202 to ERA). AC is grateful to MEC for the allocation of a PhD grant.
References
- 1.a) Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]; b) Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew Chem Int Ed. 2001;40:4570. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 2.a) Magnus KA, Ton-That H, Carpenter JE. Chem Rev. 1994;94:727. [Google Scholar]; b) Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. J Biol Chem. 2006;281:8981. doi: 10.1074/jbc.M509785200. [DOI] [PubMed] [Google Scholar]
- 3.a) Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;114:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; b) Lewis EA, Tolman WB. Chem Rev. 2004;114:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; c) Hatcher LQ, Karlin KD. J Biol Inorg Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]; d) Schindler S. Eur J Inorg Chem. 2000:2311. [Google Scholar]
- 4.a) Karlin KD, Nasir MS, Cohen BI, Cruse RW, Kaderli S, Zuberbühler AD. J Am Chem Soc. 1994;116:1324. [Google Scholar]; b) Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. J Am Chem Soc. 1998;120:7841. [Google Scholar]
- 5.Santagostini L, Gullotti M, Monzani E, Casella L, Dillinger R, Tuczek F. Chem Eur J. 2000;6:519. doi: 10.1002/(sici)1521-3765(20000204)6:3<519::aid-chem519>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 6.Costas M, Xifra R, Llobet A, Solà M, Robles J, Parella T, Stoeckli-Evans H, Neuburger M. Inorg Chem. 2003;42:4456. doi: 10.1021/ic0261833. [DOI] [PubMed] [Google Scholar]
- 7.Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbühler AD, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2002;124:4170. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
- 8.X-Ray crystal data for complex 1(BArF)2: C98H82B2Cu2F48N6, M = 2404.40, monoclinic, space group P21/n, a = 13.073(5), b = 27.978(12), c = 13.464(6) Å, β = 96.633(7), V = 4891 (3) Å3, Z = 2, T = 100(2) K, βcalc = 1.633 Mgr·M−3, μ = 0.578 mm−1, Rint = 0.0488 for 11668 independent reflections of the 69618 collected, final indices I > 2σ(I); R1 = 0.0510, WR2 = 0.1057. 2(SbF6)2: C28H56Cu2F12N6Sb2, Mf = 1075.37, monoclinic, space group P21/c, a = 16.264(4), b = 15.169(4), c = 17.642(5) Å, β = 103.776(4), V = 4227(2) Å3, Z = 4, T = 100(2) K, βcalc = 1.690 Mgr·M−3, μ = 2.337 mm−1, Rint = 0.0351 for 10415 independent reflections of the 62969 collected, final indices I > 2σ(I); R1 = 0.0305, WR2 = 0.0772.
- 9.a) Price WS. Concepts Magn Reson. 1997;9:299. [Google Scholar]; b) Price WS. Concepts Magn Reson. 1998;10:197. [Google Scholar]
- 10.L1 and L2 present a similar value of 2.51±0.17*10−10 m2*s−1
- 11.These complexes lose CO when isolated in the solid state.
- 12.Attempts to obtain a Cu(II)/Cu(I) red-ox potential by cyclic voltametry were unsuccessful.
- 13.a) Holland P, Cramer CJ, Wilkinson EC, Mahapatra S, Rodgers KR, Itoh S, Taki M, Fukuzumi S, Que L, Jr, Tolman WB. J Am Chem Soc. 2000;122:792. [Google Scholar]; b) Que L, Jr, Tolman WB. Angew Chem Int Ed. 2002;41:1114. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 14.Cu2III(μ-O)2 species have been described for xylyl-bridged TACN systems, but resulting from intermolecular interaction of Cu ions. See Mahapatra S, Kaderli S, Llobet A, Neuhold YM, Palanche T, Halfen JA, Young V, Jr, Kaden TA, Que L, Jr, Zuberbuhler A, Tolman WB. Inorg Chem. 1997;36:6343.
- 15.Halfen JA, Mahapatra S, Wilkinson E, Kaderli S, Young V, Jr, Que L, Jr, Zuberbühler AD, Tolman WB. Science. 1996;271:1397. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 16.Arii H, Saito Y, Nagatomo S, Kitagawa T, Funahashi Y, Jitsukawa K, Masuda H. Chem Lett. 2003;32:156. [Google Scholar]
- 17.From a kinetic point of view, O2 binding appears to be essentially irreversible at low temperatures and O2 release appears to be significant only at higher temperatures. However analysis of these reactions is complicated by the onset of rapid thermal decomposition.
- 18.See ref 7. Reported kinetic parameters for the oxygenation of [Cu(MeAN)]+ in CH2Cl2 are ΔH≠ = −27 kJ·mol−1 and ΔS≠ = −335 J·K−1mol−1.
- 19.a) Bol JE, Driessen WL, Ho RYN, Maase B, Que L, Jr, Reedijk J. Angew Chem Int Ed Engl. 1997;36:998. [Google Scholar]; b) Karlin KD, Dong-Heon L, Kaderli S, Zuberbühler AD. Chem Commun. 1997;5:475. [Google Scholar]; c) Tachi Y, Aita K, Teramae S, Tani F, Naruta Y, Fukuzumi S, Itoh S. Inorg Chem. 2004;43:4558. doi: 10.1021/ic049524g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full details for the preparation and characterization of 1-2, kinetic data and details on the computational calculations. CIF files and details of the crystallographic characterization of 1-2.