

Abstract
 Camptothecin (CPT), a cytotoxic natural alkaloid isolated from Camptotheca acuminata, and its derivatives represent an important class of cancer chemotherapeutic drugs that act by inhibiting topoisomerase I (top1). The mechanism of top1 inhibition by CPT has been determined by X-ray crystallography. Biochemical studies carried out both in vitro and in vivo indicated that CPT has strict DNA sequence preference for −1 T and strong preference for +1 G at the cleavage site. In order to understand the molecular determinants for the CPT binding orientation and DNA sequence selectivity, we present a quantum mechanics calculation where only π-π stacking interactions were included to shed some light on the mechanism of this sequence selectivity. This ab initio calculation can not only reproduce the experimental binding orientation of CPT at the cleavage site, but also shows very good correlation between the binding energy for different sequences and the observed frequency of CPT-stabilized sites in the SV40 viral genome. Therefore, it can be concluded that hydrogen bonding of the ligand to the surrounding amino acid residues of the protein is of minor significance. The present method should be applicable to other polycyclic top1 inhibitors.
Camptothecin (CPT), a cytotoxic natural alkaloid isolated from Camptotheca acuminata, and its derivatives represent an important class of cancer chemotherapeutic drugs that act by inhibiting topoisomerase I (top1). The mechanism of top1 inhibition by CPT has been determined by X-ray crystallography. Biochemical studies carried out both in vitro and in vivo indicated that CPT has strict DNA sequence preference for −1 T and strong preference for +1 G at the cleavage site. In order to understand the molecular determinants for the CPT binding orientation and DNA sequence selectivity, we present a quantum mechanics calculation where only π-π stacking interactions were included to shed some light on the mechanism of this sequence selectivity. This ab initio calculation can not only reproduce the experimental binding orientation of CPT at the cleavage site, but also shows very good correlation between the binding energy for different sequences and the observed frequency of CPT-stabilized sites in the SV40 viral genome. Therefore, it can be concluded that hydrogen bonding of the ligand to the surrounding amino acid residues of the protein is of minor significance. The present method should be applicable to other polycyclic top1 inhibitors.
The discovery of the cytotoxic agent camptothecin (CPT, 1) (Chart 1) from the Chinese tree Camptotheca acuminata has not only provided a lead for the development of new anticancer drugs, it has also led to the validation of topoisomerase I (top1) as a chemotherapeutic target.1,2 Top1 is a nuclear enzyme responsible for solving the topological problems associated with DNA replication and transcription by nicking and resealing one strand of DNA in a DNA duplex.3 These processes are achieved by a reversible transesterification reaction between Tyr723 of top1 and the phosphodiester bond of the DNA. The X-ray crystal structure of the ternary complex consisting of DNA, top1 and topotecan (TPT, 2), a therapeutically useful analogue of CPT, shows that the pentacyclic ring system of CPT intercalates into the DNA base pair step at the cleavage site, resulting in increased physical distance between the cleaved DNA termini and consequently inhibition of the religation step catalyzed by top1.4 Interestingly, none of the three previously proposed binding models of CPT at the cleavage site are consistent with the X-ray crystal structure.5-7
Chart 1.
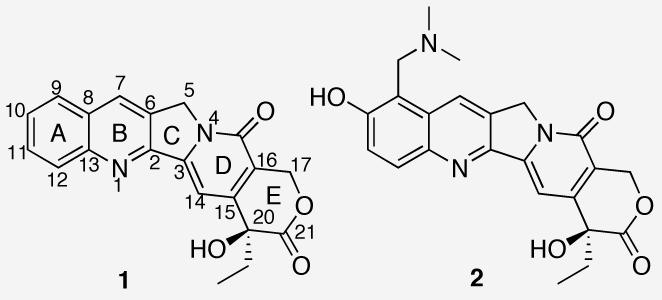
Chemical structures of CPT (1) and topotecan (2).
Previous experimental data has shown that CPT has a strict requirement for T at the −1 site and a strong preference of G at the +1 site of the cleaved strand.8 This sequence preference was observed both in vitro and in vivo.8,9 The reason for this sequence selectivity has not been explained. Since 60% of the solvent-accessible surface area of 2 is covered by base-stacking interactions and only one direct hydrogen bond is present between top1 and 2,4 we hypothesized that the primary π-π stacking interactions between CPT and the flanking base pairs would dictate the binding orientation of CPT at the cleavage site, as well as the immediate DNA sequence selectivity. The computational evidence to support this hypothesis is the subject of the present communication.
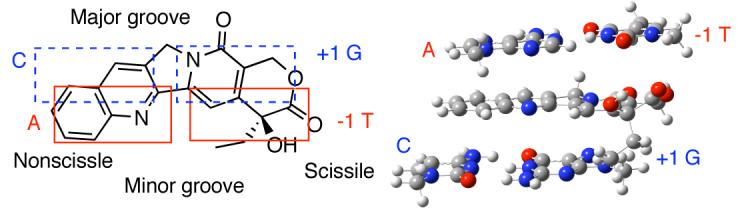
The models used in the calculations were constructed by the following steps. (1) Topotecan and the flanking base pairs were extracted from the crystal structure (PDB 1K4T). (2) The deoxyribose rings were replaced by methyl groups and the substitutents on position 9 and 10 of TPT were replaced by hydrogen atoms. (3) The energies of the camptothecin molecule, A-T base pair, and G-C base pair were each optimized separately at the HF/6-31G(d,p) level of theory in Gaussian03.10 A frequency calculation was included in each case to ensure the structural minimum. (4) The three resulting individual structures were utilized to replace the original units in the complex in step 2, resulting in model A (Figure 1).11 (5) CPT was then rotated to different orientations in the intercalation complex and the ethyl group was minimized in every orientation to avoid any steric clash using the MMFF94 force field and MMFF94 charges in Sybyl®,12 providing models B-F. (6) These six models were then subjected to a single point energy calculation at MP2/6-31G(d), where the basis set superposition error (BSSE) for the complex was calculated using the counterpoise correction method.13 It is important to include electron correlation to accurately represent the stacking interactions.14
Figure 1.
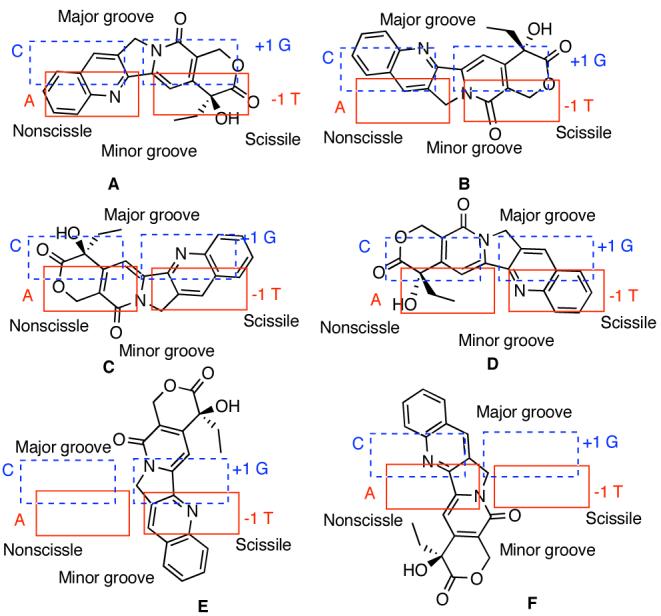
Two-dimensional schematic representation of different orientations of camptothecin (1) in the cleavage site.
The calculated MP2 energies of the six models are listed in Table 1. In contrast to the previous molecular mechanics calculations,7 this MP2 calculation, with or without BSSE, clearly demonstrates that the experimentally observed model A has the lowest energy among the six models. Although the long axis of CPT is parallel to the long axis of the base pairs in all the four models A-D, the energy differences without BSSE range from 8.87 to 42.63 kcal/mol. These energy differences can be rationalized based on electrostatic complementarity between CPT and the flanking base pairs (Figure 1S).11 Apparently, models B and C experience electrostatic repulsion between CPT and the neighboring base pairs (Figure 1S).11 The other two models E and F , having CPT's long axis perpendicular to that of the base pairs, surprisingly show relatively low energies. These results indicate that multiple factors, including dispersion forces, electrostatic attraction and charge-transfer interactions contribute to the stacking energies.15 Solvation effects were not considered because they were assumed to be similar among all the binding processes.16
Table 1.
The MP2 energies of models A-F.
| models | EMP2 (au)a | ΔEMP2a,b | EMP2' (au)c | ΔEMP2'b,c |
|---|---|---|---|---|
| A | −3188.935708 | 0.00 | −3188.903671 | 0.00 |
| B | −3188.867778 | 42.63 | −3188.828201 | 47.36 |
| C | −3188.899863 | 22.49 | −3188.860667 | 26.99 |
| D | −3188.921567 | 8.87 | −3188.888630 | 9.44 |
| E | −3188.915987 | 12.38 | −3188.891051 | 7.92 |
| F | −3188.926889 | 5.53 | −3188.899196 | 2.81 |
MP2 energies without BSSE
ΔEMP2 and ΔEMP2' refer to the energy difference (kcal/mol) between model A and other models
MP2 energies with BSSE.
Having established that the primary stacking interactions could reproduce the experimental model, we then investigated whether the stacking interactions also govern the DNA sequence preference by mutating the immediate DNA bases and calculating the interaction energies (Eint) according to Eq (1) (Figure 2). The interaction energies, after correction with BSSE for the complexes, with different DNA sequences are shown in Table 2. Among the eight different sequences, the interaction energy with −1 T-A and +1 G-C is the largest (−6.69 kcal/mol), which is consistent with observed experimental sequence preference.8,9 Although the base pair separation energies for entries 1 and 3 are larger than that required for entry 4,17 their interaction energies are larger than that in entry 4, which indicates that these interaction energy differences among different sequences are a consequence of better stacking of CPT onto certain bases instead of the differences in base pair separation energies.
Figure 2.
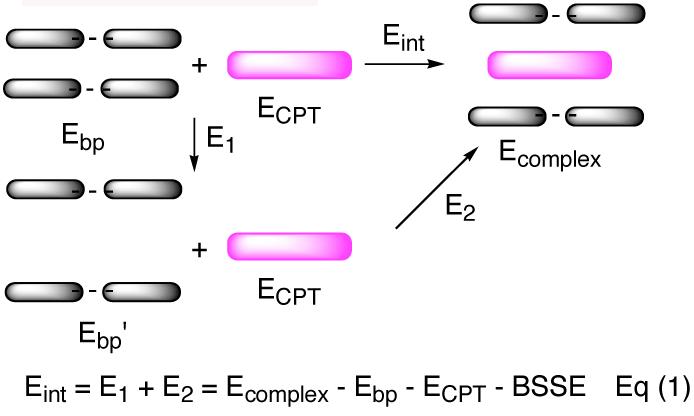
Schematic and mathematical representation of Eint. Eb p and Eb p ′ are the respective energies of the base pairs in their normal distance and their separated distance as seen in the complex.
Table 2.
The interaction energies (kcal/mol) calculated at the MP2/6-31g(d) level of theory and observed frequency of different CPT sites in the SV40 genome.
| Entry | −1a | +1a | Eint(MP2) | Frequency (P)% |
|---|---|---|---|---|
| 1 | T | G | −6.69 | 75 |
| 2 | T | A | −5.97 | 16 |
| 3 | T | C | −5.57 | 7 |
| 4 | T | T | −4.48 | 2 |
| 5 | G | T | −2.76 | NDb |
| 6 | G | G | −4.85 | NDb |
| 7 | C | G | −5.55 | NDb |
| 8 | A | G | −3.03 | NDb |
The bases listed correspond to those next to the cleavage site on the cleaved DNA strand.
Not detected.
The cleavage sites stabilized by CPT in SV40 viral genome DNA were analyzed extensively by Pommier8 and the frequency of the CPT sites for four different sequences is presented in Table 2. All of the observed CPT sites show large interaction energies (entries 1-4). Although the possible CPT sites, as demonstrated by the relatively large interaction energy, shown in entries 6 and 7 were not detected in the SV40 genome, a high correlation (r2 = 0.968) between the interaction energy and the known frequency of CPT sites was observed (Figure 2S),11 suggesting that the intercalation energy is a predominant, if not exclusive, factor governing the sequence selectivity of CPT.
In conclusion, the binding orientation of CPT in the DNA-top1 cleavage site was studied by an ab initio quantum mechanics calculation, and the calculated orientation was found to match that observed experimentally. Furthermore, the DNA sequence selectivity of CPT was studied and a good correlation was observed between the calculated interaction energy and the frequency of CPT sites in the SV40 genome. Since the calculations only involve π-π stacking interactions and are capable of predicting binding orientation and binding site selectivity, it can be concluded that hydrogen bonding of the ligand to the surrounding amino acid residues of the protein, or to the base pairs, is of minor significance. The present method should be applicable to other polycyclic top1 poisons to generate similar models for further structure-based drug design.
Supplementary Material
Acknowledgment
This work was made possible by the National Institutes of Health (NIH) through support of this work with Research Grant UO1 CA89566. We thank the Rosen Center for Advanced Computing (RCAC), Purdue University for providing computing facilities.
Footnotes
Supporting Information Available: 3-D presentation of models A-F and their Cartesian coordinates. Electrostatic potential surface maps of CPT and base pairs. Correlation figure of Eint versus frequency. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Thomas CJ, Rahier NJ, Hecht SM. Bioorg. Med. Chem. 2004;12:1585. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 2.Hsiang YH, Hertzberg R, Hecht S, Liu LF. J. Biol. Chem. 1985;260:14873. [PubMed] [Google Scholar]
- 3.Wang JC. Nat. Rev. Mol. Cell Biol. 2002;3:430. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 4.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr., Stewart L. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15387. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan Y, Weinstein JN, Kohn KW, Shi LM, Pommier Y. J. Med. Chem. 1998;41:2216. doi: 10.1021/jm9605445. [DOI] [PubMed] [Google Scholar]
- 6.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Science. 1998;279:1504. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 7.Kerrigan JE, Pilch DS. Biochemistry. 2001;40:9792. doi: 10.1021/bi010913l. [DOI] [PubMed] [Google Scholar]
- 8.Jaxel C, Capranico G, Kerrigan D, Kohn KW, Pommier Y. J. Biol. Chem. 1991;266:20418. [PubMed] [Google Scholar]
- 9.Pondarre C, Strumberg D, Fujimori A, TorresLeon R, Pommier Y. Nucleic Acids Res. 1997;25:4111. doi: 10.1093/nar/25.20.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaussian03 . Revision B.05 ed. Gaussian, Inc; Pittsburg, PA: 2003. [Google Scholar]
- 11.see Supporting Information.
- 12.Sybyl . 6.9 ed. Tripos Inc.; St. Louis, MO: 2002. [Google Scholar]
- 13.Boys SF, Bernardi F. Mol. Phys. 1970;19:553. [Google Scholar]
- 14.Hobza P, Sponer J. Chem. Rev. 1999;99:3247. doi: 10.1021/cr9800255. [DOI] [PubMed] [Google Scholar]
- 15.Reha D, Kabelac M, Ryjacek F, Sponer J, Sponer JE, Elstner M, Suhai S, Hobza P. J. Am. Chem. Soc. 2002;124:3366. doi: 10.1021/ja011490d. [DOI] [PubMed] [Google Scholar]
- 16.Solvation energies in water, calculated using PCM model, for models A, C and E are −44.11, −44.56 and −44.10 kcal/mol, respectively.
- 17.Ornstein RL, Rein R, Breen DL, Macelroy RD. Biopolymers. 1978;17:2341–2360. doi: 10.1002/bip.1978.360171005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.