

Abstract
Low-spin mononuclear alkylperoxoiron(III) complexes decompose by peroxide O-O bond homolysis to form iron(IV) species. We examined the kinetics of previously reported homolysis reactions for alkylperoxoiron(III) intermediates supported by TPA (tris-(2-pyridylmethyl)amine) in CH3CN solution and promoted by pyridine-N-oxide, by BPMCN (N,N-bis(2-pyridylmethyl)-N,N-dimethyl-trans-1,2-diaminocyclohexane) in its cis-β configuration in CH3CN and CH2Cl2, as well as for the previously unreported chemistry of TPA and 5-Me3TPA intermediates in acetone. Each of these reactions forms an oxoiron(IV) complex, except for the β-BPMCN reaction in CH2Cl2 that yields a novel (hydroxo)alkylperoxoiron(IV) product. Temperature-dependent rate measurements suggest a common reaction trajectory for each of these reactions, and verify previous theoretical estimates of a ca. 60 kJ/mole enthalpic barrier to homolysis. However, both the tetradentate supporting ligand and exogenous ligands in the sixth octahedral coordination site significantly perturb the homolyses, such that observed rates can vary over two orders of magnitude at a given temperature. This is manifested as a compensation effect in which increasing activation enthalpy is offset by increasingly favorable activation entropy. Moreover, the applied kinetic model is consistent with geometric isomerism in the low-spin alkylperoxoiron(III) intermediates, wherein the alkylperoxo ligand is coordinated in either of the inequivalent cis sites afforded by the non-heme ligands.
1. Introduction
Many monooxygenase enzymes utilize a mononuclear iron(II) center in a non-heme ligand environment to activate dioxygen and effect concomitant oxidation or oxygenation of biological substrates.1 Peroxo intermediates are often cited as mechanistically important intermediates in such processes that break down to form high-valent oxoiron species.2 Distinct oxoiron(V) and oxoiron(IV) intermediates can be generated respectively by disparate heterolytic and homolytic cleavage of the peroxide O-O bonds (eqs. 1, 2).3,4 In turn, these highly reactive species carry out the substrate oxidations.5,6
| (1) |
| (2) |
| (3) |
Our laboratory has pursued a modeling approach to this reactivity using small-molecule iron complexes.7,8 Several non-heme oxoiron(IV) complexes have been characterized;9–21 these synthetic complexes exhibit weak visible-NIR ligand field bands at ca. 700 nm, a short Fe=O bond on the order of 1.65 Å, and a low-spin (S = 1) ground state. An oxoiron(IV) intermediate also has been detected experimentally in the catalytic cycle of the α-ketoacid-dependent enzymes taurine: αKG dioxygenase (TauD)22–25 and prolyl-4-hydroxylase (P4H).26 In contrast to the synthetic ligands, the 2-His-1-carboxylate facial triad1,27 environment of the enzyme affords a weaker ligand field and gives rise to a high-spin (S = 2) ground state. However, the difference in the spin states involves only the dxy and d x2-y2 orbitals, and DFT calculations suggest the Fe=O bonding remains highly covalent and is not significantly perturbed.28,29
A number of alkylperoxoiron(III) complexes have also been generated, primarily by oxidation of synthetic iron(II) complexes with tert-butyl hydroperoxide (tBuOOH).1,2,14,15,21,30–40 These metastable species have been characterized primarily by EPR and resonance Raman spectroscopy,2 as well as by X-ray absorption spectroscopy.14 Enzymes with experimentally observable peroxo species include soybean lipoxygenase,41 superoxide reductase,42–45 and naphthalene dioxygenase.46 The synthetic peroxo complexes exhibit either low- (S = 1/2) or high-spin (S = 5/2) configurations with concomitant effects on reactivity; spectroscopic analysis and theoretical calculations indicate that low-spin species favor FeO–OR bond homolysis (eq. 2), whereas the high-spin intermediates undergo Fe–OOR bond homolysis (eq. 3).33,47,48
The TPA and BPMCN ligands (tris(2-pyridylmethyl)amine and N,N-bis(2-pyridylmethyl)-N,N-dimethyl-trans-1,2-diaminocyclohexane, respectively, Scheme 1) support low-spin alkylperoxoiron(III) intermediates, and their conversions to oxoiron(IV) species have been directly observed.15,21,38 This work examines the temperature-dependent kinetics of FeO–OR bond homolyses for the predominantly low-spin alkylperoxoiron(III) intermediates supported by these ligands (TPA, 5-Me3TPA, β-BPMCN, Scheme 1) in various solvents (acetone, CH3CN, CH2Cl2). The reactions produce oxoiron(IV) in all cases, except for the previously reported formation of a novel (hydroxo)alkylperoxoiron(IV) product derived from FeII(β-BPMCN)(OTf)2 in CH2Cl2.15 Nevertheless, some mechanistic similarities were observed in formation of this distinct product, and theoretical predictions about the common O-O bond homolyses were tested experimentally.
Scheme 1.
2. Experimental
Synthetic and general experimental procedures were described previously.15,21,38,49 Electronic absorption spectra were recorded on a Hewlett-Packard (Agilent) 8452 diode array spectrophotometer over a 190–1100 nm range in quartz cuvettes cooled to the desired temperature either in a liquid nitrogen cryostat (Unisoku) or in a methanol-filled dewar fitted with quartz windows placed in the optical path and cooled by a closed loop of chilled methanol from a circulating bath equipped with an immersion cooler under control of a thermostat (Neslab). Global kinetic fits were carried out using SPECFIT™ (Spectrum Software Associates, Chapel Hill, NC)50 on optical datasets truncated to remove initial iron(II) oxidation and terminal quenching of metastable iron(IV) products. X-band EPR spectra were obtained at 2 K on a Bruker E-500 spectrometer equipped with an Oxford ESR-10 liquid helium cryostat. Resonance Raman spectra were collected on an Acton AM-506 spectrometer (2400-groove grating) using Kaiser Optical holographic super-notch filters with a Princeton Instruments liquid N2-cooled (LN-1100PB) CCD detector with 4 cm−1 or 2 cm−1 spectral resolution. Spectra were obtained using a back-scattering geometry on liquid N 2 frozen samples with laser excitation from either a Spectra Physics 2030 argon ion laser and a 375B CW dye (Rhodamine 6G), or a Spectra Physics Beamlock 2060 krypton laser. Raman frequencies were referenced to indene.
3. Results
General remarks
Alkylperoxoiron(III) intermediates supported by TPA, 5-Me3-TPA, and BPMCN ligands (Scheme 1) are formed by treatment of iron(II) precursors with excess tBuOOH in various solvents (acetone, CH3CN, CH2Cl2).15,32,38 These complexes possess intense LMCT bands (ε = 2000–2500 M−1 cm−1), and convert to metastable oxo- or alkylperoxoiron(IV) species in near-quantitative yields (Table 1). In the present work, decay of the alkylperoxoiron(III) intermediates was monitored by visible-NIR spectroscopy, and analysis of their reaction kinetics reveals common mechanistic features among the disparate reactions. Consistent with the observed extinctions, optical data were obtained from reactions carried out using millimolar concentrations of iron(II) precursors and the minimal excess of tBuOOH empirically determined to generate optimal yields of alkylperoxoiron(III) species (i.e., 2 equivalents in CH3CN, 5–10 equivalents in CH2Cl2, and 10 equivalents in acetone, added as a concentrated stock solution, 5.5 M in nonane); conversion was found to be nearly quantitative as judged by EPR and Mössbauer spectra, except in acetone.
Table 1.
Spectroscopic data for decomposition reactions of alkylperoxoiron(III) intermediates to iron(IV) species.
| Complex | Solvent | T (K) | Other additive | FeIII-OOtBu LMCT, λmax, (εobs) | FeIII-OOtBu mole fraction | FeIV=O LF (λmax, εobs) | FeIV mole fraction | Ref. |
|---|---|---|---|---|---|---|---|---|
| Fe(5Me3TPA) | acetone | 228 | 560 nm (900) | 746 nm (160) | a | |||
| Fe(5Me3TPA) | acetone | 213 | 560 nm (1500) | 0.72b | a | |||
| Fe(TPA) | MeCN | 231 | PyO (10 mM) | 593 nm (2100) | 746 nm (340) | 0.95b | 38 | |
| Fe(BPMCN) | MeCN | 228 | H2O (300 mM) | 600 nm (2200) | 753 nm (280) | 0.90b | 15 | |
| Fe(BPMCN) | CH2Cl 2 | 206 | 566 nm (2200) | 0.86c | d | 0.94(3)e | 15 |
this work.
determined by Mössbauer spectroscopy.
determined by EPR spectroscopy.
product is an alkylperoxoiron(IV) complex.
determined by NMR spectroscopy.
The initial oxidation of iron(II) exhibited complex kinetic behavior plausibly associated with autocatalysis (Figures S1, S2 in Supporting Information); observed reaction velocities were roughly proportional to initial concentrations of iron(II) and tBuOOH, but detailed mechanistic analysis of this step is ongoing and will be addressed in a future manuscript. Nonetheless, monotonic accumulation of the alkylperoxoiron(III) CT chromophore continued until iron(II) is exhausted, and then subsequent decay to iron(IV) abruptly commenced. Therefore, the alkylperoxoiron(III) decay was cleanly separable from both the preceding rapid accumulation phase (Figures S3, S4 in Supporting Information,), as well as from the slow terminal quenching of metastable iron(IV) product. At reaction temperatures of ca. 228 K, the accumulation phase persisted for 2–5 minutes in CH3CN, tens of seconds in acetone, and seconds in CH2Cl2.
Kinetics of the subsequent alkylperoxoiron(III) decay phase(s) to iron(IV) were modeled assuming first-order kinetics for all observed phases and using global least-squares techniques. Unless otherwise noted, we have assumed irreversible unimolecular O-O bond homolyses as the rate-limiting steps, eqn. 2, yielding the rate law of eqn. 4, where we take kobs = k of reaction 2.51
| (4) |
Our interpretation of the kinetic results in this study thus depends on the assumed validity of this equation. Demonstration of the independence of kobs on tBuOOH concentration was significantly hindered by complications arising from the initial iron(II) oxidation and side reactions of excess tBuOOH with products from reaction 2, eqns. 5, 6.3,38 However, independence of kobs on iron concentration was observed for the decay of the TPA-supported alkylperoxoiron(III) intermediate in acetone at −50 °C and for the decay of the BPMCN-supported alkylperoxoiron(III) intermediate in CH2Cl2 at −67 °C (see Table S1 in Supporting Information), thereby supporting the validity of eqn 4.
| (5) |
| (6) |
Decay of [FeIII(TPA)(OOtBu)]2+ and FeII(5-Me3TPA)(OTf)2 in acetone
Treatment of either [FeII(TPA)(OTf)2] or [FeII(5-Me3TPA)(OTf)2] with 10 equiv. tBuOOH in acetone solution at 228 K resulted in rapid formation of an LMCT band (λmax = 560 nm, Table 1 and Figure 1) and low-spin iron(III) EPR and Raman spectra (Table 2, Figure S5), analogous to those reported previously for [FeII(TPA)(OOtBu)(NCMe)]2+ in CH3CN (vide infra). The LMCT band in acetone is relatively blue-shifted, presumably due to replacement of MeCN with acetone, and its absorbance is lower than expected for full formation of the intermediate, given the molar extinction coefficients typical for such complexes (ε ~ 2000 M−1cm−1). Indeed, conversion to the alkylperoxoiron(III) species is highly temperature dependent and favored at lower temperature. A Mössbauer spectrum obtained of a frozen sample prepared at 213 K from the addition of 10 equiv tBuOOH to a 1.67 mM [FeII(5-Me3-TPA)(OTf)2] solution in acetone (A560 = 2.5) shows 72% of the iron in the sample corresponding to the expected low-spin iron(III) center, with remaining components associated with a high-spin iron(III) center (20%) and a low-spin iron(IV) center (<10%) (Figure S6). If it is assumed that the low-spin alkylperoxoiron(III) intermediate contributes almost all of the absorbance at 560 nm, a molar extinction coefficient of 2100 M−1cm−1 is calculated, which is fully consistent with previous work (Table 1), so the remaining components are essentially transparent in this region. This initial oxidation product is accordingly formulated as a six-coordinate solvated complex dication [FeIII(L)(acetone)(OOtBu)]2+, L = TPA or 5-Me3TPA.
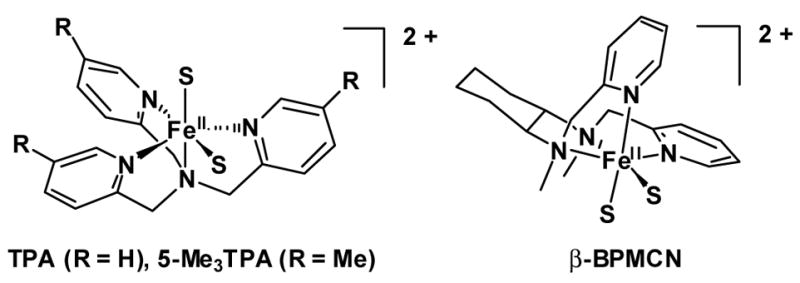
Figure 1.
Time dependent evolution visible-NIR absorption for decay of the alkylperoxoiron(III) intermediate supported by 5-Me3TPA to oxoiron(IV) in acetone at 228 K ([FeII]0 = 1.1 mM, [tBuOOH]0 = 11 mM).
Table 2.
Spectroscopic characterization of alkylperoxoiron(III) species.
| [(L)Fe(OOtBu)(X)]2+ Complex: L = | Solvent | Spin state | UV-vis (nm) | Resonance Raman (cm−1) | Ref | ||
|---|---|---|---|---|---|---|---|
| δ(tBu) | ν(Fe-O) | ν(O-O) | |||||
| TPA (X = NCMe) | CH3CN | S = 1/2 | 600 | 490 | 696 | 796 | 35 |
| 6-Me3-TPA (no X) | CH3CN | S = 5/2 | 562 | 468 | 637 | 842,877 | 35 |
| TPA (X = PyO) | CH3CN | S = 1/2 | 593 | 38 | |||
| TPA (X = acetone) | acetone | S = 1/2 | 560 | 489 | 693 | 788 | a |
| 5-Me3-TPA (X = acetone) | acetone | S = 1/2 | 560 | 488 | 691 | 789 | a |
| β-BPMCN (X = NCMe) | CH3CN | S = 1/2 | 600 | 490 | 685 | 793 | 15 |
| β-BPMCN (X = OTf and/or ROOH?) | CH2Cl2 |
S = 1/2 (minor);
S = 5/2 (major) |
566 | –
474 |
680
628,637,647,653 |
789
832,851,882 |
15 |
This work.
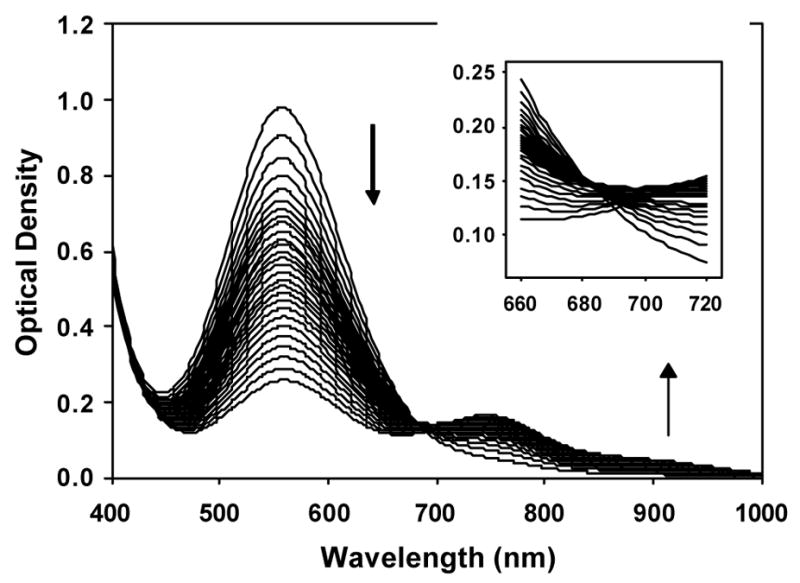
Subsequent decay of the 5-Me3TPA-supported intermediate yielded a ligand field chromophore (λmax = 746 nm) consistent with its proportional conversion to an oxoiron(IV) complex (Figure 1). However, this reaction failed to conform to simple exponential kinetics, clearly exhibiting two distinct phases for decay of the LMCT band (Figure 2), as well as growth of the ligand field band (Figure S7 in Supporting Information). An isosbestic point initially observed at 682 nm between the bleaching LMCT and growing ligand field chromophores shifted to 716 nm as the decomposition progressed (Figure 1 inset); furthermore, the tailing absorption of UV-active π-π* bands also shifted, and a second isosbestic point eventually developed on the shoulder of the high-energy bands at 437 nm. Both the time-dependent optical data observed at 690 nm between the shifting isosbestic points and at 410 nm on the high-energy tail above the late-developing second isosbestic point clearly exhibited biphasic kinetics of opposing directions (Figures S8, S9 in Supporting Information). Accordingly, the data were globally fit to double exponential models (kobs = 1.18(2) × 10−1 s−1, 2.86(6) × 10−4 s−1, Table S1 and Figure S10 in Supporting Information), with rapid convergence of the least-squares refinement to milli-AU residuals (Figure 2 and Figure S11 in Supporting Information).
Figure 2.
Observed (black) vs. calculated (red) optical density at 560 nm for conversion of 5-Me3TPA-supported alkylperoxoiron(III) to oxoiron(IV) shown in Figure 1.
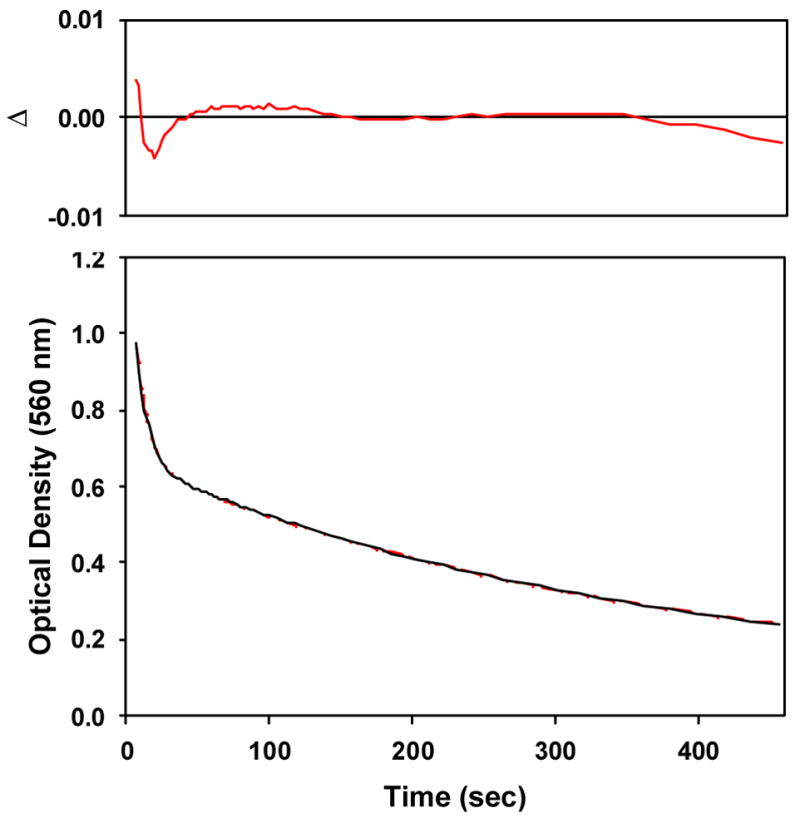
A parallel, convergent reaction scheme ( i.e., A → C, B → C, Figure 3) suggests that the 560 nm LMCT feature is a superposition of two bands that bleach at different rates to form the oxoiron(IV) ligand field spectrum. The LMCT absorption can partitioned between a continuum of mole fractions representing A and B with concordant effects on their relative extinctions. However, inspection of the 560 nm decay trace clearly shows the fast phase comprises only one- third of the total amplitude (Figure 2). Degenerate sequential models ( i.e., A → B → C) must also exist (Figure S12 in Supporting Information),57 but the calculated intermediate (“B”) spectrum contains both alkylperoxoiron(III) LMCT and oxoiron(IV) ligand field absorption features, and the former is inverted for one permutation of the observed rates. Hence, we take the sequential model to be nonsensical.
Figure 3.
Calculated spectra (top) and time-dependent concentrations (bottom) for an A (red) → C (green), B (blue) → C (green) kinetic model applied to time-dependent optical data shown in Figure 1 for conversion of 5-Me3TPA-supported alkylperoxoiron(III) to oxoiron(IV) in acetone at 228K, with initial mole fractions arbitrarily set to [A]0 = 0.52, [B]0 = 0.33, [C]0 = 0.15.
Completely analogous results were obtained for the TPA-supported intermediate (Figures S13-S17 in Supporting Information), although the observed rates decreased slightly (kobs = 3.17(2) × 10−2 s−1, 9.5(1) × 10−4 s−1 at 223 K). Furthermore, variation of the initial iron and tBuOOH concentrations by a factor of 4 gave rate constants that were independent of these concentrations. These results support our assumption that the decay of the alkylperoxoiron(III) intermediate is a first-order process (eqn 4).
Despite the successful fitting of parallel first-order reactions to the optical data, the presence of a significant fraction of the charged iron in an unreactive and spectroscopically transparent state that forms during the initial oxidation of the starting iron(II) complex (Table 1) may raise concern that the complicated kinetics observed might arise from the presence of this mass balance. However the various isosbestic points observed during decay of the alkylperoxoiron(III) species strongly suggest that this side product is fully inert. To test the generality of our kinetic analysis, we reexamined the previously reported reactions of alkylperoxoiron(III) intermediates supported by β-BPMCN in CH3CN solution,15 as well as TPA in CH3CN assisted by added pyridine-N-oxide,38 which produce much higher LMCT extinctions and yield oxoiron(IV) in essentially quantitative yield as demonstrated by Mössbauer spectroscopy (Table 1). As described in the following sections, we found evidence of comparable kinetic behavior for these reactions as well.
Decay of [FeIII(TPA)(OOtBu)]2+ in MeCN
[FeII(TPA)(NCMe)2]2+ reacts with excess tBuOOH in MeCN solvent at −42 °C to form an intensely blue metastable S = ½ FeIII-OOtBu intermediate with λmax at 600 nm (Table 1). As previously reported, conversion of [FeII(TPA)(OOtBu)]2+ to oxoiron(IV) in CH3CN solution was sluggish, preventing accumulation of the metastable product. However, fast conversion to a nearly quantitative yield of oxoiron(IV) was obtained upon addition of pyridine-N-oxide to the preformed alkylperoxoiron(III) intermediate at 231 K, which was postulated to bind to the alkylperoxoiron(III) species as the sixth ligand, promoting relatively rapid O-O bond homolysis.38 Decay of the alkylperoxoiron(III) LMCT band in the presence of 10 mM pyridine-N-oxide (λmax = 593 nm, ε = 2100 M −1 cm−1) was coincident with the appearance of a ligand field band, indicative of the formation of [FeIV(O)(TPA)(pyO)]2+ (λmax = 746 nm, ε = 340 M−1 cm−1, Figure 4).
Figure 4.
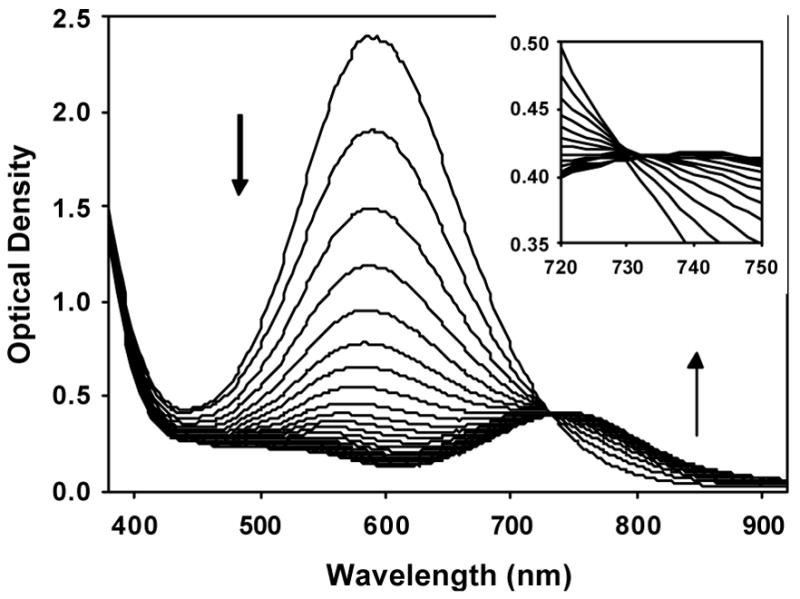
Time dependent evolution visible-NIR absorption for decay of the alkylperoxoiron(III) intermediate supported by TPA to oxoiron(IV) in CH3CN with added pyridine-N-oxide at 228 K ([FeII]0 = 1.3 mM, [tBuOOH]0 = 2.6 mM, [PyNO] = 10 mM).
Decay of the LMCT absorption proceeds with an apparent isosbestic point near 730 nm (Figure 4), and these optical data were previously fit to first-order observed reaction kinetics using initial rate techniques.38 In fact, we reconfirmed that the optical data can be fit to a simple exponential decay (A → B), either using a single wavelength coincident with the absorption maximum (kobs = 1.29(1) × 10−1 s−1, R2 = 0.9991) or globally (kobs = 1.32(1) × 10−1 s−1, σ= 1.08 × 10−2, Figure 5). However, in both cases the exponential fits showed significant structured residuals. Close inspection of the optical data revealed the apparent isosbestic point again shifted during the reaction (Figure 4 inset), and optical density changes over the bracketed wavelength interval was biphasic (Figure S20 in Supporting Information). These observations closely resembled the spectroscopic results obtained in acetone ( vide supra), although the red-shift of the LMCT band in CH3CN nearly disguised the more complex kinetics. Fitting of a parallel A → C, B → C model resulted in significant attenuation and randomization of the residuals (Figure 5 and Figures S18, S19 in Supporting Information). Calculated spectra for species A and B again featured coincident alkylperoxoiron(III) LMCT features; arbitrary assignment of a 1:1 mixture led to approximately degenerate calculated extinctions (Figure 6). Moreover, calculated rates (kobs = 2.5(2) × 10−1 s−1, 1.02(3) × 10−1 s−1, σ = 4.42 × 10−3) differ by a factor less than two about a single exponential fit, which further conceals the kinetic complexity.
Figure 5.
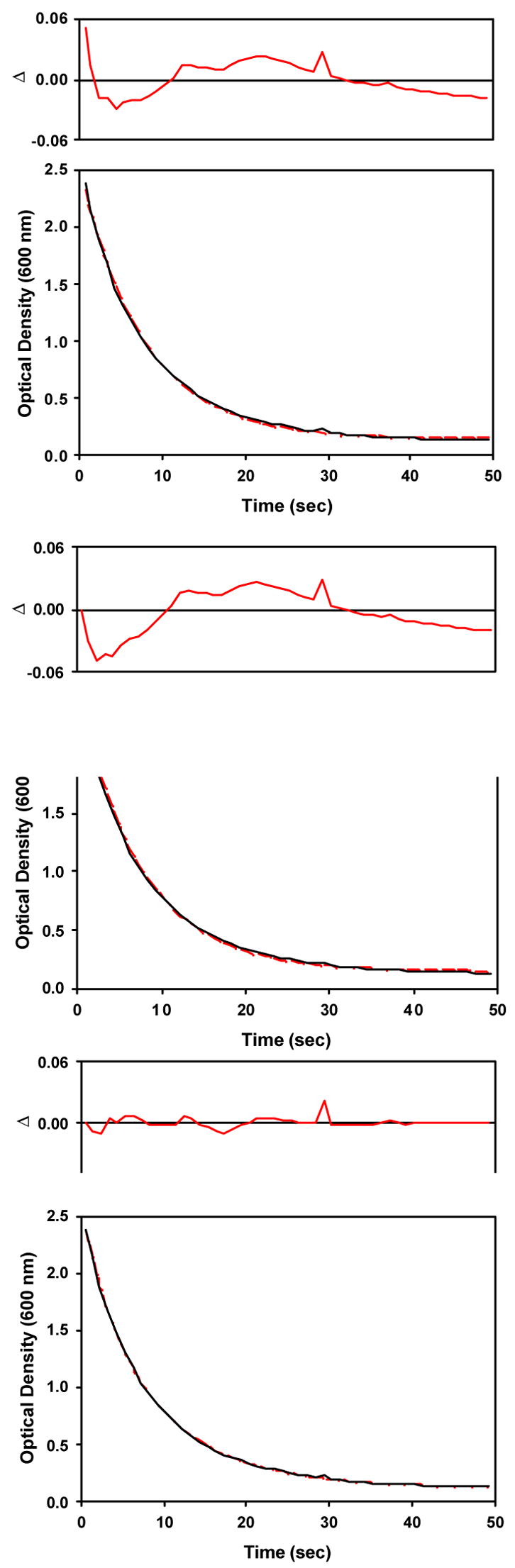
Observed (black) and calculated (red) optical densities at 600 nm calculated using a single exponential (top), a global A → B model (middle) and a global A → C, B → C model (bottom) for decomposition of the alkylperoxoiron(III) intermediate supported by TPA to oxoiron(IV) in CH3CN in the presence of pyridine-N-oxide at 231 K (data shown in Figure 4).
Figure 6.
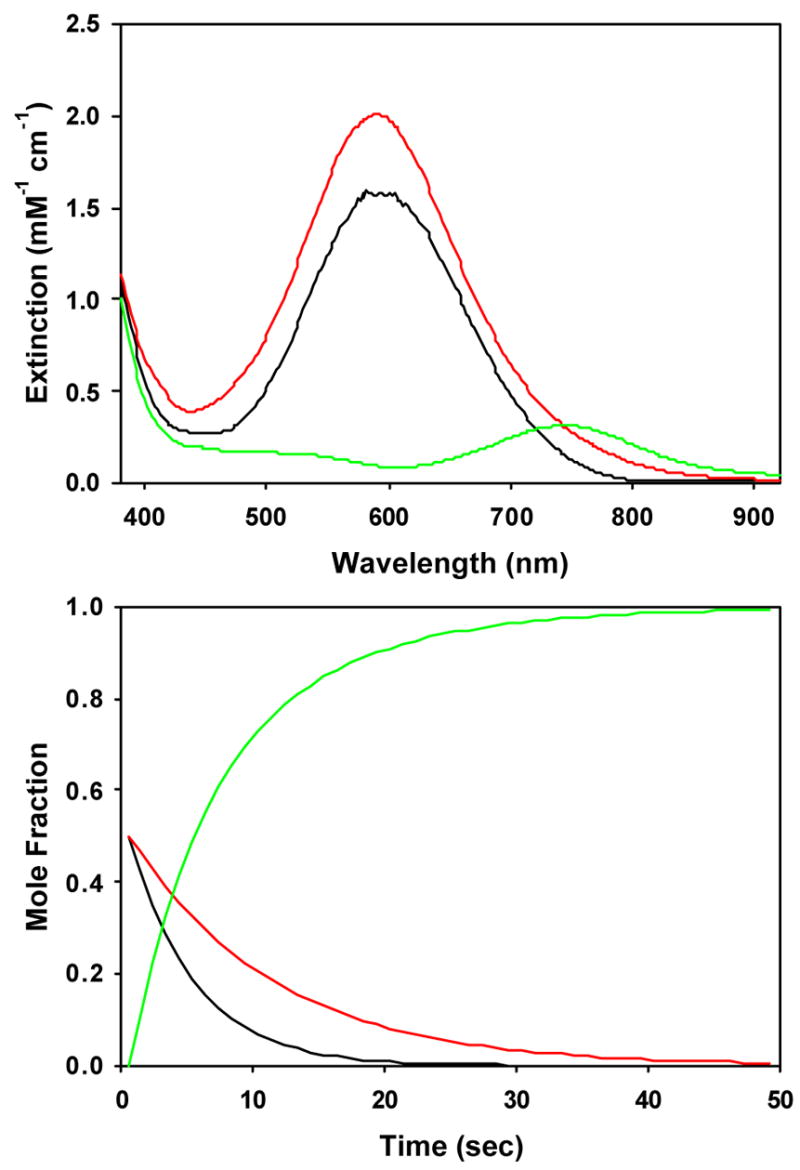
Calculated spectra (top) and time-dependent concentrations (bottom) for A → C, B → C kinetic model of time-dependent optical data shown in Figure 4 for conversion of the alkylperoxoiron(III) intermediate supported by TPA to oxoiron(IV) in CH3CN with added pyridine-N-oxide, with initial mole fractions arbitrarily set to [A]0 = 0.50, [B]0 = 0.50, [C]0 = 0.
Decay of [FeIII(β-BPMCN)OOtBu)]2+ in CH3CN
We recently reported the reaction chemistry of alkylperoxoiron(III) intermediates supported by the BPMCN ligand in its cis-beta topology.15 Interestingly, this chemistry exhibited a marked solvent dependence, with distinct decomposition reactions forming an oxoiron(IV) complex in CH3CN and a (hydroxo)alkylperoxoiron(IV) complex in CH2Cl2 (Figure 7), both in essentially quantitative yields as demonstrated by Mössbauer spectroscopy. EPR and Raman spectral characterization of the alkylperoxoiron(III) intermediates revealed two overlapping low-spin signals in organonitrile solvents, but significant crossover to a mixture of high-spin species in CH2Cl2 (Table 2). Kinetic analyses of time-dependent visible-NIR spectrophotometric data for conversion of the alkylperoxoiron(III) intermediates to the disparate iron(IV) products were undertaken to gain insight into the solvent-dependent decay mechanisms.
Figure 7.
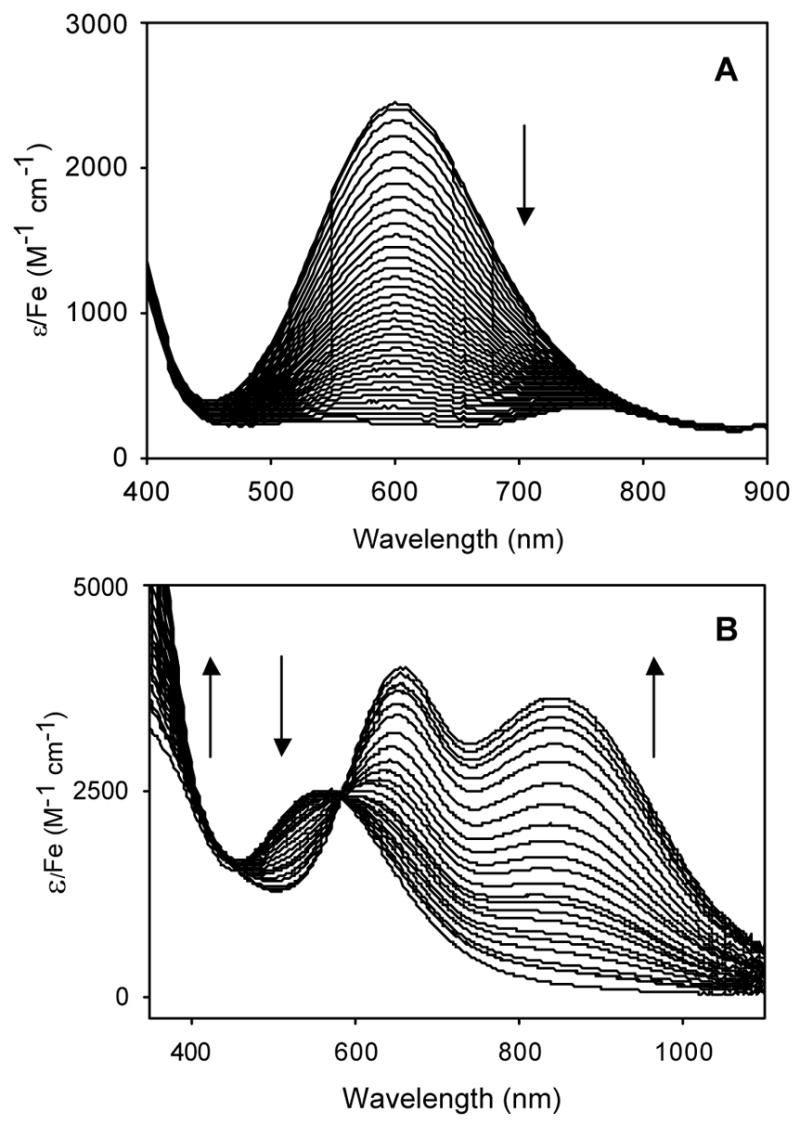
Time dependent evolution of visible-NIR spectra for decomposition of the alkylperoxoiron(III) intermediates supported by β-BPMCN to oxoiron(IV) in CH3CN/H2O at 228 K (A; [FeII]0 = 0.58 mM, [tBuOOH]0 = 1.2 mM, [H2O] = 0.3 M), and to alkylperoxoiron(IV) in CH2Cl2 at 206 K (B; [FeII]0 = 1.8 mM, [tBuOOH]0 = 9.0 mM), as monitored by visible-NIR spectroscopy.
Spectra obtained during decay of the alkylperoxoiron(III) intermediate to an oxoiron(IV) species at 228K in CH3CN resembled the TPA reaction (Figure 7A). For this reaction, it was necessary to introduce 0.3 mM H2O to accelerate the initial oxidation of iron(II), presumably converting the kinetically inert low-spin [FeII(BPMCN)(NCMe)2]2+ precursor to the more labile high-spin [FeII(BPMCN)(OH2)2]2+; therefore, the obtained alkylperoxoiron(III) species might have retained an aqua ligand in the sixth site. An intense LMCT chromophore centered at 600 nm (ε = 2500 M−1 cm−1) bleached monotonically to form an oxoiron(IV) ligand field band (λmax = 753 nm, ε = 280 M−1 cm−1); with this red-shifted LMCT, no isosbestic points were evident. A single wavelength trace coincident with the LMCT maximum deviated from a simple exponential (Figure 8). Again, the data were successfully fit to a parallel (A → C, B → C) reaction scheme (Figures S21, S22 in Supporting Information), with kobs = 7.88(8) × 10−3 s−1, 9.2(1) × 10−4 s−1 (Figure 9).
Figure 8.
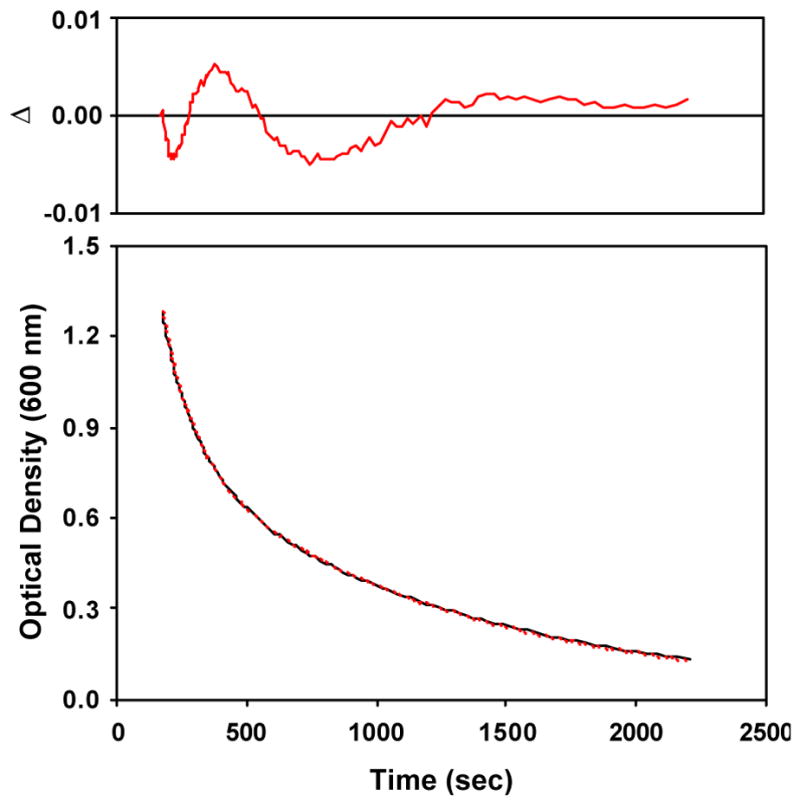
Observed (black) vs. calculated (red) optical density at 600 nm for conversion of -supported alkylperoxoiron(III) intermediate supported by β-BPMCN to oxoiron(IV) in CH3CN/H2O at 228 K for data shown in Figure 7A.
Figure 9.
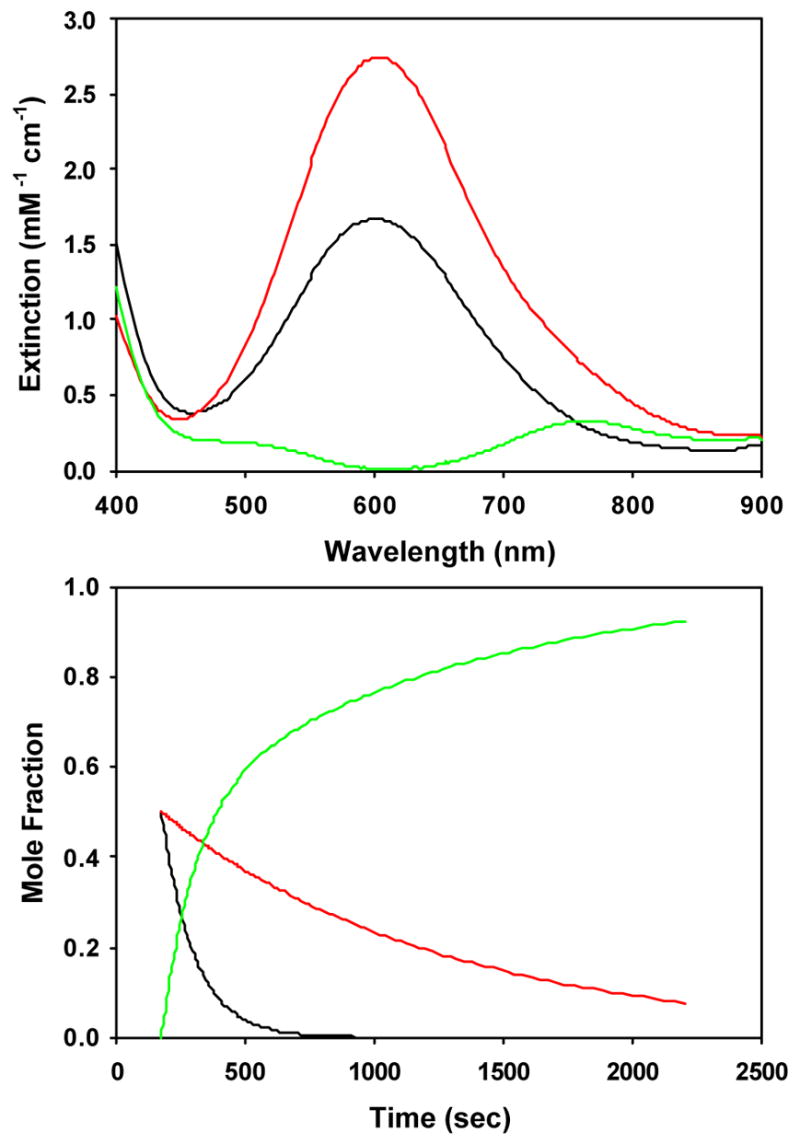
Calculated spectra (top) and time-dependent concentrations (bottom) for A → C, B → C kinetic model for decomposition of the alkylperoxoiron(III) intermediate supported by β-BPMCN to oxoiron(IV) in CH3CN/H2O at 228 K, with initial mole fractions arbitrarily set to [A]0 = 0.50, [B]0 = 0.50, [C]0 = 0.
Decay of [FeIII(β-BPMCN)OOtBu)]2+ in CH2Cl2
The alkylperoxoiron(III) intermediate of β-BMPCN in CH2Cl2 exhibited a distinct purple chromophore (λmax = 566 nm, ε = 2500 M−1cm−1, Figure 7B) at 206 K, and bleaching of this chromophore produced a unique product spectrum (λmax = 656, 845 nm; ε = 4000, 3600 M −1cm−1) that could not be assigned to an oxoiron(IV) complex. Raman and NMR spectroscopy were consistent with the presence of an alkylperoxo ligand, and hence the product was assigned as an unprecedented (hydroxo)alkylperoxoiron(IV) complex;15 this product formulation is equivalent to a tBuOOH adduct of an oxoiron(IV) complex. The time course of this reaction was most obvious at long wavelength, where the distinctive product bands dominate the spectrum; the reaction kinetics were biphasic at 840 nm, with a brief induction period preceding first-order growth of the product chromophore (Figure 10). Furthermore, the optical data failed to exhibit clean isosbestic points throughout the reaction, clearly indicating the presence of at least one intermediate. The time-dependent optical data were accordingly fit using global least-squares techniques to a scheme of consecutive first-order reactions, A → B → C (kobs = 1.73(6) × 10−3 s−1, 5.22(7) × 10−4 s−1, Figure 11 and Figures S23, S24 in Supporting Information). The two rate constants from the kinetic analysis of data obtained at −67 °C were found to be independent of the starting iron concentration used (0.07 to 0.33 mM, Table S1), supporting the validity of eqn 4 for this transformation. The order of the two calculated rate constants could not be determined, as plausible spectra for the intermediate (“B”) were calculated for either permutation. In fact, both calculated spectra for B retained a significant CT band only slightly blue-shifted from the initial chromophore, suggesting that the intermediate is a distinct alkylperoxoiron(III) complex.
Figure 10.
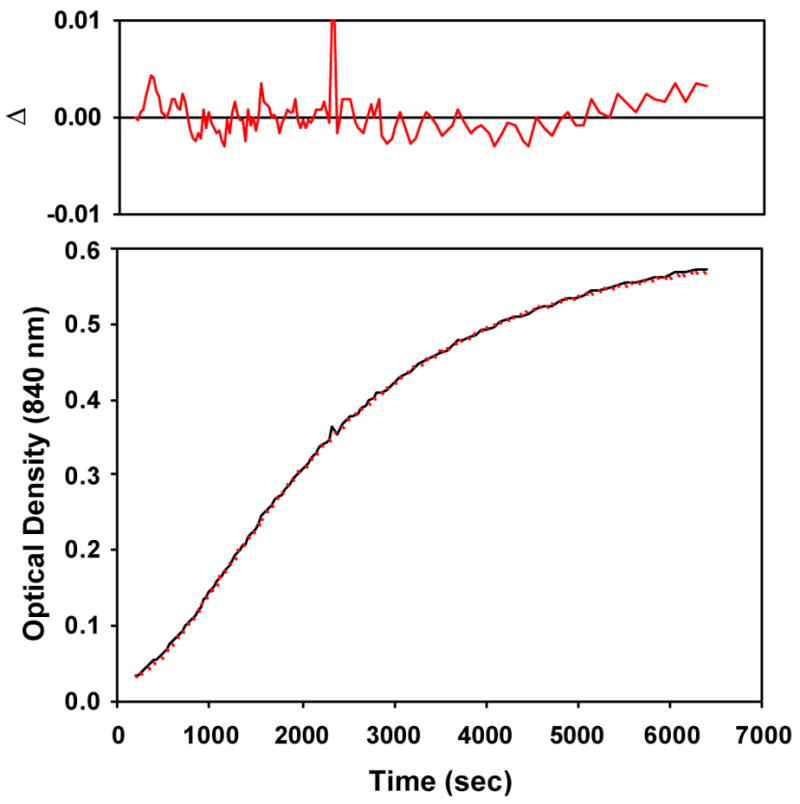
Observed (black) vs. calculated (red) optical density at 840 nm for conversion of alkylperoxoiron(III) intermediate supported by β-BPMCN to an (hydroxo)alkylperoxoiron(IV) product at 206 K for data shown in Figure 7B (pathlength = 0.1 mm).
Figure 11.
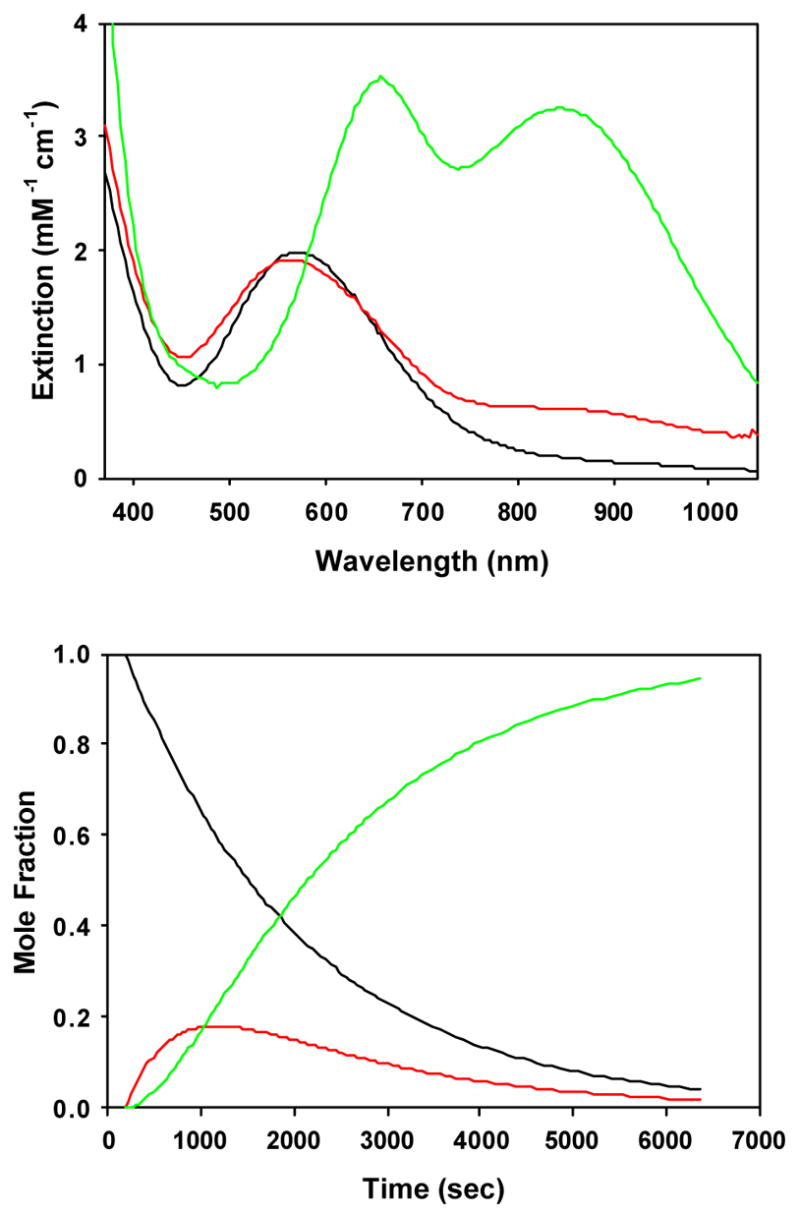
Calculated spectra (top) and time-dependent concentrations (bottom) for an A → B → C kinetic model to the time-dependent optical data shown in Figure 7B for decomposition of the alkylperoxoiron(III) intermediate supported by β-BPMCN in CH2Cl2 at 206 K.
The reaction was monitored independently at −67 °C by periodically freeze-quenching aliquots into EPR tubes (Figure 12). Three different signals (g = 1.96–2.18, g = 4.3, g = 8) corresponding to an 86% yield of spin-isomeric alkylperoxoiron(III) species were observed;15 their monotonic decay to EPR-silent iron(IV) product paralleled a single exponential corresponding to the slower rate determined optically under comparable conditions (Figure 12). Because the brief induction period was not resolved in this time course, a more detailed analysis was not warranted, notwithstanding the more complex kinetic model applied to the optical data. Qualitative monitoring of the reaction by resonance Raman spectroscopy revealed only minor initial changes in the alkylperoxoiron(III) ν(O-O) modes, followed by their subsequent decay. Finally, the rates of reaction of all the alkylperoxoiron(III) complex intermediates to iron(IV) species were determined over a range of experimentally accessible temperatures (Table S1 in Supporting Information). Arrhenius plots are shown in Figure 13 and derived activation parameters are collected in Table 3. The temperature dependences are all very similar, except for that of the slower observed rate for decay of the alkylperoxoiron(III) intermediate supported by β-BPMCN in CH2Cl2, which forms the unique alkylperoxoiron(IV) product. Its smaller slope compared to the others suggests a different chemical transformation from that associated with the other reactions. Most importantly, the applied kinetic models are robust over a wide range of temperatures and observed rates.
Figure 12.
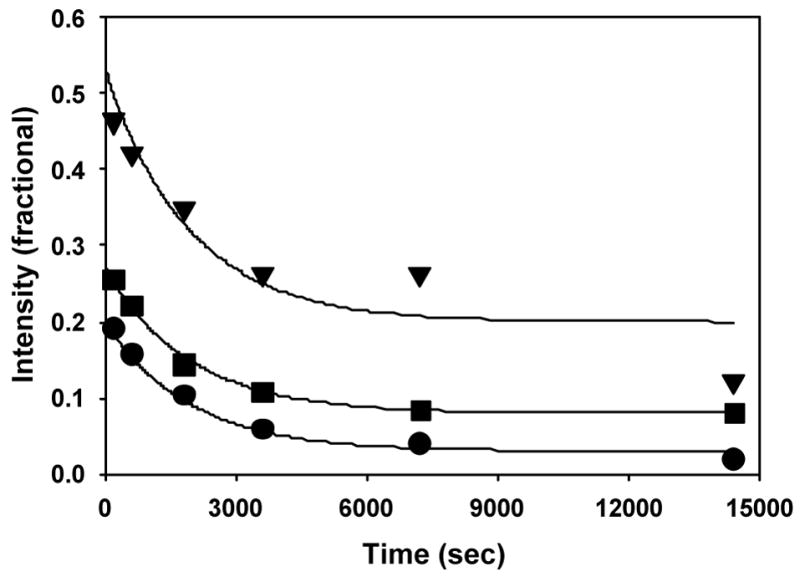
Time course for decay of the three EPR signals associated with the alkylperoxoiron(III) intermediate supported by β-BPMCN in CH2Cl2 at 206 K ([FeII]0 = 1.0 mM, [tBuOOH]0 = 10 mM; circle, g = 2.2; square, g= 4.3; triangle, g = 8). Exponentials correspond to the slower observed rate constant of 5.2 × 10−4 s−1 obtained from the A → B → C kinetic model fit to optical data (Figure 11).
Figure 13.
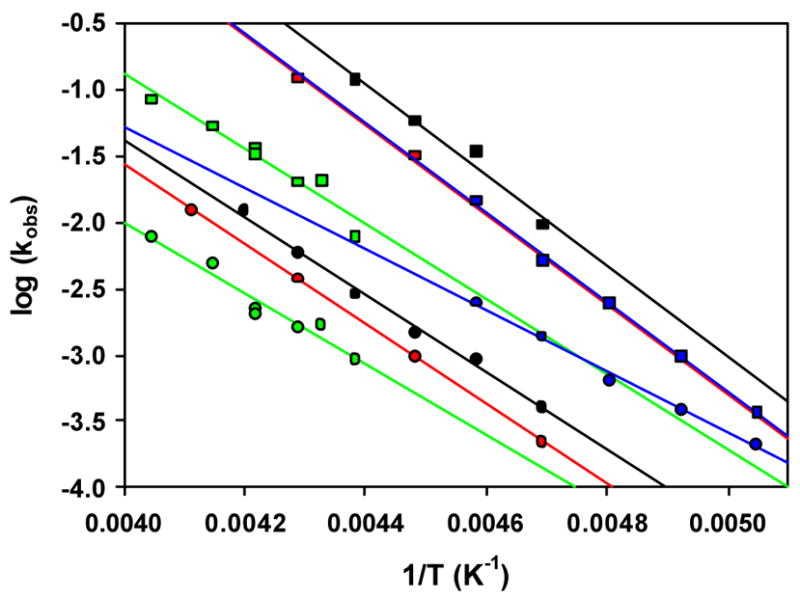
Arrhenius plots for the decay reactions of alkylperoxoiron(III) intermediates to iron(IV) products for the following ligands and solvents: TPA/acetone (red); 5-Me3TPA/acetone (black); β-BPMCN/CH3CN/H2O (green); and β-BPMCN/CH2Cl2 (blue).
Table 3.
Observed temperature dependence for reaction steps forming iron(IV) species.
| Ligand, solvent | R2 | ΔH‡obs (kJ/mol) | ΔS‡obs (J/molK) | Reference |
|---|---|---|---|---|
| 5Me3TPA, acetone | ||||
| k1 | 0.970 | 64(8) | +19(37) | a |
| k2 | 0.994 | 54(2) | −54(10) | |
|
| ||||
| TPA, acetone | ||||
| k1 | 0.997 | 63(4) | +9(17) | a |
| k2 | 0.999 | 56(1) | −50(3) | |
|
| ||||
| BPMCN, CH3CN | ||||
| k1 | 0.942 | 52(6) | −51(26) | a |
| k2 | 0.941 | 49(6) | −85(24) | |
|
| ||||
| BPMCN, CH2Cl2 | ||||
| kslow | 0.998 | 42(2) | −98(11) | a |
| kfast | 0.993 | 63(2) | +9(9) | |
|
| ||||
| TPA, CH3CN/PyNO | 52(1) | −74(3) | 38 | |
|
| ||||
| TPA, acetone/H2O2 | 54(3) | −39(12) | 49 | |
This work.
4. Discussion
This study has examined the decomposition of low-spin alkylperoxoiron(III) intermediates supported by the three closely related tetradentate non-heme ligands shown in Scheme 1 to metastable iron(IV) products. The study included reactions in several solvents, so that the sixth solvento ligand site was also varied. The alkylperoxoiron(III) intermediates were formed in situ by rapid oxidation of iron(II) complexes with excess tBuOOH; this process is essentially quantitative in CH3CN and CH2Cl2 but somewhat less efficient in acetone, particularly at higher temperatures. Subsequent decomposition reactions involve O -O bond homolysis to form oxoiron(IV) complexes in three cases and a distinct alkylperoxoiron(IV) species in the case of the β-BPMCN complex in CH2Cl2. One plausible interpretation of the results would associate all observed reaction rates with FeIIIO-OR bond homolyses of alkylperoxoiron(III) complex intermediates, except for the distinctive slower rate for β-BPMCN in CH2Cl2, which might reflect a different sort of transformation (vide infra).
An unexpected complication was the general observation of biphasic kinetics for the conversion of alkylperoxoiron(III) intermediates to oxoiron(IV), which was interpreted for all these cases to involve two parallel decay pathways. A plausible origin for the convergent parallel reaction scheme is isomerism of the alkylperoxoiron(III) reactants. The most straightforward rationale for this situation would be geometric isomerism of the alkylperoxoiron(III) complex, in which the peroxide is coordinated in either of the two inequivalent sites trans to the amine or a pyridine donor of the tetradentate supporting ligand (Scheme 1). Isomers decomposing by O-O bond homolysis would yield two distinct first-order traces under the differential trans influence, provided that the low spin state of iron(III) prevented facile interconversion of the isomers on the reaction timescale. The ratio of observed rates is consistent with a modest trans influence, Δ ΔG# = 2–7 KJ/mole. DFT calculations on isomers of [FeIII(TPA)(OH)(OOH)]+ suggest the ground state arrangement of the peroxo ligand trans to the amine would be more stable by only 11 KJ/mole,58 and a recent DFT study of low-spin alkylperoxoiron(III) complexes of a related tetradentate ligand suggest the isomeric Raman spectra would be very similar.40 Indeed, the presumptive isomers are not obviously resolved in EPR and Raman spectra, so independent corroboration of this hypothesis has not been possible.
Other forms of isomerism might include five- vs. six-coordinate species, different solvento co-ligands, and different spin states. Coincident solvent effects on both observed rates clearly preclude a common five-coordinate species, and significantly different sixth-site ligands might be expected to shift the relative LMCT energies. Moreover, five-coordinate alkylperoxoiron(III) complexes are expected to be high-spin, and such species are not observed for the TPA and BPMCN intermediates in MeCN.48 We also discount the possibility of cis- and trans-folded peroxide ligands; for example, calculations on tBuOOH predict that the cis structure is coincident with the global maximum of the energy profile for rotation about the O-O bond.59
A theoretical study of FeO–OR bond homolysis in hypothetical model complexes [(H3N)4FeIII(OHx)(OOtBu)]x+ (x = 1, 2) concluded that a low-spin (S = ½) ground state facilitates dissociation of the alkoxyl radical and formation of the oxoiron(IV) center.47,48 The activation barrier was estimated to be 60–80 KJ/mole with minimal entropic contribution. In contrast, a high-spin (S = 5/2) state presents a significant orbital barrier to FeO–OR bond homolysis and instead leads to Fe–OOR bond homolysis.48 Use of the temperature-dependent rates observed in this study to derive experimental activation parameters for O -O bond homolysis may be problematic, given the possibilities of spin and sixth-site ligand equilibria. Notwithstanding the kinetic complexities already described, observed activation enthalpies obtained for decomposition of the various alkylperoxoiron(III) intermediates closely approach the theoretical estimate of the activation energy for homolytic O-O bond cleavage in a model low-spin alkylperoxoiron(III) complex (Table 3). Hence, we invoke a coincident mechanism of rate-limited, irreversible O-O bond homolysis for these reactions, eqns. 2,4 except as noted for the formation of alkylperoxiron(IV) in CH2Cl2 (vide infra).
There appears to be a compensation effect within the temperature-dependent rate data, such that increasing activation enthalpies are offset by increasingly positive entropies (Figure 14). Arbitrary fitting of a linear relationship to available data, including the previously reported pyridine-N-oxide promoted and acetone/H2O2 condensate reactions of TPA complexes,38,49 yields an isokinetic temperature β = 143 ± 12 K and ΔH‡ = 61.3(6) KJ/mole at the intercept. Similar trends reported for metal-alkyl bond homolyses of alkylcobalt(III) complexes were attributed to more product-like transition states resulting from increased endothermicity of the bond homolysis, with concomitantly positive activation entropy.60 The less compelling range of the postulated trend in the present work may reflect the relatively weak O-O bonds and attenuated steric effects on their cleavage. Nevertheless, the negative entropies observed herein are striking for a unimolecular fragmentation.61,62
Figure 14.
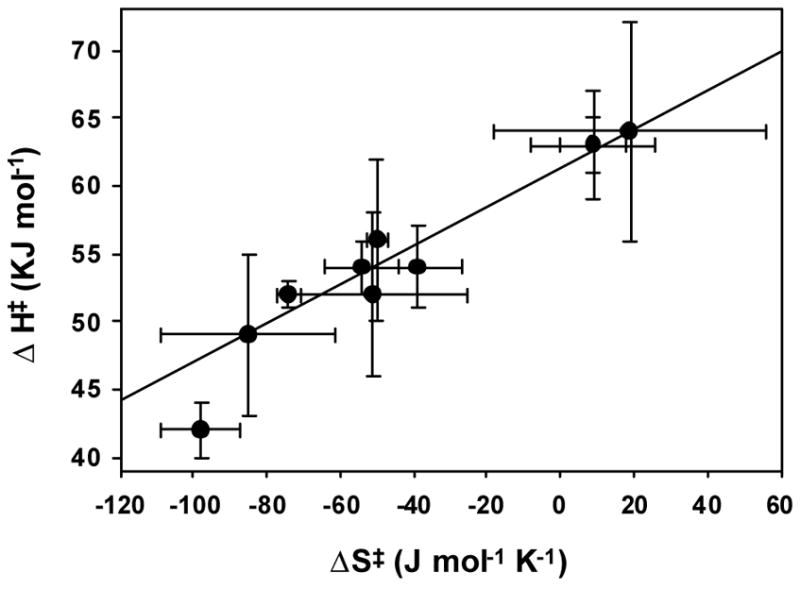
Isokinetic plot for decompositions of alkylperoxoiron(III) intermediates.
The decomposition of the β-BPMCN-supported alkylperoxoiron(III) complex in CH2Cl2 (Figure 7B) differs from the other reactions in several respects.15 First of all, instead of an oxoiron(IV) complex, a unique (hydroxo)alkylperoxoiron(IV) product is formed, which entails the interaction of the iron center with a second equivalent of tBuOOH. Secondly, the kinetics of its formation exhibits biphasic kinetics that requires a sequential (A → B → C) scheme (rather than a parallel scheme as found for the others), in which the faster step has a rate constant about three times larger than that of the slower step. Thirdly, the faster observed rate has a temperature dependence similar to the other reactions and activation parameters that fit into the line drawn for the isokinetic plot (Figures 13 and 14), but the slower observed rate is distinct and has a smaller activation enthalpy. Given the product formulation for this reaction, the mechanism of product formation likely involves the combination of distinct adduct formation and O-O bond homolysis steps, but our kinetic analysis does not allow the order of the observed rates to be unambiguously established. Therefore, four permutations are possible for assignment of the two observed rate constants to the two mechanistic steps (Scheme 2).
Scheme 2.

The most straightforward sequence involves initial O-O bond homolysis of the FeIII-OOR intermediate to form an FeIV =O species followed by 1,2-capture of excess tBuOOH to form the (hydroxo)alkylperoxoiron(IV) product (permutations i and iii in Scheme 2). However, the fact that the calculated spectrum for intermediate B is only slightly shifted from the initial alkylperoxoiron(III) chromophore suggests that B is still an alkylperoxoiron(III) species and O-O bond cleavage to form an iron(IV) species has not occurred at this stage. Thus, permutations i and iii can be excluded by this argument.
Permutations ii and iv involve initial formation of the FeIII(OOR)(ROOH) adduct followed by O-O bond homolysis; they differ only in the assignments of the faster and the slower steps. On the assumption that the FeIII(OOR)(ROOH) adduct is the only plausible low-spin iron(III) species that can form in CH2Cl2 solution, the observation that the low-spin iron(III) species represents only a small fraction (20%) of the iron(III) species present in both EPR and Raman spectra is inconsistent with permutation iv where the adduct would be expected to accumulate and become the major component. Furthermore the EPR time course clearly shows the simultaneous decay of both high-spin and low-spin iron(III) signals to silent oxoiron(IV) species with the same slow rate constant (Figure 12), which is inconsistent with permutation iv. On the other hand, the temperature dependence of the faster step matches those of the other transformations discussed in this paper, supporting its assignment to the O-O bond homolysis step, as in permutation ii. This leads to the assignment of the adduct formation step to the slower observed rate, which has the more negative entropy of activation that would be associated with a bimolecular reaction. Taken together, these arguments favor assignment of permutation ii, in which a slow initial step forms an FeIII(OOR)(ROOH) adduct, presumably including spin crossover to a reactive low-spin state that subsequently forms a (alkylhydroperoxide)oxoiron(IV) complex by O-O bond homolysis at the faster observed rate, with final, rapid tautomerization to the final (hydroxo)alkylperoxoiron(IV) product.
Two problems remain unresolved regarding this mechanistic assignment. First, the fast observed rate assigned to alkylperoxoiron(III) homolysis should also exhibit complications of geometric isomerism, but the reaction kinetics are fit well by sequential single exponentials. Unlike the alkylperoxoiron(III) intermediates, clear spectroscopic evidence for isomerism was obtained for the alkylperoxoiron(IV) product: two distinct, paramagnetically shifted 2H NMR signals were observed in a 60:40 ratio for the product derived from a reaction using (CD3)3COOH, and two sets of Raman modes exhibited different excitations using 632.8 and 752.5 nm laser irradiation.15 There are two possible explanations for this discrepancy. First, the iron(IV) isomers might indeed form by overlapping reactions of alkylperoxoiron(III) isomers, but the isomeric rates might be accidentally degenerate, as for the TPA-supported intermediate in the presence of pyridine-N-oxide. Secondly, rapid tautomerization within the FeIII(OOR)(ROOH) adduct would give exclusive reaction by the faster isomer, but the iron(IV) product might be configurationally unstable on the reaction timescale. A second issue is whether a [tBuOOH] dependence can be observed for the slower observed rate assigned to adduct formation. Bimolecular adduct formation should exhibit first-order dependence, but not the reverse unimolecular dissociation, spin crossover, or subsequent homolysis step. Therefore, the kinetic dependence on [tBuOOH] will fall between zero- and first-order limits. Unfortunately, time dependent concentrations of alkyl hydroperoxide could not be determined, and the large, 5- to 10-fold stoichiometric excess of tBuOOH required to obtain full accumulation of the alkylperoxoiron(III) and alkylperoxoiron(IV) chromophores did not allow an experimental dependence on [tBuOOH] to be established for phases subsequent to the initial iron(II) oxidation.
Despite the significant remaining mechanistic uncertainties regarding formation of the alkylperoxoiron(IV) complex, a close relationship to formation of the other oxoiron(IV) products is clearly demonstrated, as the limiting rates of O-O bond homolyses leading to the disparate products are nearly coincident. Moreover, the overall kinetic data further substantiate the influence of the supporting ligation on the rate of O-O bond homolysis. Observed rates are clearly affected by the supporting and solvento ligands, as the combined kinetic effects exceed two orders of magnitude. Thus, the 5-Me3TPA reaction is slightly faster than TPA reaction in acetone, and the reaction in CH3CN promoted by pyridine-N-oxide is faster still. The β-BPMCN reaction is relatively slow in CH3CN/H2O, but matches the TPA/acetone rate in CH2Cl2, notwithstanding despite the formation of divergent iron(IV) products. Notwithstanding the unanticipated complexity in the observed kinetics, the experimental observations substantiate earlier theoretical calculations that predicted the observed enthalpic barrier to O-O bond homolyses of low-spin alkylperoxoiron(III) intermediates,46,47 while further elucidating an unexpectedly rich reaction landscape.
Supplementary Material
Kinetic data, resonance Raman and EPR spectra of TPA and 5-Me3TPA alkylperoxoiron(III) intermediates in frozen acetone, details of the kinetic fits. These data are available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the National Institutes of Health (GM-33162 and GM-38767 to LQ and EB-001475 to EM). M.C. thanks Fundacio La Caixa for a postdoctoral fellowship, and A.S. thanks the National Science Foundation for a Graduate Research Fellowship.
References
- 1.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 2.Girerd JJ, Banse F, Simaan AJ. Struct Bonding. 2000;97:145–177. [Google Scholar]
- 3.MacFaul PA, Ingold KU, Wayner DD, Que L., Jr J Am Chem Soc. 1997;119:10594–10598. [Google Scholar]
- 4.Nielsen A, Larsen FB, Bond AD, McKenzie CJ. Angew Chem Int Ed. 2006;45:1602–1606. doi: 10.1002/anie.200502656. [DOI] [PubMed] [Google Scholar]
- 5.Fitzpatrick PF. Biochemistry. 2003;42:10483–10491. [Google Scholar]
- 6.Bassan A, Blomberg MRA, Siegbahn PEM. Eur J Chem. 2003;9:4055–4067. doi: 10.1002/chem.200304768. [DOI] [PubMed] [Google Scholar]
- 7.Costas M, Chen K, Que L., Jr Coord Chem Rev. 2000;200–202:517–544. [Google Scholar]
- 8.Shan X, Que L., Jr J Inorg Biochem. 2006;100:421–433. doi: 10.1016/j.jinorgbio.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Grapperhaus CA, Mienert B, Bill E, Weyhermüller T, Wieghardt K. Inorg Chem. 2000;39:5306–5317. doi: 10.1021/ic0005238. [DOI] [PubMed] [Google Scholar]
- 10.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 11.Lim MH, Rohde JU, Stubna A, Bukowski MR, Costas M, Ho RYN, Münck E, Nam W, Que L., Jr Proc Nat’l Acad Sci USA. 2003;100:3665–3670. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaizer J, Klinker EJ, Oh NY, Rohde JU, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L., Jr J Am Chem Soc. 2004;126:472–473. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 13.Balland V, Charlot MF, Banse F, Girerd JJ, Mattioli TA, Bill E, Bartoli JF, Battioni P, Mansuy D. Eur J Inorg Chem. 2004:301–308. [Google Scholar]
- 14.Rohde JU, Torelli S, Shan X, Lim MH, Klinker EJ, Kaizer J, Chen K, Nam W, Que L., Jr J Am Chem Soc. 2004;126:16750–16761. doi: 10.1021/ja047667w. [DOI] [PubMed] [Google Scholar]
- 15.Jensen MP, Costas M, Ho RYN, Kaizer J, Mairata i Payeras A, Münck E, Que L, Jr, Rohde J-U, Stubna A. J Am Chem Soc. 2005;127:10512–10525. doi: 10.1021/ja0438765. [DOI] [PubMed] [Google Scholar]
- 16.Bukowski MR, Koehntop KD, Stubna A, Bominaar EL, Halfen JA, Münck E, Nam W, Que L., Jr Science. 2005;310:1000–1002. doi: 10.1126/science.1119092. [DOI] [PubMed] [Google Scholar]
- 17.Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L., Jr Angew Chem Int Ed. 2005;44:3690–3694. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]
- 18.Kim SO, Sastri CV, Seo MS, Kim J, Nam W. J Am Chem Soc. 2005;127:4178–4179. doi: 10.1021/ja043083i. [DOI] [PubMed] [Google Scholar]
- 19.Sastri CV, Park MJ, Ohta T, Jackson TA, Stubna A, Seo MS, Lee J, Kim J, Kitagawa T, Münck E, Que L, Jr, Nam W. J Am Chem Soc. 2005;127:12494–12495. doi: 10.1021/ja0540573. [DOI] [PubMed] [Google Scholar]
- 20.Suh Y, Seo MS, Kim KM, Kim YS, Jang HG, Tosha T, Kitagawa T, Kim J, Nam W. J Inorg Biochem. 2006;100:627–633. doi: 10.1016/j.jinorgbio.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Paine TK, Costas M, Kaizer J, Que L., Jr J Biol Inorg Chem. 2006;11:272–276. doi: 10.1007/s00775-006-0089-6. [DOI] [PubMed] [Google Scholar]
- 22.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 23.Price JC, Barr EW, Glass TW, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 24.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 25.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 26.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Krebs C. Proc Nat’l Acad Sci USA. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Que L., Jr Nat Struct Biol. 2000;7:182–184. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 28.Decker A, Rohde J-U, Que L, Jr, Solomon EI. J Am Chem Soc. 2004;126:5378–5379. doi: 10.1021/ja0498033. [DOI] [PubMed] [Google Scholar]
- 29.Pestovsky O, Stoian S, Bominaar EL, Shan X, Münck E, Que L, Jr, Bakac A. Angew Chem Int Ed. 2005;44:6871–6874. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- 30.Ménage S, Wilkinson EC, Que L, Jr, Fontecave M. Angew Chem Int Ed. 1995;34:203–205. [Google Scholar]
- 31.Kim J, Larka E, Wilkinson EC, Que L., Jr Angew Chem Int Ed. 1995;34:2048–2051. [Google Scholar]
- 32.Zang Y, Kim J, Dong Y, Wilkinson EC, Appelman EH, Que L., Jr J Am Chem Soc. 1997;119:4197–4205. [Google Scholar]
- 33.Wada A, Ogo S, Watanabe Y, Mukai M, Kitagawa T, Jitsukawa K, Masuda H, Einaga H. Inorg Chem. 1999;38:3592–3593. doi: 10.1021/ic9900298. [DOI] [PubMed] [Google Scholar]
- 34.Ogihara T, Hikichi S, Akita M, Uchida T, Kitagawa T, Moro-oka Y. Inorg Chim Acta. 2000;297:162–170. [Google Scholar]
- 35.Sobolev AP, Babushkin DE, Talsi EP. J Mol Cat A. 2000;159:233–245. [Google Scholar]
- 36.Kim J, Zang Y, Costas M, Harrison RG, Wilkinson EC, Que L., Jr J Biol Inorg Chem. 2001;6:275–284. doi: 10.1007/s007750000198. [DOI] [PubMed] [Google Scholar]
- 37.Jensen MP, Lange SJ, Mehn MP, Que EL, Que L., Jr J Am Chem Soc. 2003;125:2113–2128. doi: 10.1021/ja028478l. [DOI] [PubMed] [Google Scholar]
- 38.Kaizer J, Costas M, Que L., Jr Angew Chem Int Ed. 2003;42:3671–3673. doi: 10.1002/anie.200351694. [DOI] [PubMed] [Google Scholar]
- 39.Lehnert N, Fujisawa K, Solomon EI. Inorg Chem. 2003;42:469–481. doi: 10.1021/ic020496g. [DOI] [PubMed] [Google Scholar]
- 40.Bautz J, Comba P, Que L., Jr Inorg Chem. doi: 10.1021/ic0602401. in press. [DOI] [PubMed] [Google Scholar]
- 41.Skrzypczak-Jankun E, Bross RA, Carroll RT, Dunham WR, Funk MO., Jr J Am Chem Soc. 2001;123:10814–10820. doi: 10.1021/ja011759t. [DOI] [PubMed] [Google Scholar]
- 42.Coulter ED, Emerson JP, Kurtz DM, Jr, Cabelli DE. J Am Chem Soc. 2000;122:11555–11556. [Google Scholar]
- 43.Emerson JP, Coulter ED, Cabelli DE, Phillips RS, Kurtz DM., Jr Biochemistry. 2002;41:4348–4357. doi: 10.1021/bi0119159. [DOI] [PubMed] [Google Scholar]
- 44.Mathé C, Mattioli TA, Horner O, Lombard M, Latour JM, Fontecave M, Nivière V. J Am Chem Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]
- 45.Nivière V, Asso M, Weill CO, Lombard M, Guigliarelli B, Favaudon V, Houée-Levin C. Biochemistry. 2004;43:808–818. doi: 10.1021/bi035698i. [DOI] [PubMed] [Google Scholar]
- 46.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Science. 2003;299:1039–1042. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 47.Lehnert N, Ho RYN, Que L, Jr, Solomon EI. J Am Chem Soc. 2001;123:8271–8290. doi: 10.1021/ja010165n. [DOI] [PubMed] [Google Scholar]
- 48.Lehnert N, Ho RYN, Que L, Jr, Solomon EI. J Am Chem Soc. 2001;123:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 49.Mairata i Payeras A, Ho RYN, Fujita M, Que L., Jr Eur J Chem. 2004;10:4944–4953. doi: 10.1002/chem.200400480. [DOI] [PubMed] [Google Scholar]
- 50.Maeder M, Zuberbühler AD. Anal Chem. 1990;62:2220–2224. [Google Scholar]
- 51.Trapping of the reactive tBuO• byproduct was further considered. This radical can decay either by β-scission of tBuO• to form acetone and CH3•, or by H-atom abstraction from solvent and excess tBuOOH to form relatively unreactive alkylperoxyl radicals, tBuOO• decomposing in turn to O2 and stable tBuOOtBu. Comparable rates for these processes are expected at room temperature (e.g., 6 × 104 s−1 and 1 × 107 M−1 s−1 for β-elimination and H-atom abstraction from tBuOOH, respectively in CH3CN).52–55 However, the reaction barrier to bimolecular H-atom abstraction is expected to be largely entropic,56 whereas unimolecular β-scission exhibits a relatively favorable activation entropy and a largely enthalpic barrier.54,55 So the bimolecular decay pathway is expected to dominate trapping at the lower temperatures used in this study. This notion is corroborated by reaction of [FeII(β-BPMCN)](OTf)2 with (CD3)3COOH in CH2Cl2 at −77 °C; integrity of the d9-tBu substituent during the course of the reaction was demonstrated, as the resonance of d6-acetone was not observed by 2H NMR spectroscopy.15
- 52.Paul H, Small RD, Jr, Scaiano JC. J Am Chem Soc. 1978;100:4520–4527. [Google Scholar]
- 53.Snelgrove DW, Lusztyk J, Banks JT, Mulder P, Ingold KU. J Am Chem Soc. 2001;123:469–477. [Google Scholar]
- 54.Tsentalovich YP, Kulik LV, Gritsan NP, Yurkovskaya AV. J Phys Chem A. 1998;102:7975–7980. [Google Scholar]
- 55.Weber M, Fischer H. J Am Chem Soc. 1999;121:7381–7388. [Google Scholar]
- 56.Finn M, Friedline R, Suleman NK, Wohl CJ, Tanko JM. J Am Chem Soc. 2004;126:7578–7584. doi: 10.1021/ja0493493. [DOI] [PubMed] [Google Scholar]
- 57.Vajda S, Rabitz H. J Phys Chem. 1988;92:701–707. [Google Scholar]
- 58.Bassan A, Blomberg MRA, Siegbahn PEM, Que L., Jr J Am Chem Soc. 2002;124:11056–11063. doi: 10.1021/ja026488g. [DOI] [PubMed] [Google Scholar]
- 59.Khursan SL, Antonovsky VL. Russian Chem Bulletin, International Ed. 2003;52:1312–1325. [Google Scholar]; Izvestiya Akademii Nauk Seriya Khimicheskaya. 2003:1241–1253. [Google Scholar]
- 60.Halpern J. Bull Chem Soc Jpn. 1988;61:13–15. [Google Scholar]
- 61.Huang KW, Han JH, Cole AP, Musgrave CB, Waymouth RM. J Am Chem Soc. 2005;127:3807–3816. doi: 10.1021/ja044512f. [DOI] [PubMed] [Google Scholar]
- 62.DiPasquale AG, Hrovat DA, Mayer JM. Organometallics. 2006;25:915–924. doi: 10.1021/om050818z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kinetic data, resonance Raman and EPR spectra of TPA and 5-Me3TPA alkylperoxoiron(III) intermediates in frozen acetone, details of the kinetic fits. These data are available free of charge via the internet at http://pubs.acs.org.