

Abstract
The indeno[1,2-c]isoquinolines are an important class of topoisomerase I inhibitors with anticancer activity. The condensation of Schiff bases with homophthalic anhydrides provides a mixture of cis- and trans-4-carboxy-3,4-dihydro-3-phenyl-1(2H)isoquinolones. Although the cis products can be readily converted to indeno[1,2-c]isoquinolines with thionyl chloride, the trans products do not afford indeno[1,2-c]isoquinolines using this method. The present report describes a route for conversion of the trans diastereomers to indeno[1,2-c]isoquinolines using selenoxide elimination and Friedel-Crafts cyclization chemistry.
Indenoisoquinoline 4 (NSC 314622) was initially isolated as a byproduct during a total synthesis of the antileukemic agent nitidine chloride.1 Treatment of cis-3 with thionyl chloride unexpectedly resulted in the formation of 4 (Scheme 1) instead of the anticipated acid chloride.2 This thionyl chloride-mediated reaction is stereospecific, since similar treatment of trans-3 gave the corresponding acid chloride trans-5 instead of the indenoisoquinoline 4.1 Since the discovery that indenoisoquinoline 4 is a non-camptothecin topoisomerase I inhibitor,3 a variety of its analogs have been synthesized and evaluated for their potentials as novel topoisomerase I inhibitors with anticancer activity.4–16
SCHEME 1.
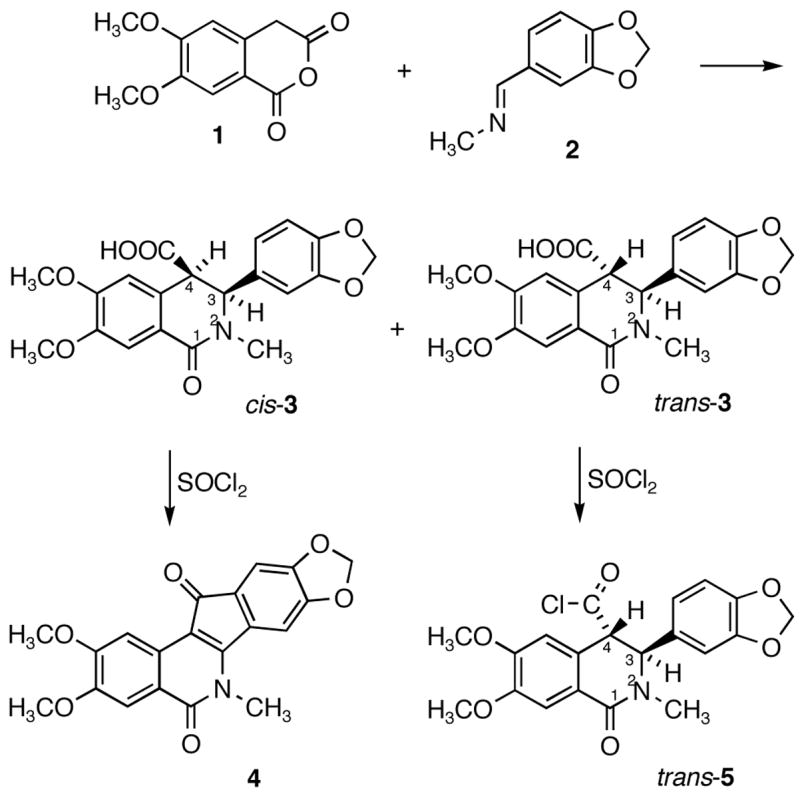
Stereospecific Reactions of cis-3 and trans-3 with Thionyl Chloride
The condensation of Schiff bases (e.g. 2) with homophthalic anhydrides (e.g. 1) usually results in the formation of a diastereomeric mixture of 3-aryl-4-carboxyisoquinolones, as exemplified by cis-3 and trans-3.17 This fact, coupled with the failure to convert the trans isomers (e.g. trans-3) to indenoisoquinolines (e.g. 4) by SOCl2, compromises the efficiency to make indenoisoquinolines of general structure 4 by this route because the trans products are "thrown out". In order to increase the efficiency of this approach to indenoisoquinolines, a method was sought to transform trans-3 to indenoisoquinoline 4.
The initial step in the conversion of cis-3 to indenoisoquinoline 4 by thionyl chloride is assumed to be conversion to the acid chloride cis-5 (Scheme 2). Acid-catalyzed enolization would provide the intermediate enol 6, which could react with thionyl chloride to afford the sulfinyl chloride 7. Loss of hydrochloric acid and sulfur monoxide would then generate the α,β-unsaturated acid chloride 8.2 Alternatively, reaction of thionyl chloride with the lactam carbonyl of cis-5 and deprotonation of H-4 could lead to intermediate 9, which may form the unsaturated acid chloride 8 through loss of hydrochloric acid and sulfur monoxide. Intramolecular Friedel-Crafts reaction of 8 could afford the product 4.
SCHEME 2.
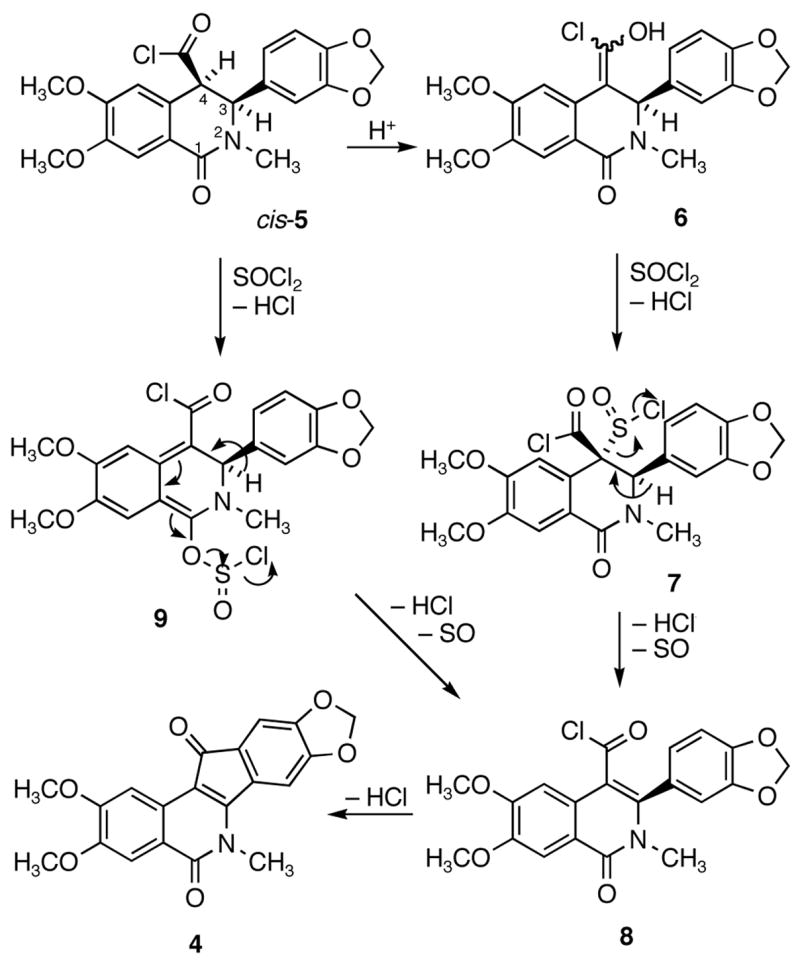
Possible Mechanisms for the Conversion of cis Acid Chloride 5 to Indenoisoquinoline 4
According to the possible mechanisms proposed for this SOCl2-mediated reaction of cis-3 to form 4 outlined in Scheme 2, deprotonation of H-4 from cis-5 is required.2 If a similar deprotonation were possible with trans-5, both of them would be able to deliver indenoisoquinoline 4 after SOCl2 treatment. The stereospecificity of the SOCl2-mediated reaction of acid 3 is therefore likely to be due to a difference in the kinetic acidity of H-4 in trans-5 vs. cis-5. Due to the A-strain present between the 3-phenyl ring and 2-methyl group, the 3-phenyl substituent is pseudoaxial in both diastereomers.15,17 Thus, H-4 in cis-5 is pseudoaxial, whereas in trans-5 it is pseudoequatorial. This prediction is consistent with the AM1 optimized geometries of trans-3 and cis-3 implemented in Gaussian03 (Figure 1).18 As a consequence, the C4-H4 bond in cis-5 has more orbital overlap with the adjacent aromatic ring than it does in trans-5, which renders H-4 kinetically more acidic in cis-5 than in trans-5.
FIGURE 1.
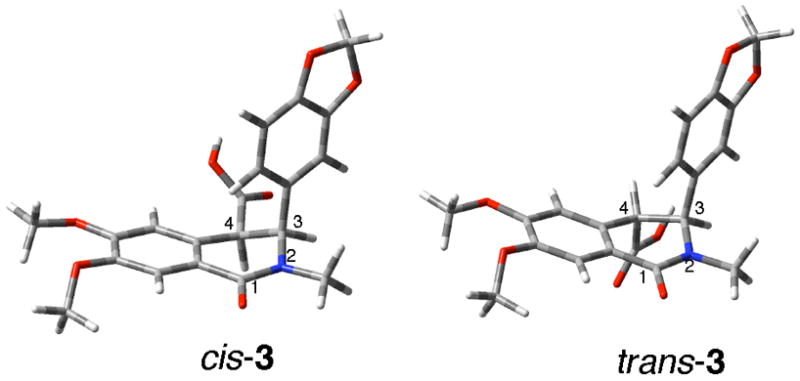
AM1 optimized geometries of cis-3 and trans-3.
Based on this kinetic acidity analysis, a stronger base would be required to deprotonate H4 in the trans series than in the cis series. Conversion of trans-3 to ester 10, followed by trapping the corresponding enolate with a selenium species and oxidation to the selenoxide, should then result overall in dehydrogenation,19 a key step in the formation of indenoisoquinoline 4 from cis-3. To this end, trans-3 was methylated with TMSCHN2 in MeOH-benzene to provide trans ester 10 (Scheme 3), the structure of which was confirmed by X-ray crystallography.15 Deprotonation of ester 10 with n-BuLi, followed by the treatment with phenylselenyl chloride, did not result in completion of the desired reaction, even after prolonged reaction time. Consistent with the recognition of the soft nature of selenium in terms of the HSAB (hard soft acid base) theory,20 a softer sodium enolate, formed by deprotonation with NaHMDS, was employed instead of the hard lithium enolate. To our delight, complete transformation and high yield (85%) of the dehydrogenated compound 11 was obtained after oxidative elimination. It should be noted that direct conversion of trans ester 10 to dehydrogenated compound 11 using a variety of oxidants (DDQ, CAN, SeO2) in different solvents (CH3CN, 1,4-dioxane, benzene, toluene) failed to yield complete transformation. This is in strong contrast to the corresponding cis ester, which was shown to be completely dehydrogenated in the presence of DDQ.2 Ester hydrolysis of 11 under basic conditions afforded acid 12. Prolonged heating at reflux is essential for complete saponification, which may be due to stabilization of the ester carbonyl by its incorporation into a vinylogous imide system. Acid chloride formation from 12 with SOCl2, followed by Friedel-Crafts cyclization, provided an 84% yield of indenoisoquinoline 4, which displayed physical and spectral data that were identical with that obtained from cis-3 by treatment with SOCl2. This represents an efficient procedure to convert trans-3 into a medicinally relevant molecule 4.
SCHEME 3.
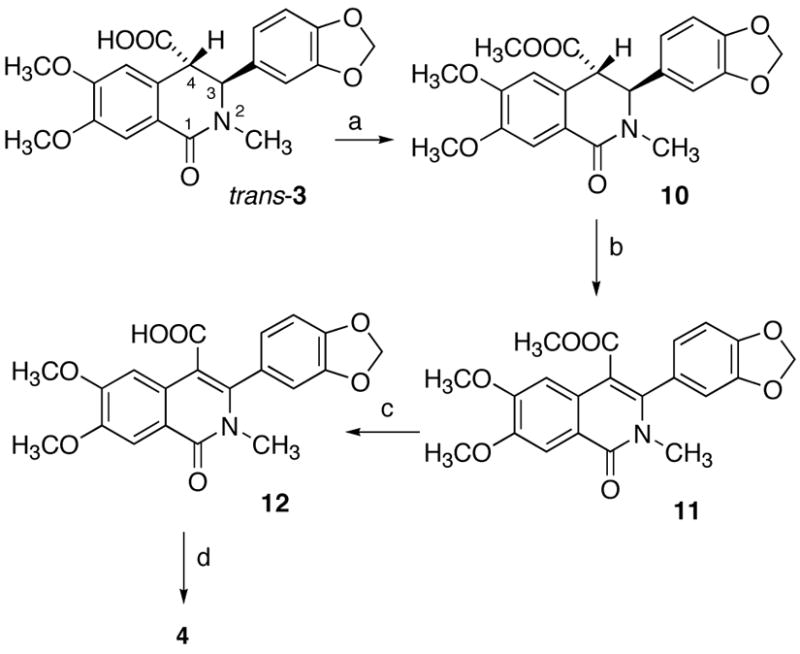
Synthesis of Indenoisoquinoline 4 from trans-3a
aReagents and conditions: (a) TMSCHN2, MeOH-benzene (2:7), room temperature, 30 min (99%); (b) (1) NaHMDS, PhSeCl, –78 ºC to room temperature, 12 h, (2) H2O2, AcOH, room temperature, 12 h (85%); (c) LiOH•H2O, THF-MeOH-H2O, reflux, 36 h (91%); (d) SOCl2, room temperature, 12 h (84%).
In conclusion, a method has been developed for converting trans-3 to indenoisoquinoline 4. This increases the efficiency of preparation of indenoisoquinolines from Schiff bases and homophthalic anhydrides in general. It has recently been reported that the three-component reaction of homophthalic anhydrides with amines and aldehydes in the presence of KAl(SO4)2·12H2O21 or in ionic liquid solvents22 stereoselectively affords high yields of cis-4-carboxy-3,4-dihydro-3-phenyl-1(2H)isoquinolones, and therefore these approaches should also be considered for maximizing the yields of indeno[1,2-c]isoquinolines in cases in which the yields of cis diastereomers are low from the Schiff base-anhydride approach. On the other hand, the present approach from the trans diastereomers offers another alternative for cases in which the yields of indeno[1,2-c]isoquinolines from the cis diastereomers are low, as it is in the synthesis of nitrated indeno[1,2-c]isoquinolines.10
Experimental Section
5,6-Dihydro-5,11-diketo-2,3-dimethoxy-6-methyl-8,9-methylenedioxy-11H-indeno[1,2-c]isoquinoline (4)
SOCl2 (0.2 mL) was added to acid 12 (15 mg, 0.039 mmol) at room temperature. The resulting mixture was stirred at room temperature for 4 h. The excess SOCl2 was evaporated yielding a residue, which was treated with benzene (2 × 3 mL) and evaporated. The remaining residue was subjected to flash column chromatography on silica gel, eluting with CHCl3, yielding a dark red solid (12.0 mg, 84%), which displayed identical physical data with authentic 4 obtained from cis-3.2
trans-3,4-Dihydro-6,7-dimethoxy-4-methoxycarbonyl-N-methyl-3-(3',4'-methylenedioxyphenyl)-1(2H)-isoquinolone (10)
TMSCHN2 (2.0 M in hexane, 0.65 mL, 1.3 mmol) was added to a stirred suspension of trans-3 (385 mg, 1 mmol) in MeOH/benzene (2 mL:7 mL) at room temperature. The resulting mixture was stirred at room temperature for 30 min and the solution became clear. The solvent was removed under reduced pressure and the residue was subjected to flash column chromatography, eluting with CHCl3-MeOH (20:1), yielding a white solid 395 mg (99%): mp 185–186 ºC. 1H NMR (300 MHz, CDCl3) δ 7.63 (s, 1 H), 6.66 (d, J = 8.1 Hz, 1 H), 6.57 (s, 1 H), 6.53 (dd, J = 7.8, 1.8 Hz, 1 H), 6.48 (d, J = 1.5 Hz, 1 H), 5.89 (d, J = 1.2 Hz, 1 H), 5.87 (d, J = 1.2 Hz, 1 H), 5.06 (brs, 1 H), 3.93 (s, 3 H), 3.84 (s, 3 H), 3.73 (brs, 1 H), 3.68 (s, 3 H), 3.08 (s, 3 H); ESIMS m/z (rel intensity) 400 (100, MH+). Anal. Calcd for C21H21NO7·0.3H2O: C, 62.31; H, 5.38; N, 3.46. Found: C, 62.13; H, 5.35; N, 3.23. The crystal for X-ray analysis was obtained from a solution of 10 in CHCl3. Summary of X-ray crystal data: C21H21NO7; FW = 399.40; a = 11.0652(13) Å; b = 14.1943(19) Å; c =13.049(2) Å; β = 113.227(11)o; vol = 1883.4(4) Å3; monoclinic; space group P21/c; Z =4; crystal size = 0.30 × 0.23 × 0.06 mm; GOF = 1.318; R (Fo) = 0.091, Rw(Fo2) = 0.179.
6,7-Dimethoxy-4-methoxycarboxy-N-methyl-3-(3',4'-methylenedioxyphenyl)-1(2H)-isoquinolone (11)
NaHMDS (14.8 mL, 1.0 M in THF, 14.8 mmol) was added slowly to a stirred solution of ester 10 (4.54 g, 11.4 mmol) in THF (20 mL) at –78 ºC. The reaction mixture was stirred at –78 ºC for 30 min, and then a solution of phenylselenyl chloride (2.83 g, 14.8 mmol) in THF (5.0 mL) was added and the mixture was stirred at –78 ºC for 1 h. The reaction mixture was allowed to warm to room temperature and stirred at room temperature overnight. The reaction was quenched by slow addition of 1 N HCl (20 mL) at 0 ºC. CHCl3 (3 × 100 mL) was used to extract the product. The combined organic layers were washed with H2O (2 × 30 mL) and brine (2 × 30 mL). The resulting organic solution was dried over anhydrous Na2SO4, filtered, and concentrated to afford the selenide as a residue that was used without further purification in the next operation. The residue was dissolved in THF (100 mL). Acetic acid (3.0 mL) and H2O2 (30%, 27 mL) were added sequentially to the stirred solution at 0 ºC. The reaction mixture was allowed to warm to room temperature and stirred at room temperature overnight. Saturated NaHCO3 (30 mL) was added to the reaction mixture at 0 ºC. CHCl3 (3 × 100 mL) was used to extract the product. The combined organic layers were washed with H2O (2 × 30 mL) and brine (2 × 30 mL). The resulting organic solution was dried over anhydrous Na2SO4, filtered, and concentrated to afford a residue. The residue was subjected to flash column chromatography on silica gel, eluting with CHCl3, to yield a light yellow solid (3.8 mg, 85%): mp 158–159 ºC. 1H NMR (300 MHz, CDCl3) δ7.52 (s, 1 H), 6.81 (s, 1 H), 6.67 (s, 1 H), 6.66 (d, J = 8.1 Hz, 1 H), 6.10 (d, J = 8.1 Hz,1 H), 5.82 (s, 2 H), 3.79 (s, 3 H), 3.72 (s, 3 H), 3.29 (s, 3 H), 3.12 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.0, 160.8, 152.7, 148.5, 147.5, 147.0, 141.8, 127.8, 127.6, 122.2, 117.8, 110.7, 108.8, 107.6, 107.0, 103.5, 100.9, 55.3, 55.2, 51.1, 33.2; IR (film) 2950,1716, 1644, 1489, 1242, 1036, 928, 759 cm−1; ESIMS m/z (rel intensity) 398 (MH+, 100); HRESIMS m/z calcd for C21H19NO7 + H 398.1240, found 398.1237.
6,7-Dimethoxy-3-(3',4'-methylenedioxyphenyl)-4-carboxy-N-methyl-1(2H)-isoquinolone (12)
LiOH•H2O (145 mg, 3.5 mmol) was added to a stirred solution of ester 11 (137 mg, 0.35 mmol) in THF-MeOH-H2O (2:2:1, 5 mL) at room temperature. The resulting mixture was then heated at reflux for 36 h. The reaction mixture was then cooled to room temperature and the organic solvent was removed under reduced pressure. The residue was neutralized with 1 N HCl (5 mL). The precipitate was collected by filtration and washed with H2O and CHCl3, yielding a white powder (120 mg, 91%): mp 256–258 ºC. 1H NMR (300 MHz, DMSO-d6) δ 7.66 (s, 1 H), 7.06 (s, 1 H), 7.04 (s, 1 H), 7.02 (d, J = 7.5 Hz, 1 H), 6.86 (d, J = 8.1 Hz, 1 H), 6.11 (s, 2 H), 3.89 (s, 3 H), 3.84 (s, 3 H), 3.20 (s, 3 H); 13C NMR (75 MHz, DMSO-d6) δ 167.9, 160.5, 153.6, 149.0, 147.9, 147.1, 140.8, 128.2, 127.8, 123.2, 117.9, 112.3, 109.9, 108.3, 107.4, 104.6, 101.5, 55.6 (2 C), 33.5; ESIMS m/z (rel intensity) 384 (MH+, 100); HRESIMS m/z calcd for C20H17NO7+ H 384.1083, found 384.1084.
Supplementary Material
1H NMR spectra of compounds 4, 11, and 12. X-ray crystallographic information file and ORTEP drawing of compound 10. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support of this work with Research Grant UO1 CA89566. This research was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06-14499 from the National Center for Research Resources of the National Institutes of Health.
References
- 1.Cushman M, Cheng L. J Org Chem. 197843:286–288. [Google Scholar]
- 2.Cushman M, Cheng L. J Org Chem. 197843:3781–3783. [Google Scholar]
- 3.Kohlhagen G, Paull K, Cushman M, Nagafuji P, Pommier Y. Mol Pharmacol. 199854:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 4.Strumberg D, Pommier Y, Paull K, Jayaraman M, Nagafuji P, Cushman M. J Med Chem. 199942:446–457. doi: 10.1021/jm9803323. [DOI] [PubMed] [Google Scholar]
- 5.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. J Med Chem. 200043:3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 6.Jayaraman M, Fox BM, Hollingshead M, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 200245:242–249. doi: 10.1021/jm000498f. [DOI] [PubMed] [Google Scholar]
- 7.Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker BL, Stewart L, Cushman M. J Med Chem. 200346:3275–3282. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]
- 8.Nagarajan M, Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 200346:5712–5724. doi: 10.1021/jm030313f. [DOI] [PubMed] [Google Scholar]
- 9.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Cancer Res. 200363:7428–7435. [PubMed] [Google Scholar]
- 10.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Bioorg Med Chem Lett. 200414:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 200447:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 12.Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Bioorg Med Chem. 200412:5147–5160. doi: 10.1016/j.bmc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 13.Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Org Chem. 200469:7495–7501. doi: 10.1021/jo048808f. [DOI] [PubMed] [Google Scholar]
- 14.Xiao X, Antony S, Pommier Y, Cushman M. J Med Chem. 200548:3231–3238. doi: 10.1021/jm050017y. [DOI] [PubMed] [Google Scholar]
- 15.Xiao X, Miao ZH, Antony S, Pommier Y, Cushman M. Bioorg Med Chem Lett. 200515:2795–2798. doi: 10.1016/j.bmcl.2005.03.101. [DOI] [PubMed] [Google Scholar]
- 16.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Mol Pharmacol. 200567:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 17.Cushman M, Gentry J, Dekow FW. J Org Chem. 197742:1111–1116. doi: 10.1021/jo00427a001. [DOI] [PubMed] [Google Scholar]
- 18.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Revision B.05 ed. Pittsburg: [Google Scholar]
- 19.Reich HJ, Reich IL, Renga JM. J Am Chem Soc. 197395:5813–5815. [Google Scholar]
- 20.Lowry TH, Richardson KS. Mechanism and Theory in Organic Chemistry. Harper and Row; New York: 1987. pp. 318–322. [Google Scholar]
- 21.Azizian J, Mohammadi AA, Karimi AR, Mohammadizadeh MR. J Org Chem. 200570:350–352. doi: 10.1021/jo049138g. [DOI] [PubMed] [Google Scholar]
- 22.Yadav JS, Reddy BVS, Raj KS, Prasad AR. Tetrahedron. 200359:1805–1809. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H NMR spectra of compounds 4, 11, and 12. X-ray crystallographic information file and ORTEP drawing of compound 10. This material is available free of charge via the Internet at http://pubs.acs.org