

Abstract
Tuberculosis (TB) is the leading cause of infectious disease mortality in the world by a bacterial pathogen. We previously demonstrated that a bisubstrate inhibitor of the adenylation enzyme MbtA, which is responsible for the second step of mycobactin biosynthesis, exhibited potent antitubercular activity. Here we systematically investigate the structure activity relationships of the bisubstrate inhibitor glycosyl domain resulting in the identification of a carbocyclic analogue that possesses a KIapp value of 2.3 nM and MIC99 values of 1.56 μM against M. tuberculosis H37Rv. The SAR data suggest the intriguing possibility that the bisubstrate inhibitors utilize a transporter for entry across the mycobacterial cell-envelope. Additionally, we report improved conditions for the expression of MbtA and biochemical analysis demonstrating that MbtA follows a random sequential enzyme mechanism for the adenylation half-reaction.
Keywords: Mycobacterium tuberculosis, tuberculosis, adenylation inhibitor, siderophore biosynthesis, mycobactin, nonribosomal peptide synthetase
Introduction
Tuberculosis (TB) is the leading cause of infectious disease mortality in the world by a bacterial pathogen and an estimated 8 million people are diagnosed each year with TB.1 The emergence of extensively-drug-resistant (XDR) TB strains that are refractory to treatment by the first-line agents isoniazid and rifampin as well as three or more of the six second-line drugs is cause for grave concern. The Centers of Disease Control (CDC) found that from 2000 to 2004, XDR-TB increased from 5% of multidrug resistant TB cases to 6.5% worldwide.2 In the industrialized nations (including the United States) in the survey, XDR-TB increased from 3 to 11% during this same five-year period. Consequently, the development of new drugs is urgently needed for this virtually unbeatable form of TB.
The acquisition of iron by M. tuberculosis and other pathogenic bacteria is an essential process. In a mammalian host the concentration of free iron in serum and body fluids is too low to support bacterial growth.3 The extracellular iron of the host is primarily bound by iron transport proteins such as the transferrins and lactoferrins, while intracellular iron is sequestered by heme compounds and iron-sulfur clusters in various proteins and in the iron storage protein ferritin.4 Therefore, bacteria have evolved a number of mechanisms to obtain this vital micronutrient. The most prevalent mechanism involves the synthesis, secretion, and reuptake of small molecule iron chelators termed siderophores.4,5 Siderophores extract iron from host proteins and the resulting ferric-siderophore complex is actively imported by dedicated bacterial membrane transporters.6 M. tuberculosis produces two series of structurally related peptidic siderophores known as mycobactin-T7 and carboxymycobactins8 that vary by the appended lipid residue and will be hereafter collectively referred to as the ‘mycobactins’ (Figure 1).9,10 Initially, the mycobactins were regarded as an “essential substance” since they were found to be required for growth by Mycobacterium paratuberculosis.11 The mycobactins were first recognized as potential chemotherapeutic targets as early as 1945 when J. Francis pointed out that they would provide a good model for the development of chemotherapeutic agents, since if antagonists could be found, then they would be highly selective.9 Indeed derivatives of mycobactins produced through chemical synthesis have been found to have antituberculosis activity and may likely act as mycobactin antagonists.12–14 Recent advances in the understanding of mycobactin biosynthesis have provided new opportunities to antagonize siderophore production in M. tuberculosis. The corresponding biosynthetic genes for these siderophores are clustered in two different regions of the M. tuberculosis genome.15–17 The mbt-1 cluster (mbtA-mbtJ) encodes for a set of proteins that builds the mycobactin core scaffold.16 The mbt-2 locus (mbtK-mbtN) encodes for four gene products responsible for activation and attachment of the lipid residue.17 Both gene clusters are transcriptionally regulated by the IdeR repressor protein that binds Fe(II).18 During iron-limitation IdeR dissociates from the promoter region enabling transcription of the mbt operons.
Figure 1.

Biosynthesis of the mycobactins by a mixed NRPS-PKS assembly line initiated by MbtA.
The crucial role of the mycobactins for virulence was demonstrated through a targeted genetic disruption of mbtB, which blocked mycobactin production.19 Significantly, the resulting mutant strain was restricted for growth in iron-limiting media and impaired for growth in macrophage THP-1 cells.19 The siderophore knockout strain of M. tuberculosis also exhibited retarded iron acquisition in the phagosomal compartment of macrophages compared to wild-type M. tuberculosis.20 The finding that mycobactins directly acquire intracellular iron through lipid trafficking provided the first evidence that these siderophores are important in vivo.21 Furthermore p-aminosalicylic acid (PAS), a drug used for the treatment of TB, has been shown to inhibit mycobactin synthesis; however, the large doses (up to 12 g/day) and resulting gastrointestinal side-effects have relegated PAS to a second-line agent.22,23 In addition several other observations have indirectly provided evidence for the importance of mycobacterial iron metabolism. Thus, iron overload and active TB infection are inversely correlated24 and M. tuberculosis containing a constitutively expressed IdeR iron repressor homologue exhibited attenuated virulence in a BALB/c mouse model.25 Collectively these findings have established the mycobactins and iron acquisition as critical for pathogenesis of M. tuberculosis.
Mycobactins are mixed-ligand siderophores that are characterized by an aromatic hydroxy acid, which caps the N-terminus of a highly modified peptidic scaffold.5 These are biosynthesized by large modular enzymes termed the non-ribosomal peptide synthetases (NRPSs) since they operate independently of the mRNA templated ribosomal machinery and function analogously to the well-studied type I polyketide synthases (PKSs) with their modular organization and use of a thiotemplate mechanism.26,27 The enzymology of NRPS-catalyzed siderophore biosynthesis has been intensively investigated over the last decade, enabling the rational design of small molecule inhibitors towards these proteins.28
The mycobactin core scaffold is synthesized through the activity of six enzymes MbtA-F that comprise a mixed NRPS-PKS assembly line.15–17 The salicylic acid starter unit is prepared by a dedicated enzyme, MbtI, which utilizes the primary metabolite chorismate as a substrate.29–31 Biosynthesis is initiated by the stand-alone adenylation enzyme MbtA which activates salicylic acid at the expense of ATP and loads the adenylated intermediate onto the thiolation domain of MbtB where it undergoes sequential elongation by serine (catalyzed by MbtB), lysine (catalyzed by MbtE), two malonyl CoA’s (to form the (β-hydroxybutyrate residue – catalyzed by the PKS enzymes MbtC and MbtD), and another molecule of lysine (catalyzed by MbtF).16 Additional modifications through lipidation (catalyzed by MbtK) and N-hydroxylation (catalyzed by MbtG) of the lysine residues provides the mycobactins.17,32,33
The second biosynthetic step, catalyzed by MbtA, is an attractive point to block for the following reasons. First, the adenylating enzyme MbtA is a member of the well-studied adenylate-forming enzyme superfamily, and inhibitors of the functionally related aminoacyl tRNA synthetases have already been developed and are used clinically (e.g. mupirocin- a topical antibiotic, an inhibitor of isoleucyl tRNA synthetase).34,35 Second, the crystal structures of the adenylation domains DhbE determined by Stubbs and co-workers allowed the development of a homology model of MbtA.36 Third, comparison of mbtA to the human proteome showed no functional homologs. Fourth, inhibitors towards the adenylation protein MbtA are expected to be useful against the biosynthesis of a wide array of structurally diverse siderophores that are capped with an aryl acid.28 Finally, the mycobactin auxotrophy of M. paratuberculosis was recently shown to be due a truncation of mbtA.37
MbtA incorporates salicylic acid into the mycobactin core scaffold, using a two-step reaction that is mechanistically similar in most members of the adenylate-forming enzyme superfamily and the functionally related aminoacyl tRNA synthetases.27,38 In the adenylation or first half-reaction, binding of ATP and salicylic acid 1 is followed by nucleophilic attack of the substrate carboxylate on the α-phosphate of ATP to generate a tightly bound acyl adenylate 2 and the release of pyrophosphate (Figure 1). In the acylation or second half-reaction, the enzyme binds the phosphopantetheine (ppan) cofactor of the N-terminal thiolation domain of MbtB and transfers the acyl adenylate 2 onto the nucleophilic sulfur atom of this cofactor moiety to provide salicyl-bound-MbtB 3 which is ultimately elaborated to the mycobactins.
The reaction mechanism catalyzed by MbtA presents several opportunities to develop inhibitors against MbtA. The finding that acyl adenylate species bind several orders of magnitude more tightly than the substrate acids suggests that analogues incorporating stable linkers as bioisosteres of the labile acylphosphate function will provide potent enzyme inhibitors.34,39 This tight binding is essential to ensure that the acyl adenylate is not lost to the bulk solvent through diffusion before this is channeled to the corresponding acceptor residue. Additionally, sequestering the acyl adenylate by the enzyme may reduce the likelihood of adventitious hydrolysis of the mixed phosphoric-carboxylic acid anhydride. Inhibitors based on this design principle are considered bisubstrate inhibitors since they are expected to interact with both substrate binding pockets.40 Bisubstrate inhibitors that simply mimic the acyl adenylate intermediate can be further classified as intermediate mimetics.35 The inhibitor scaffold is thus comprised of four domains (Figure 2). Modifications of the linker region have been most extensively investigated.39,41–44 Previously, we synthesized acylsulfamate, acylsulfamide, (β-ketophosphonate, acyltriazole, sulfamate, β-ketosulfonamide, and α,α-difluoro-β-ketosulfonamide linkages as surrogates for the labile acylphosphate linkage.44,45 Among these salicyl-AMS inhibitors 6 and 7 incorporating the acylsulfamate and acylsulfamide linkages respectively exhibited the highest activity (see Figure 2). In this article, we have systematically examined the role of the ribose subunit of the salicyl-AMS inhibitor scaffold through the preparation of inhibitors 8–13 (Figure 2). Additionally, we report the cloning, overexpression, and purification of MbtA. Biochemical analysis of MbtA using a bisubstrate inhibitor has provided details into the reaction mechanism of the first half-reaction catalyzed by a NRPS adenylating enzyme. Comparison of the in vitro enzyme inhibition of MbtA as well as in vivo activity against whole-cell M. tuberculosis has yielded important insight into specificity of inhibitor uptake by M. tuberculosis.
Figure 2.
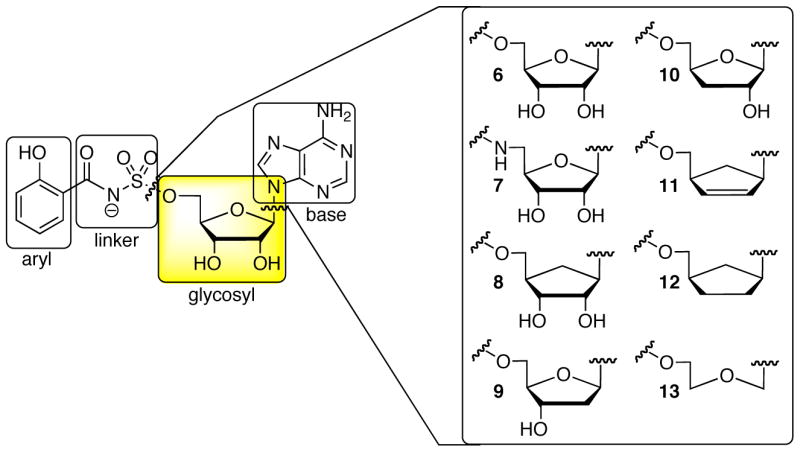
The bisubstrate inhibitor template is comprised of four domains: aryl, linker, glycosyl, and base. The acylphosphate linkage of the acyl adenylate intermediate 2 is mimicked by an acylsulfamate linkage in salicyl-AMS 6. The expanded portion of the figure shows the glycosyl modifications described herein.
Results
Chemistry
Inspired by the work of Vince and co-workers, we initially investigated the synthesis of carbocyclic analog 8, wherein the ribofuranose ring oxygen is replaced by a CH2. Removal of the labile glycosidic linkage is expected to increase both the lipophilicity and metabolic stability.46 The synthesis of 8 was most efficiently carried out from aristeromycin 14, a nucleoside antibiotic produced by Streptomyces citricolor.47 Protection of 14 as the acetonide48 followed by sulfamoylation afforded 15.49 Salicylation with NHS ester 16 in the presence of DBU provided 17, which was deprotected with 80% aqueous TFA to furnish 8.
The importance of the 2′- and 3′-hydroxyls of the ribofuranose moiety was explored by the preparation of analogues 9 and 10. Initially, the chemistry was optimized on the 2′-deoxy nucleoside. Consistent with the greater instability of 2′-deoxy nucleosides, we found that 9 was unstable under acidic conditions (50% aq TFA, 3 h) previously employed for the synthesis of several analogues and for the conversion of 20 to 21 (vide infra).44 The choice of the benzyl ether to protect the salicyl group was guided by this consideration. The synthesis of analogue 9 began with bis-silylation of 18 to afford 20 followed by selective removal (50% aq TFA, 0 °C, 3 h)42 of the 5′-O-TBS to provide 21 which was sulfamoylated to yield 24 (Scheme 2). Our established salicylation protocol with 26 employing DBU as base afforded a recalcitrant DBU salt of 27, which was deprotected to afford the DBU salt of 9. The DBU could not be removed by ion-exchange or by chromatography (co-eluting with 1% Et3N) that had previosly been successful in cases where the DBU salt was obtained. We believe that the DBU salt forms a very tight complex as we also observed the [M+DBU+H]+ ion by mass spectrometry. Additionally, we observed that the DBU salt of 9 was inactive in both the in vitro enzyme assay and against a whole-cell assay of M. tuberculosis (data not shown) while the triethylammonium salt of 9 displayed potent activity (vide infra). The lack of activity of the DBU salt also provides strong corroborating evidence that this forms a tight-complex with the inhibitor. Thus, we turned to our complementary method employing Cs2CO3 as base. Successful coupling of 24 to NHS ester 2644 was realized employing 3 equivalents of Cs2CO3 to provide 27. Sequential deprotection of the benzyl ether by catalytic hydrogenation to 29 and TBS ether with TBAF afforded 9. Synthesis of inhibitor 10 was initiated from the nucleoside antibiotic cordycepin 19 using an analogous series of reactions (Scheme 2). Protection as the di-O-TBS ether 22, followed by the selective cleavage of the 5′-O-TBS, optimally proceeded using p-TsOH in methanol to provide 23. Sulfamoylation of the 5′-OH afforded 25 that was coupled to 26 employing DBU to provide 28, which was isolated as the triethylammonium salt illustrating the capricious nature of this reaction. Sequential deprotection of the benzyl ether by catalytic hydrogenation to 30 and TBS ether with 80% aq TFA afforded 10.
Scheme 2a.

aReaction conditions: (a) TBSC1, imidazole, cat. DMAP, DMF, 84% (20); (b) 50% aq TFA, 60%; (c) p-TsOH, MeOH, 78% over 2 steps (23); (d) NH2SO2C1, NaH, DME, 38% (24), 82% (25); (e) Cs2CO3, DMF,; (f) DBU, DMF, 87% (28); (g) Pd/C, H2, MeOH, 17% over 2 steps (29); (h) TBAF, THF, 50%; (i) 80% aq TFA, 53% over 2 steps.
We also examined analogues 11 and 12 lacking both the 2′ and 3′ hydroxyl groups. For these analogues the carbocyclic sugar analogue was employed due to its greater stability. Aristeromycin 14 was transformed to a cyclic orthoester 31 using triethyl orthoformate and perchloric acid (Scheme 3).50 Treatment of the resulting orthoester in refluxing Ac2O followed by 4 N aq HC1 afforded 32 with a overall yield of 48% from 14.50 Compound 32 was sulfamoylated to yield 33, which was coupled to NHS ester 16 mediated by DBU to afford 34. Deprotection of 34 with 80% aqueous TFA provided analogue 11. Catalytic hydrogenation of 11 provided the fully reduced dideoxy analog 12.
Scheme 3a.

aReaction conditions: (a) HC(OMe)3, pTsOH; (b) 0 Ac2O, h) 6N HC1, 48% from 14; (c) NH2SO2C1, NaH, DME, 55%; (d) 16, DBU, DMF, 81%; (e) 80% aq TFA, 80%; (f) Pd/C, H2, MeOH, 36%.
Additionally, the acyclo analogue 13 was prepared starting from the acyclo nucleoside 3551, which was sulfamoylated to provide 36 (Scheme 4).49 Coupling of 36 with NHS ester 26 provided 37. Catalytic hydrogenation of 37 afforded the acyclo analog 13.
Scheme 4a.

aReaction conditions: (a) NH2SO2C1, NaH, DME, 45%; (b) 26, DBU, DMF, 44%; (c) Pd/C, H2, MeOH, 98%.
Biochemistry/Enzymology
Walsh and co-workers previously overexpressed MbtA as a N-terminal maltose binding protein (MBP) fusion with a C-terminal hexa-his tag; however, the total yield of protein obtained after purification and removal of the MBP was approximately 0.1 mg protein per L of culture.16 In order to obtain adequate amounts of protein for biochemical analysis, MbtA was subcloned from BAC143Rv (kindly provided by Dr. Stewart Cole, Institute Pasteur15) into pET-SUMO which incorporates an N-terminal hexa-his SUMO tag onto the corresponding gene product.42 Co-expression with the chaperones GroEL and GroES from the plasmid pGRO7 (Takara) were found to further improve expression levels 10–20 fold. Expression with these chaperones necessitated additional processing to remove GroEL from MbtA before subsequent Ni affinity purification. This was accomplished employing a modified protocol using ATP and denatured E. coli proteins to cause GroEL to release MbtA and bind to the denatured proteins.52 Recombinant MbtA was purified by Ni-NTA affinity chromatography. Cleavage of the (his)6-SUMO tag with SUMO protease and subsequent Ni-NTA affinity chromatography to remove the SUMO fragment and his-tagged protease provided native MbtA (2 mg/L) in approximately 95% purity as determined by SDS-PAGE (Supporting Information, Figure S1). For each batch of enzyme prepared, the amount of active enzyme was determined by titrating with tight-binding inhibitor 8 (For a representative plot see Supporting Information, Figure S2).53
Determination of the steady-state kinetic parameters of salicylic acid and ATP as well as inhibition of MbtA by the bisubstrate inhibitors was measured using a [32P]PPrATP exchange assay.54 The assay exploits the equilibrium nature of the adenylation reaction which can be summarized as follows: E + S + ATP ⇆[Emiddot;S-AMP] + PPt. Since the product S-AMP remains tightly bound to the enzyme, a steady-state kinetic analysis in the forward direction cannot be performed due to product inhibition. However, measurement in the reverse direction is readily performed using [32P]PPj and following its incorporation into γ-[32P]ATP.
The KM of ATP with MbtA has not been previously reported and was experimentally determined as 184 ± 24 μM by measuring v0 as a function of [ATP] to provide a saturation curve, which was fit by non-linear regression analysis to the Michaelis-Menten equation (Supporting Information, Figure S3). Analogously, the KM of salicylic acid was determined as 3.3 ± 0.5 μM (literature16 9.0 μM: MbtA with a C-terminal His-tag) by measuring v0 as a function of salicylic acid [Sal] (Supporting Information, Figure S4).
In order to evaluate whether the tight-binding inhibitors exhibited time-dependent inhibition, a time-course analysis was performed. The reaction velocity remained linear from 2–20 minutes when both 6 and [32P]PPi were added at t0. Thus, the inhibitor reached a rapid equilibrium consistent with earlier reports of acylsulfamate bisubstrate inhibitors with isoleucine tRNA synthetase.55,56 Nevertheless, in order to ensure that the reaction was at steady-state, all substrates (ATP, PPi, salicylic acid) and inhibitor were preincubated for 10 minutes and observation of the residual enzyme activity was measured by addition of [32P]PPi at t0. The initial rates, v0, at a given [I] were determined by single time-point stopped-time incubations at 20 minutes. Since the inhibitors exhibited tight-binding behavior (KIapp < 200·[E]) the fractional initial velocities (vi/v0) and [I] were fit to the Morrison equation (equation 1, see Experimental Section) by non-linear regression analysis constraining [E] where vi is the initial velocity with inhibitor and v0 represents the initial velocity for a DMSO control to obtain KIapp values for 8–13.40,57 A representative plot is shown in Figure 3 (See Supporting Information, Figures S5–S9). The [E] was experimentally determined by active-site titration with 8.
Figure 3.

Dose-response of fractional initial velocity of γ-[32P]-ATP formation catalyzed by MbtA as a function of inhibitor 8 concentration. The curve represents the best non-linear fit of the data to the Morrison equation. The data points represent the mean with standard error of duplicate experiments.
The KIapp of the parent ribose inhibitor 6 was found to be 6.6 nM (Table 1). Replacement of the 5′-oxygen atom of the ribose moiety with a nitrogen atom in analogue 7 led to a 1.8-fold increase in activity (7 vs. 6). Carbocyclic analogue 8 displayed potent inhibition with a KIapp of 2.3 nM, which represents a 3-fold increase in activity (8 vs. 6) suggesting that the ring-oxygen does not participate in H-bonding. In order to more precisely map out the structural requirements of the ribofuranose subunit analogues 9 and 10 were evaluated. Deletion of the 2′-alcohol in 9 led to an approximately 2-fold increase in activity (9 vs. 6) while deletion of the 3′-alcohol in analogue 10 also resulted in a 2-fold increase in potency (10 vs. 6). The dideoxy-dehydro carbocyclic analogue 11 exhibited a 126-fold loss in binding affinity (11 vs. 6), but still maintained submicromolar enzyme inhibition. Dideoxy carbocyclic analogue 12 was found to possess a mere 9-fold decrease in binding affinity (12 vs. 6) despite the removal of both the 2′ and 3′ alcohols and the ribofuranose ring oxygen. Thus, an approximately 14-fold was regained by simply reducing the unsaturation in 11 (12 vs. 11). The acyclo analogue 13 exhibited a profound 2,530-fold decrease in activity (13 vs. 6) clearly demonstrating the importance of the conformational rigidity of the parent ribose moiety.
Table 1.
SAR of the Glycosyl Domain.
 | |||||
|---|---|---|---|---|---|
| Inhibitor | Structure, R = | ClogPa | KIapp (μM) MbtA | MIC99 (μM)bM. tuberculosis H37Rv | MIC99 (μM)cY. pseudotuberculosis |
| isoniazid | nad | nde | na | 0.18f | na |
| 6 |

|
−0.89 | 0.0066 ± 0.0015g | 0.29f | 20 |
| 7 |

|
nd | 0.0037 ± 0.0006g | 0.19f | nd |
| 8 |

|
−0.26 | 0.0023 ±0.0004 | 1.56 | >100 |
| 9 |

|
−0.10 | 0.0035 ±0.0004 | 1.56 | nd |
| 10 |

|
−0.09 | 0.0032 ±0.0005 | 1.56 | 80 |
| 11 |

|
1.42 | 0.830 ±0.078 | >200 | >100 |
| 12 |

|
1.53 | 0.061 ± 0.005 | >200 | nd |
| 13 |
|
0.59 | 16.7 ± 0.5 | >200 | >100 |
Carbocyclic inhibitor 8, which represents the most potent inhibitor, was selected for further biochemical evaluation in order to determine the modality of inhibition and hence true inhibition constant. The apparent inhibition constant (KIapp of 8 was determined as a function of [ATP] from 5–55·KM(ATP) The KIapp varied linearly with [ATP] demonstrating that this inhibitor is competitive with respect to ATP (Figure 4A). Based on the Cheng-Prusoff equation (see equation 2, Experimental Section) for a competitive tight-binding inhibitor, the inhibition constant ki′ = 0.038 ± 0.007 nM with respect to the varied substrate (ATP) at a given concentration of the nonvaried substrate (Salicylic acid).58 For a bisubstrate inhibitor, ki′ also depends on the concentration of the nonvaried substrate because of overlapping binding sites.59,60 Next, the ki′ of 8 was determined as a function of the nonvaried substrate salicylic acid [Sal] from 5–50 ·KM(Sal) at a fixed concentration of ATP. The ki′ was also found to vary linearly with [Sal] demonstrating that this inhibitor is also competitive with respect to salicylic acid (Figure 4B). Extrapolation to zero concentration of salicylic acid provides the true inhibition constant. However, the inability to accurately determine the small y-intercept due to experimental error precluded measurement of KI Alternatively, for a rapid-equilibrium random sequential mechanism (vide infra) the true KI can be determined from equation 3 (see Experimental Section); however, this requires knowledge of the dissociation constant of salicylic acid (KD(Sal)).60
Figure 4.

A) KIapp as a function of ATP concentration. B) KIapp as a function of salicylic acid concentration.
Molecular Modeling
To investigate the structural basis for the in vitro activity results, we docked each compound into a homology model based on the X-ray structure of the homolog DhbE.36 Key interactions of the nucleoside region of 6 appear in Figure 5. The hydrogen bonding pattern of the lowest energy structure was identical to that seen for the natural phosphate intermediate in the DhbE X-ray structure. The sugar pucker, 3′ endo, was also conserved, and the lowest energy conformation deviated from the X-ray structure’s ligand by only 0.22 Å RMS. The deletion of the 2′ or 3′ hydroxyl in 9 and 10 did not alter the C3′ endo pucker of the lowest-energy structure, despite the loss of two hydrogen bonds in each case. The small change in the in vitro activity of 8 relative to the parent compound was consistent with the DhbE structure, where the 4′ oxygen was 3.6 Å from the nearest possible hydrogen bonding donor, the side chain of Lys519. Our modeling of 6 and 7 confirmed this lack of a hydrogen bonding partner for ribose-based ligands. The other single-heteroatom change, in 7, also resulted in no significant structural changes, although the 5′ NH to Lys519 hydrogen bond had a slightly less favorable geometric configuration than the parent compound. However, the 5′ NH was able to form an internal hydrogen bond with the aryl carbonyl.
Figure 5.

Schematic diagram of key nucleoside/protein interactions for analog 6, based on docking. For comparison with the DhbE X-ray structure, residues are numbered according to PDB entry 1MDB.36
With most of the single-heteroatom substitutions, two conformations of the C4′-C5′-O5′-S linker were observed, one matching the DhbE X-ray structure and the other pivoting the two central atoms and breaking the O5′-Lys519 hydrogen bond. However, for the carbocycle 8, only the alternate conformation was observed, suggesting that this hydrogen bond is not critical for activity.
Compounds 11-13 involved more significant chemical changes. The 3 ′ endo pucker was maintained in 12, but as with the carbocycle 8, the lowest energy structure showed the loss of the O5′-Lys519 hydrogen bond. Although the ~10-fold lower in vitro activity of this compound likely resulted from the loss of hydrogen bonding interactions, the X-ray structure of DhbE in the absence of a ligand showed a slight movement of Arg428, allowing it to form a salt bridge with Asp413. Therefore, 12 may bind without a large energy penalty for desolvating Asp413. The more significant (~ 100-fold) loss of activity for 11 was likely due to strain, as the experimentally observed C3′ endo pucker would distort planarity around the double bond. The lowest energy structure was indeed distorted, with a C4′ exo pucker, although the pucker amplitude was reduced relative to 6 (16.4 vs. 39.4, calculated using PROSIT).61 Finally, 13 docked with the conformation expected for active compounds (0.51 Å RMS relative to the DhbE x-ray structure), but the remaining sugar atoms shifted ~ 0.75 A into the space generated by deleting C2′ and C3′.
Biological Activity Against Whole-cell Mycobacterium tuberculosis and Yersinia pseudotuberculosis
All inhibitors synthesized above were evaluated against whole-cell M. tuberculosis H37Rv under iron-limiting conditions. The whole-cell assay also measures inherent resistance, such as membrane permeability, a factor that is not assessed with the in vitro enzyme assay.19 The minimum inhibitory concentrations (MIC99) that inhibited complete growth of M. tuberculosis are shown in Table 1. Isoniazid was used as a positive control while DMSO was employed as a negative control. The previously determined MIC99 values of compounds 6 and 7 are shown for comparison.44 Both of these compounds display activity rivaling the first line antitubercular agent isoniazid. The carbocyclic analogue 8 displayed an MIC99 of 1.56 μM, which represents a 5-fold loss of activity relative to the parent compound 6. Similarly the 2′ and 3′ deoxy derivatives 9 and 10 displayed MIC99 values of 1.56 μM. However, removal of both the 2′ and 3′ alcohols in cyclopentene analogue 11 resulted in complete loss of activity consistent with its poor enzyme inhibition (KIapp = 830 nM). The removal of the 2′- and 3′-oxygen atoms in 12 resulted in total loss of in vivo activity despite its moderate in vitro activity (KIapp = 61 nM). The acyclo adenosine analog 13 was inactive in accord with its weak enzyme inhibition (KIapp = 16.7 μM). Additionally, several of the compounds were assessed for activity against an isolate of Yersinia pseudotuberculosis which, like Y. pestis, requires the salicyl-capped NRPS-PKS derived siderophore known as yersiniabactin for growth under iron-limiting conditions.62 Biosynthesis of yersiniabactin is initiated by the adenylating enzyme YbtE which shows 39% amino acid identity to MbtA, but absolute conservation of putative nucleoside binding residues.42,44 The parent compound 6 exhibited moderate inhibition with a MIC99 of 20 μM and 3-deoxy analogue 10 possessed an MIC99 of 80 μM. Carbocyclic 8, cyclopentene analogue 11 and acyclo analogue 13 displayed no inhibition of Y. pseudotuberculosis growth up to 100 μM.
Discussion
Inhibition of siderophore biosynthesis has emerged as an attractive strategy to develop novel antibiotics against pathogens that require siderophores for virulence.42–44,63–65 In this report we have systematically investigated the SAR of the glycosyl domain of our salicyl-nucleoside bisubstrate inhibitor. In particular, in vitro results against MbtA showed that deletion of the 2′, 3′, and 4′ oxygen atoms were generally well tolerated, while modifications making the sugar more (13) or less (11) flexible were detrimental. The activity of carbocyclic analog 8 was especially significant. A major metabolic and decomposition pathway of nucleosides involves expulsion of the nucleobase as a result of the chemically and enzymatically susceptible glycosidic bond, thus carbocyclic analog 8 is noteworthy since this glycosidic bond is replaced by a stable linkage. Overall, in vitro enzyme inhibition and in vivo antimycobacterial activity were well correlated except for analogue 12, which displayed potent enzyme inhibition toward MbtA, yet had no antimycobacterial activity. The parent acylsulfamate 6 and 3-deoxy analogue 10 were also found to be moderate inhibitors of Y. pseudotuberculosis growth demonstrating that these inhibitors are effective against other siderophore-producing bacteria under iron-limiting conditions. The reasons for the reduced activities of these compounds against Y. pseudotuberculosis are not clear, but suggest that in vitro comparisons of YbtE and MbtA as well as other physiological studies are needed for development of analogs active against other pathogens.
We postulate that the lack of bioactivity of 12 is a result of hindered transport and by corollary propose that the sugar moiety of the nucleoside subunit is critical for recognition by a putative transporter(s). In general, acyl adenylate intermediate mimetics developed for the functionally related aminoacyl tRNA synthetases were found to display excellent enzyme inhibition, but were inactive in vivo against whole-cell microorganisms as a result of limited membrane permeability, a problem that was expected to be exacerbated with the imposing mycobacterial cell envelope.35 The findings that the bisubstrate inhibitors such as 6 possess potent submicromolar MIC99 values provides strong evidence that these compounds are using a transporter for uptake. The mycobacterial cell-wall provides a permeability barrier to hydrophilic solutes that results in intrinsic resistance to many antibacterial agents.66 In order to obtain vital nutrients and co-factors, M. tuberculosis possesses an astonishing 37 ABC (ATP dependent binding cassette) transporters of which 16 have been unambiguously assigned as importers responsible for assimilation of amino acid, nucleotides, and other essential co-factors.67 Rodriguez and co-workers recently reported that the gene products of Rvl345 and Rvl347 encode for an ABC transporter responsible for transport of carboxymycobactin.68 Additionally, M. tuberculosis transports many hydrophilic solutes via facilitated diffusion mediated by a class of proteins known as the porins (the major porin is known as MspA), which have also been shown to play an important role in transport of antibiotics such as β-lactams and aminoglycosides.69 The availability of targeted disruption mutants of several ABC transporters and MspA should enable the identification of the transporter(s) responsible for uptake of the bisubstrate inhibitors. Alternatively, identification of resistant-conferring mutations in M. tuberculosis towards the nucleoside bisubstrate inhibitors described herein may also lead to the identification of the transporter of these antibacterial agents.
Inhibition of complex biosynthetic pathways is most effective when targeting the rate-limiting biosynthetic step. However, neither the rate of mycobactin synthesis by M. tuberculosis nor the rate limiting biosynthetic step is known. Thus far only MbtA, the N-terminal carrier domain of MbtB, the phosphopantetheinyl transferase MbtT, and the tailoring enzymes MbtK-MbtN, have been functionally characterized.16,17 The approximately 2–3 log difference between KIapp and the MIC99 value could be partially due to the requirement to fully abrogate MbtA activity, if this is not the rate limiting step. Walsh and co-workers have measured the rates of aryl acid activation by EntE, the adenylation enzyme involved in synthesis of the siderophore enterobactin.70 EntE catalyzes the activation and loading of 2,3-dihydroxybenzoic acid onto the cognate carrier domain of EntB and proceeds with kcat ~ 100 min−1.70 Thus, it is quite likely that the rate of activation catalyzed by MbtA is fast with respect to the overall rate of mycobactin synthesis. A greater understanding of the enzymology of the other proteins (MbtB-MbtI) and determination of the rate of mycobactin synthesis in vivo will be necessary in order to establish whether MbtA catalyzes the rate-limited step. The recently reported crystal structures of MbtI and MbtK pave the way for the rational design of inhibitors towards these enzymes.30,31,33 Since MbtI catalyzes the very first biosynthetic step, inhibitors towards MbtI could display synergy with those of MbtA since these enzymes catalyze consecutive biosynthetic steps.
Another important consideration for inhibitor design is an understanding of enzyme reaction mechanism. Despite a preponderance of bioinformatic,71,72 biochemical,70,73,74 and structural studies36,75 of NRPS adenylation domains, the detailed reaction mechanism of these adenylation domains has not been fully elucidated.27,38 Bisubstrate inhibitors can serve as powerful mechanistic probes for multisubstrate reactions and allow one to discriminate among related reaction mechanisms.60 Presuming a rapid equilibrium process, the competitive inhibition pattern observed in the adenylation half-reaction towards both ATP and salicylic acid for the bisubstrate inhibitor 8 provides unequivocal support for a sequential random mechanism.60 Quadri and co-workers observed that YbtE and bisubstrate inhibitor 6 display competitive behavior with respect to ATP and uncompetitive with respect to salicylic acid.42 However, as discussed extensively by Fromm this inhibition pattern for a bisubstrate inhibitor can arise from either a sequential- ordered or random mechanism.60 The crystal structure of the adenylating enzyme DhbE, which activates 2,3-dihydroxybenzoic acid, shows that the aryl acid substrate binding pocket is disordered in the absence of a ligand, but highly ordered when the ATP binding pocket is occupied, which suggests a sequential ordered reaction mechanism for this enzyme.36 In the absence of kinetic data one cannot infer the kinetic mechanism based solely on structural data.
The significance of an understanding of the reaction mechanism is two-fold. First, it provides a model for inhibitor binding with either the nucleoside or salicyl domain of the bisubstrate inhibitor first docking into the active site followed by binding of the other respective domain. Second, the inhibitors must compete with both ATP and salicylic acid. Salicylic acid is an abundant metabolite of M. tuberculosis as well as many other pathogens. In fact, the concentration of salicylic acid has been measured as high as 200 μM or approximately 40-.KMSal in growing cultures of M. smegmatis.76 Consequently, the KIapp values measured herein may represent an accurate measure of in vivo inhibitor efficacy since these were measured under supersaturating substrate conditions (salicylic acid was 250 μM or 50· KMSal and ATP was set at 10 mM or 55·KMATP).
Conclusion
In conclusion, we have reported the systematic evaluation of the structure activity relationships of the bisubstrate inhibitor glycosyl domain that govern binding towards MbtA and in vivo activity against M. tuberculosis. Molecular modeling has been used to interpret the SAR data. These studies have provided a foundation for future SAR campaigns and we have importantly identified several modifications to the glycosyl inhibitor template that can be used to modulate the pharmacodynamic and pharmacokinetic properties of the inhibitors. The intriguing possibility that the bisubstrate inhibitors utilize a transporter for entry across the mycobacterial cell-envelope is supported by SAR studies. Efforts are now underway to examine the importance of the aryl and base domains of the bisubstrate template to identify sites amenable to modification. Detailed pharmacokinetic, toxicity, and mechanism of action studies of the designed bisubstrate inhibitors are underway and the results will be reported in due course.
Experimental Section
Chemistry General Procedures
All commercial reagents (Sigma-Aldrich, Acros) were used as provided unless otherwise indicated. Aristeromycin (14) and acycloadenosine (34) were kindly provided by Prof. Robert Vince (University of Minnesota, Minneapolis, MN). 2-Deoxyadenosine and 3-deoxyadenosine were obtained from Berry and Associates (Dexter, MI). An anhydrous solvent dispensing system (J. C. Meyer) using 2 packed columns of neutral alumina was used for drying THF, DMF and CH2Cl2 and the solvents were dispensed under argon. Anhydrous DME was purchased from Aldrich and used as provided. Flash chromatography was performed with Silia P grade silica gel 60 (Silicycle) with the indicated solvent system. All reactions were performed under an inert atmosphere of dry Ar or n2 in oven-dried (150 °C) glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26 ppm) or methanol (3.31 ppm), and carbon chemical shifts are reported using an internal standard of residual chloroform (77.0 ppm) or methanol (49.1 ppm). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad), coupling constant, integration. High resolution mass spectra were obtained on Agilent TOP II TOF/MS instrument equipped with either an ESI or APCI interface. Optical rotations were measured on Rudolph Autopol III polarimeter. Melting points were measured on electrothermal Mel-Temp manual melting point apparatus and are uncorrected.
General procedure for sulfamoylation.77
To a solution of 2′,3′-O-isopropylidene protected nucleoside (1 mmol, 1 equiv) in DME (50 mL) at 0 °C was added NaH (60 % suspension in mineral oil, 1.5 mmol, 1.5 equiv). After 30 min a solution of sulfamoyl chloride (1.5 mmol, 1.5 equiv) in DME (10 mL) was added drop wise over 5 min and the reaction stirred 16 at rt. The reaction was quenched at 0 °C with MeOH (10 mL) then concentrated under reduced pressure. Purification by flash chromatography afforded the title compound. Due to the instability of the products HRMS data could not be obtained using either ESI(+/−) or APCI(+/−) to identify the molecular ion peak. Mass peaks corresponding to loss of the sulfamoyl moiety [M-SO2NH2]+ were observed in some cases (data not shown).
General procedures for salicylation
Procedure A
To a solution of 2′,3′-O-isopropylidene protected 5′-O-sulfamoylated nucleoside (1 mmol, 1 equiv) in DMF (30 mL) at 0 °C was added NHS ester (3.0 mmol, 3.0 equiv) followed by DBU (1.5 mmol, 1.5 equiv) and the reaction was stirred 16 h at rt. The reaction mixture was concentrated under reduced pressure. Purification by flash chromatography using a mixture of MeOH and EtOAc containing 1% Et3N afforded the title compounds. Procedure B. To a solution of 2′,3′-O-isopropylidene protected 5′-O-sulfamoylated nucleoside (1 mmol, 1 equiv) in DMF (30 mL) at 0 °C was added NHS ester (3.0 mmol, 3.0 equiv) followed by Cs2CO3 (3.0 mmol, 3.0 equiv) and the reaction was stirred 16 h at rt. The reaction mixture was filtered to remove solids and washed with a small quantity of DMF. Concentration under reduced pressure followed by purification by flash chromatography using a mixture of MeOH and EtOAc containing 1% Et3N afforded the title compounds.
2′,3-O-isopropylidene-5′-O-(sulfamoyl)aristeromycin (15)
This was prepared from 2′,3′-0-isopropylidenearisteromycin48 (290 mg, 0.95 mmol, 1.0 equiv) using the general procedure for sulfamoylation. Purification by flash chromatography (4:1 EtOAc/MeOH) afforded the title compound as a thick oil (230 mg, 63%): Rf 0.70 (3:1 EtOAc/MeOH); [α]20D-6.6 (c 1.8, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.29 (s, 3H), 1.54 (s, 3H), 2.37 (q, J= 12.0 Hz, 1H), 2.46–2.54 (m, 1H), 2.54–2.64 (m, 1H), 4.24 (dd, J = 16.2, 6.6 Hz, 1H), 4.28 (dd, J = 16.2, 6.6 Hz, 1H), 4.70 (t, J = 6.0 Hz, 1H), 4.88–4.96 (m, 1H), 5.09 (t, J = 6.6 Hz, 1H), 8.19 (s, 1H), 8.21 (s, 1H); 13C NMR (150 MHz, CDC13) δ 25.4, 27.7, 34.9,44.5, 62.6, 71.1, 82.2, 84.8, 115.3,120.6, 141.6,150.7, 153.6,157.4.
N-Hydroxysuccinimidyl 2-(methoxymethyloxy)benzoate (16)
A solution of LiOH (660 mg, 27.5 mmol, 3.0 equiv) in MeOH (18 mL) and water (2 mL) was added to methyl 2-(methoxymethyloxy)benzoate78 (1.81 g, 9.17 mmol, 1.0 equiv) and the reaction mixture was refluxed for 4 h. The reaction mixture was concentrated and the residue was dissolved in H2O (20 mL) and the pH was adjusted to 3 then extracted with EtOAc (3x50 mL). The combined organic extracts were concentrated to afford 2-(methoxymethyloxy)benzoic acid (1.56 g, 93%) which was directly carried onto the next step: 1H NMR (600 MHz, CDCl3) δ 3.54 (s, 3H), 5.34 (s, 2H),7.15 (t, J = 7.8 Hz, 1H), 7.26 (d, J= 8.4 Hz, 1H), 7.51 (t, J= 9.0 Hz, 1H), 8.14 (d, J= 7.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) 5 57.0, 95.8,115.1, 118.4,122.9, 133.5,134.9, 156.2,166.0.
To a solution of the crude product (1.56 g, 8.56 mmol, 1.0 equiv) from above in THF (80 mL) at 0 °C was added N-hydroxysuccinimide (0.988 g, 8.56 mmol, 1.0 equiv) and DCC (1.76 g, 8.56 mmol, 1.0 equiv). The resulting mixture was stirred for 30 min at 0 °C then 2 h at rt. The reaction mixture was filtered to remove the DCU precipitate and the filtrate was concentrated under reduced pressure. Purification by flash chromatography (4:1 EtOAc/hexanes) afforded the title compound 16 (2.02 g, 85%): Rf= 0.85 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 2.86 (br s, 4H), 3.50 (s, 3H), 5.26 (s, 2H), 7.08 (t, J = 7.8 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.55 (t, J = 8.4 Hz, 1H), 8.02 (d, J = 7.8 Hz, 1H); 13C NMR (150 MHz, CDC13) δ 25.7, 56.5, 94.8, 115.3, 121.5, 132.4, 135.7, 158.1, 160.3, 169.4; unable to obtain a HRMS using either ESI(+/−) or APCI (+/−).
5′-O-(N-(2-Hydroxybenzoyl)sulfamoyl)aristeromycin triethylammonium salt (8)
This was prepared from 15 (90 mg, 0.23 mmol, 1.0 equiv) and 16 (196 mg, 0.70 mmol) using the general salicylation procedure A. Purification by flash chromatography (85:15:1 EtOAc/MeOH/Et3N) provided the salicylated adduct as a DBU salt (40 mg, 25%), further elution afforded compound 17 (120 mg, 60%) as a triethylammonium salt.
The triethylammonium salt of compound 17 (70 mg, 0.11 mmol) prepared above was treated with 80% aq TFA (1.0 mL) at rt for 4 h. The reaction was thoroughly concentrated in vacua to remove all residual TFA. Purification of the residue by flash chromatography (70:30:1 EtOAc/MeOH/Et3N) afforded the title compound 8 (40 mg, 66%): Rf = 0.20 (7:3 EtOAc/MeOH); [α]20D –4.9 (c 0.78, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.23 (t, J = 7.2 Hz, 9H), 1.95–2.05 (m, 1H), 2.40–2.55 (m, 2H), 3.07 (q, J = 7.2 Hz, 6H), 4.12 (dd, J = 5.4, 3.0 Hz, 1H), 4.27 (d, J = 5.0 Hz, 2H), 4.50 (dd, J = 9.0, 5.4 Hz, 1H), 4.80–4.95 (m, 1H), 6.70–6.85 (m, 2H), 7.27 (t, J = 7.2 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 8.13 (s, 1H), 8.31 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 9.5, 30.2, 44.3, 47.7, 61.0, 71.5, 73.6, 76.8, 117.9, 119.3, 120.4, 120.7, 131.3, 134.3, 141.6, 151.2, 153.4, 157.2, 162.0, 174.8; HRMS (ESI+) calcd for C18H21N6O7S [M+H]+ 465.1187, found 465.1204 (error 3.7 ppm).
3′,5′-O-bis(tert-Butyldimethylsilyl)-2-deoxyadenosine (20)
To a solution of 2-deoxyadenosine (3.00 g, 11.1 mmol, 1.0 equiv) in DMF (16 mL) at 0 °C were added imidazole (4.54 g, 66.8 mmol, 6.0 equiv) and DMAP (200 mg, 1.6 mmol, 0.15 equiv). Next, a solution of TBSC1 (4.20 g, 27.9 mmol, 2.5 equiv) in DMF (8.0 mL) was added dropwise at 0 °C and the reaction stirred 30 min. The reaction mixture was diluted with saturated aq NaHCO3 (100 mL) and extracted with EtOAc (3×100 mL). The combined organic extracts were dried (Na2SO4, filtered and concentrated under reduced pressure. Purification by flash chromatography (6:1 EtOAc/hexanes) afforded the title compound (4.5 g, 84%) as a white solid: mp = 123–125 °C; Rf= 0.7 (1:3 EtOAc/hexanes); [α]20D–2.7 (c 0.96, CHC13); 1H NMR (600 MHz, CDC13) δ 0.00 (s, 6H), 0.01 (s, 6H), 0.82 (s, 18H), 2.34 (ddd, J = 13.2, 6.0, 4.2 Hz, 1H), 2.50–2.58 (m, 1H), 3.68 (dd, J = 10.8, 3.0 Hz, 1H), 3.78 (dd, J = 11.4, 4.2 Hz, 1H), 3.92 (dd, J = 6.6, 3.0 Hz, 1H), 4.52 (dd, J = 9.0, 3.6 Hz, 1H), 5.70 (br s, 2H), 6.36 (t, J = 6.6 Hz, 1H), 8.05 (s, 1H), 8.26 (s, 1H); 13C NMR (150 MHz, CDC13) δ −5.5,−5.4, −4.1, −4.7, 18.0, 18.4, 25.8, 26.0, 41.3, 62.8, 71.9, 84.3, 87.9, 120.0, 139.1, 149.6, 152.8, 155.3; HRMS (ESI+) calcd for C22H40N5O3Si2 [M-H]+ 478.2664, found 478.2656 (error 1.7 ppm).
3′-O-tert-Butyldimethylsilyl-2-deoxyadenosine (21)
To a solution of 20 (1.0 g, 2.08 mmol, 1.0 equiv) in THF (25 mL) was added 50% aq TFA (12 mL).42 After 3 h, the reaction mixture was quenched with aq 10 M NH4OH until the pH was basic (~10). The reaction mixture was concentrated under reduced pressure and the solid obtained was dried, dissolved in ethyl acetate and washed with brine. The aqueous layer was extracted with EtOAc (3×50 mL). The combined EtOAc extracts were dried (Na2SO4, filtered and concentrated. Purification by flash chromatography (EtOAc) afforded the title compound (0.45 g, 60%) as a white solid: mp = 178–182 °C; Rf = 0.2 (EtOAc); [α]20D –4.8 (c 0.89, CH3OH); 1H NMR (600 MHz, CDC13) δ 0.14 (s, 6H), 0.94 (s, 9H), 2.37 (ddd, J = 13.2, 6.0, 2.4 Hz, 1H), 2.80 (ddd, J = 13.2, 7.8, 5.4 Hz, 1H), 3.70 (dd, J = 12.0, 3.0 Hz, 1H), 3.81 (dd, J = 12.0, 3.0 Hz, 1H), 4.03 (d, J = 2.4 Hz, 1H), 4.68 (t, J = 3.0 Hz, 1H), 6.42 (t, J = 6.6 Hz, 1H), 8.17 (s, 1H), 8.31 (s, 1H); 13C NMR (150 MHz, CDC13) δ −4.6 (2C), 18.7, 26.3, 42.1, 63.4, 74.4, 87.1, 90.4, 120.8, 141.6, 150.0, 153.4,157.4; HRMS (ESI+) calcd for C16H28N5O3Si [M+H]+ 366.1956, found 366.1984 (error 7.7 ppm).
3′-O-tertButyldimethylsilyl-2′-deoxy-5′-O-(sulfamoyl)adenosine (24)
This was prepared from 21 (1.81 g, 4.92 mmol, 1.0 equiv) using the general procedure for sulfamoylation. Purification by flash chromatography (19:1 EtOAc/MeOH) afforded the title compound (0.80 g, 38%) as a viscous colorless oil: Rf= 0.55 (9:1 EtOAc/MeOH); [[α]20D −41.7 (c 0.690, CH3OH); 1H NMR (600 MHz, CDC13) δ 0.09 (s, 6H), 0.98 (s, 9H), 2.43 (ddd, J = 13.2, 6.0, 3.6 Hz, 1H), 2.60–2.75 (m, 1H), 4.18 (dd, J = 9.0, 3.0 Hz, 1H), 4.25 (dd, J = 10.8, 4.2 Hz, 1H), 4.20 (dd, J = 10.8, 3.0 Hz, 1H), 4.60–4.70 (m, 1H), 6.43 (t, J = 6.6 Hz, 1H), 8.16 (s, 1H), 8.20 (s, 1H); 13C NMR (150 MHz, CDC13) δ –5.6, –5.5, 17.4, 25.1, 40.4, 68.1, 72.0, 84.1, 84.8,118.8, 138.8,148.7, 152.3,155.3.
5 ′-O-(N-(2-Benzyloxybenzoyl)sulfamoyl)-3 ′-O-tert-butyldimethylsilyl-2 ′-deoxyadenosine 1,8-diazabicyclo[5.4.0]undec-7-ene salt (27·DBU)
Compound 24 (150 mg, 0.34 mmol) was coupled with 2644 (329 mg, 1.012 mmol, 1.5 equiv) using the general salicylation procedure A. Purification by flash chromatography (90:10:1 EtOAc/MeOH/Et3N) afforded the title compound (120 mg, 44%) as a viscous oil: Rf= 0.3 (EtOAc/MeOH); [[α]20D+7.1 (c 0.22, CH3OH); 1H NMR (600 MHz, CD3OD) δ 0.00 (s, 3H), 0.01 (s, 3H), 0.80 (s, 9H), 1.35–1.60 (m, 8H), 2.20–2.35 (m, 1H), 2.60–2.75 (m, 1H), 2.76 (t, J = 5.4 Hz, 2H), 3.00–3.20 (m, 4H), 3.25–3.32 (m, 2H), 3.86 (q, J = 3.6 Hz, 1H), 4.03 (dd, J = 10.8, 4.2 Hz, 1H), 4.13 (dd, J = 10.8, 3.6 Hz, 1H), 4.55–4.64 (m, 1H), 5.02 (s, 2H), 6.26 (t, J = 6.6 Hz, 1H), 6.89 (t, J = 7.2 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 7.15–7.25 (m, 3H), 7.26–7.36 (m, 3H), 7.69 (d, J= 7.8 Hz, 1H), 8.04 (s, 1H), 8.15 (s, 1H); 13C NMR (150 MHz, CD3OD) δ − 4.7, − 4.6, 18.8, 24.2, 26.3, 27.8, 27.9, 30.0, 32.0, 38.1, 41.2, 50.1, 52.5, 69.8, 72.2, 73.8, 85.6, 86.4, 114.2, 120.4, 122.1, 123.9, 129.5, 129.6, 129.8, 131.8, 133.9, 137.7, 141.0, 150.4, 153.9, 157.3, 158.1, 168.3, 174.2; HRMS (ESI+) calcd for C39H55N8O7SSi [M+DBU+H]+ 807.3678, found 807.3674 (error 0.5 ppm).
2 ′-Deoxy-5 ′-O-(N-(2-hydroxybenzoyl)sulfamoyl)adenosine l,8-diazabicyclo[5.4.0]undec-7-ene salt (9·DBU)
To a solution of 27-DBU· (80 mg, 0.099 mmol, 1.0 equiv) in MeOH (10 mL) was added 10% Pd/C (20 mg) and the reaction stirred under an H2 atm for 8 h. The reaction mixture was filtered through a plug of Celite and the residue was washed with MeOH (4×10 mL). The combined filtrates were concentrated and the crude obtained was treated with TBAF (0.5 mL, 1.0 M THF solution, 0.50 mmol, 5.0 equiv) for 3 h. The reaction was concentrated in vacua. Purification by flash chromatography (15:85:1 MeOH/EtOAc/Et3N) afforded the title compound (16 mg, 27% over all yield for two steps) as thick oil: Rf = 0.2 (1:5 MeOH/EtOAc); [[α]20D +9.0 (c 0.51, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.35–1.60 (m, 8H), 2.47 (ddd, J= 13.8, 7.2, 4.8 Hz, 1H), 2.79–2.83 (m, 1H), 3.81 (dt, J= 13.8, 7.2 Hz, 2H), 3.34 (t, J = 6.0 Hz, 2H), 3.50–3.60 (m, 4H), 4.15 (q, J = 3.6 Hz, 1H), 4.30 (dd, J = 10.8, 4.2 Hz, 1H), 4.37 (dd, J = 10.8, 3.0 Hz, 1H), 4.60–4.68 (m, 1H), 6.43 (t, J = 6.6 Hz, 1H), 6.80–6.90 (m, 2H), 7.32 (t, J = 8.4 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), 8.29 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 24.2, 27.9, 28.0, 30.1, 31.9, 38.0, 40.9, 50.3, 52.6, 70.5, 72.5, 85.8, 86.2, 116.7, 118.5, 120.0, 120.4, 128.7, 134.7, 140.9, 150.4, 153.9, 157.3, 161.4, 171.2, 174.6; HRMS (ESI+) calcd for C26H35N8O7S [M+DBU+H]+ 603.2344, found 603.2363 (error 3.2 ppm).
2′-Deoxy,3′-O-tert-butyldimethylsilyl-5′-O-(N -(2-hydroxybenzoyl)sulfamoyl)adenosine (29)
Compound 24 (l00mg, 0.22 mmol, 1.0 equiv) was coupled to 26 (215 mg, 0.66 mmol, 3.0 equiv) using the general salicylation procedure B. The reaction mixture was filtered then concentrated in vacuo. Purification by flash chromatography afforded 27 as the triethylammonium salt.
Compound 27 prepared above was treated with Pd/C (15 mg) in MeOH (15 mL) for 6 h. The reaction mixture was filtered, washed with MeOH and the filtrate concentrated. Purification by flash chromatography (80:20:1 EtOAc/MeOH/Et3N) afforded the title compound (21 mg, 17% over two steps) as a thick oil: Rf = 0.5 (4:1 EtOAc/MeOH); [α]20D −93 (c 0.88, CH3OH); 1H NMR (600 MHz, CD3OD) δ0.08 (s, 3H), 0.10 (s, 3H), 0.90 (s, 9H), 2.39 (ddd, J = 13.2, 6.0 Hz, 1H), 2.80–2.90 (m, 1H), 4.16–4.24 (m, 1H), 4.32 (dd, J= 10.8, 3.6 Hz, 1H), 4.36 (dd, J = 10.8, 3.6 Hz, 1H), 4.70–4.80 (m, 1H), 6.48 (t, J = 6.6 Hz, 1H), 6.70–6.84 (m, 2H), 7.29 (t, J = 8.4 Hz, 1H), 7.95 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), 8.47 (s, 1H); 13C NMR (150 MHz, CD3OD) δ− 4.6, −4.5, 18.9, 26.4, 42.2, 69.9, 74.9, 85.9, 87.5, 118.1, 119.4, 120.3, 120.6, 131.6, 134.6, 141.3, 150.5, 153.9, 157.4, 162.3, 175.4; HRMS (ESI+) calcd for C23H33N6O7SSi [M+H]+ 565.1895, found 565.1817 (error 13.8 ppm).
2′-Deoxy-5′-O-(N -(2-hydroxybenzoyl)sulfamoyl)adenosine triethylammonium salt (9·Et3N)
To a solution of 29 (10.5 mg, 0.018 mmol, 1.0 equiv) in THF (2.0 mL) was added TBAF (1.0 M solution in THF, 0.1 mL, 6.0 equiv) and the solution stirred 3 h at rt. The reaction mixture was concentrated and purification of the residue by flash chromatography (65:35:1 EtOAc/MeOH/Et3N) afforded the title compound (5.1 mg, 50%): Rf= 0.15 (3:1 EtOAc/MeOH); [α]20D−99 (c 0.25, CH3OH) 1H NMR (600 MHz, CD3OD) δ1.29 (t, J= 7.2 Hz, 9H), 2.44 (ddd, J= 13.8, 5.4, 2.4 Hz, 1H), 2.70–2.90 (m, 1H), 3.18 (q, J = 7.2 Hz, 6H), 4.24 (s, 1H), 4.32 (dd, J = 11.4, 3.6 Hz, 1H), 4.35 (dd, J = 10.8, 3.6 Hz, 1H), 4.62–4.70 (m, 1H), 6.50 (t, J = 6.6 Hz, 1H), 6.75–6.84 (m, 2H), 7.29 (t, J = 7.8 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 8.16 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ9.4, 41.6, 48.1, 70.0, 73.5, 85.9, 87.0, 118.0, 119.4, 120.3, 120.8, 131.5, 134.5, 141.3, 150.6, 153.9, 157.4, 162.2, 175.1; HRMS (ESI+) calcd for C17H19N6O7S [M+H]+ 451.1030, found 451.1014 (error 3.5 ppm).
2′-O-tert-Butyldimethylsilyl-3-deoxyadenosine (23)
Compound 19 (100 mg, 0.37 mmol, 1.0 equiv) was bis-silylated with TBSCl (140 mg, 0.93 mmol, 2.5 equiv) to afford 22 using the procedure described for the preparation of 20. The di-TBS product obtained was used directly for the next step.
To a solution of crude di-TBS product prepared above (170 mg, 0.37 mmol, 1.0 equiv) in MeOH/EtOAc (1:1, 10 mL) at 0 °C was added pTsOH·H2O (0.35 g, 1.83 mmol, 2.8 equiv). After 5 h the reaction was complete and the reaction mixture was quenched using an excess of solid K2CO3 (500 mg), and stirred for 1 h, filtered and concentrated under reduced pressure. Purification by flash chromatography (1:20 MeOH/EtOAc) afforded the title compound (106 mg, 78% over two steps) as a white solid: mp = 154–156 °C; Rf = 0.2 (EtOAc); [α]20D −49.4 (c 0.890, CH3OH); 1H NMR (600 MHz, CDC13) δ0.00 (s, 3H), 0.14 (s, 3H), 1.06 (s, 9H), 2.44–2.56 (m, 1H), 2.83 (ddd, J = 10.8, 7.2, 2.4 Hz, 1H), 3.82 (d, J = 12.6 Hz, 1H), 4.26 (d, J = 12.6 Hz, 1H), 4.77 (d, J = 9.0 Hz, 1H), 5.33 (q, J = 7.2 Hz, 1H), 5.87 (d, J = 6.0 Hz, 1H), 6.11 (br s, 2H, NH2), 6.49 (br s, 1H, OH), 8.10 (s, 1H), 8.62 (s, 1H); 13C NMR (150 MHz, CDC13) δ− 5.4, −5.2, 17.8, 25.5, 34.8, 69.4, 73.9, 80.4, 93.6, 121.1, 140.5, 148.6, 152.5, 155.9; HRMS (ESI+) calcd for C16H28N5O3Si [M+H]+ 366.1956, found 366.1976 (error 5.5 ppm).
2′-O-tert -Butyldimethylsilyl-3′-deoxy-5′-O-(sulfamoyl)adenosine (25)
This was prepared from 23 (90 mg, 0.246 mmol, 1.0 equiv) using the general procedure for sulfamoylation. Purification by flash chromatography (30:1 EtOAc/MeOH) afforded the title compound (90 mg, 82%): mp = 238–240 °C melted with charring; Rf = 0.40 (97:3 EtOAc/MeOH); [α]20D +13.7 (c 0.980, CH3OH); 1H NMR (600 MHz, CD3OD) δ0.03 (s, 3H), 0.06 (s, 3H), 0.87 (s, 9H), 2.11 (ddd, J = 13.2, 6.0, 3.0 Hz, 1H), 2.35 (ddd, J = 13.2, 8.4, 6.0 Hz, 1H), 4.29 (dd, J = 10.8, 3.6 Hz, 1H), 4.43 (dd, J = 10.8, 2.4 Hz, 1H), 4.65–4.75 (m, 1H), 4.75–4.85 (m, 1H), 5.98 (d, J = 2.4 Hz, 1H), 8.19 (s, 1H), 8.30 (s, 1H); 13C NMR (150 MHz, CD3OD) δ− 4.9, −4.8, 18.8, 26.1, 35.8, 70.9, 77.7, 79.2, 92.8, 120.3, 140.4, 150.5, 152.9, 157.3; MS (ESI+) calcd for C16H26N5O2Si [M−SO2NH2]+ 348.2, found 348.1.
5′-O-(N -(2-Benzyloxybenzoyl)sulfamoyl)-2′-O-tert-butyldimethylsilyl-3′-deoxyadenosine triethylammonium salt (28)
This was prepared from 25 (70 mg, 0.157 mmol, 1.0 equiv) and 26 (154 mg, 0.47 mmol. 3.0 equiv) using the general salicylation procedure A. Purification by flash chromatography (90:10:1 EtOAc/MeOH/Et3N) afforded the title compound (90 mg, 87%) as thick oil: Rf = 0.55 (1:9 MeOH/EtOAc); [α]20D +19 (c 0.70, CH3OH); 1H NMR (600 MHz, CD3OD) δ0.01 (s, 3H), 0.05 (s, 3H), 0.86 (s, 9H), 1.20 (t, J= 7.2 Hz, 9H), 1.85–1.95 (m, 1H), 2.20–2.40 (m, 1H), 3.07 (q, J= 7.2 Hz, 6H), 4.15 (d,J= 11.4 Hz, 1H), 4.30 (d, J= 10.8 Hz, 1H), 4.40–4.50 (m, 1H), 4.73 (d, J = 2.4 Hz, 1H), 5.10 (s, 2H), 5.93 (s, 1H), 6.93 (t, J = 7.2 Hz, 1H), 7.06 (d, J = 9.0 Hz, 1H), 7.20–7.40 (m, 4H), 7.35 (d, J = 7.8 Hz, 1H), 7.48 (d, J = 7.8 Hz, 2H), 8.18 (s, 1H), 8.47 (s, 1H); 13C NMR (150 MHz, CD3OD) δ −4.8, −4.7, 9.2, 18.8, 26.1, 35.7, 47.7, 70.6, 71.6, 78.1, 79.6, 92.5, 114.3, 120.2, 121.5, 128.8, 128.9, 129.4, 129.7, 131.2, 131.9, 138.7, 140.8, 150.5, 153.8, 157.1, 157.2, 176.7; HRMS (ESI+) calcd for C30H39N6O7SSi [M+H]+ 655.2365, found 655.2389 (error 3.7 ppm).
3′-Deoxy-5′-O-(N-(2-hydroxybenzoyl)sulfamoyl)adenosine triethylammonium salt (10)
To a solution of 28 (70 mg, 0.092 mmol, 1.0 equiv) in MeOH (10 mL) was added 10% Pd/C (20 mg) and the reaction was stirred under a H2 atm for 8 h. The reaction mixture was filtered through a plug of Celite and the residue was washed with MeOH (4x10 mL). The combined filtrates were concentrated and the crude obtained was treated with 80% aq TFA (2.0 mL) for 8 h. The reaction was concentrated in vacuo. Purification by flash chromatography (10:90:1 MeOH/EtOAc/Et3N) afforded the title compound (27 mg, 53%) as thick glassy oil: Rf = 0.3 (1:4 MeOH/EtOAc); [α]20D−10 (c 0.28, MeOH); 1H NMR (600 MHz, CD3OD) δ1.26 (t, J = 7.2 Hz, 9H), 2.05–2.20 (m, 1H), 2.40–2.55 (m, 1H), 3.15 (q, J = 7.2 Hz, 6H), 4.32 (dd, J = 11.4, 4.2 Hz, 1H), 4.46 (d, J= 11.4 Hz, 1H), 4.60–4.80 (m, 2H), 6.00 (s, 1H), 6.70–6.85 (m, 2H), 7.27 (t, J = 7.2 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 8.12 (s, 1H), 8.47 (s, 1H); 13C NMR (150 MHz, CD3OD) δ9.3, 34.9, 47.9, 70.7, 76.9, 79.9, 92.9, 117.9, 119.3, 120.3, 120.7, 131.3, 134.3, 140.7, 150.2, 153.7, 157.3, 162.0, 174.9; HRMS (ESI+) calcd for C17H19N6O7S [M+H]+451.1030, found 451.1053 (error 5.1 ppm).
(1′R, 4′S)-9-[4-(Hydroxymethyl)cyclopent-2-en-1 -yl]adenine (32).50
To a solution of aristeromycin 14 (0.7 g, 2.63 mmol, 1.0 equiv) and trimethyl orthoformate (7.0 mL, 64 mmol, 24.3 equiv) in DMF (2.1 mL) was added pTsOH.H2O (720 mg, 3.81 mmol, 1.45 equiv). After 36 h at rt, anhydrous K2CO3 (1.05 g, 7.27 mmol) was added and the mixture stirred for 2 h. The reaction mixture was filtered and the solids were washed with a minimum amount of trimethyl orthoformate (2.0 mL). The combined filtrates and washings were concentrated under reduced pressure to provide 31 as a gummy residue, which was azeotropically dried with toluene (20 mL) on a rotary evaporator.
Crude 31 from above was refluxed 16 h in Ac2O (10 mL). The reaction was cooled to rt, filtered through a plug of silica gel, and concentrated under reduced pressure to afford a dark yellow solid, which was treated with 6 N aq HCl (6 mL) at rt for 12 h. The reaction was concentrated in vacuo. Purification by flash chromatography (3:7 MeOH/EtOAc) afforded the title compound (150 mg, 48%): Rf = 0.3 (3:7 MeOH/EtOAc); [α]20D −6.2 (c 0.59, MeOH); 1H NMR (600 MHz, CD3OD) δ1.73 (dt, J = 13.8, 6.0 Hz, 1H), 2.82 (ddd, J= 13.8, 8.4, 5.4 Hz, 1H), 3.02 (br s, 1H), 3.58 (dd, J = 10.4, 4.8 Hz, 1H), 3.65 (dd, J = 10.4, 5.4 Hz, 1H), 5.68 (t, J = 6.0 Hz, 1H), 5.95 (d, J = 6.0, 1H), 6.21 (d, J = 6.0 Hz, 1H), 8.12 (s, 1H), 8.19 (s, 1H); 13C NMR (150 MHz, CD3OD) δ35.6, 49.2, 61.3, 65.4, 120.3, 130.5, 140.1, 140.9, 150.3, 153.5, 157.3; HRMS (ESI+) calcd for C11H14N5O [M+H]+ 232.1193, found 232.1197 (1.7 ppm).
(1′R, 4′S)-9-[4-((Sulfamoyl)oxymethyl)cyclopent-2-en-1-yl]adenine (33)
This was prepared from 32 (150 mg, 0.648 mmol, 1.0 equiv) using the general procedure for sulfamoylation. Purification by flash chromatography (30:1 EtOAc/MeOH) afforded the title compound (110 mg, 55%) as an oil: Rf = 0.50 (7:3 EtOAc/MeOH); [α]20D +70.8 (c 0.290, CH3OH); 1H NMR (600 MHz, CD3OD) δ1.77 (dt, J = 14.4, 6.0 Hz, 1H), 2.91 (ddd, J = 13.8, 9.0, 5.4 Hz, 1H), 3.27 (br s, 1H), 4.16 (dd, J = 9.6, 4.8 Hz, 1H), 4.22 (dd, J = 9.6, 4.8 Hz, 1H), 5.73 (t, J = 6.0 Hz, 1H), 6.01 (d, J = 6.0 Hz, 1H), 6.21 (d, J = 5.4 Hz, 1H), 8.09 (s, 1H), 8.20 (s, 1H); 13C NMR (150 MHz, CDC13) δ35.5, 46.1, 61.0, 72.3, 120.2, 131.5, 138.3,140.8, 150.4,153.6, 157.3; MS (ESI+) calcd for C11H12N5 [M-SO2NH2]+214.1, found 214.1.
(1′R,4′S)-9[4((N(2(Methoxymethyloxy)benzoyl)sulfamoyl)oxymethyl)cyclopent-2-en-1-yl]adenine triethylammonium salt (34)
This was prepared from 33 (100 mg, 0.32 mmol) and 16 (270 mg, 0.96 mmol, 3.0 equiv) using the general salicylation procedure A. Purification by flash chromatography (80:20:1 EtOAc/MeOH/Et3N) afforded the title compound (150 mg, 81%) as a viscous oil: Rf = 0.65 (7:3 EtOAc/MeOH); [α]20D+42.8 (c 0.980, CH3OH); 1H NMR (600 MHz, CD3OD) δ1.25 (t, J= 7.2 Hz, 9H), 1.85–1.95 (m, 1H), 2.85–2.95 (m, 1H), 3.14 (q,J= 7.2 Hz, 6H), 3.27 (br s, 1H), 3.43 (s, 3H), 4.20–4.32 (m, 2H), 5.25 (s, 2H), 5.73 (t, J = 6.0 Hz, 1H), 5.97 (d, J = 5.4 Hz, 1H), 6.24 (d, J = 6.0 Hz, 1H), 6.95 (t, J = 7.2 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 7.25 (t, J = 8.4 Hz, 1H), 7.38 (d, J = 7.2 Hz, 1H), 8.19 (s, 1H), 8.23 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 34.5, 45.2, 55.5, 59.9, 71.1, 95.3, 115.9, 119.0, 121.3, 121.4, 128.6, 129.9, 130.0, 131.3, 137.6, 139.9, 149.2, 152.4, 154.5, 156.1, 175.2; HRMS (ESI+) calcd for C20H23N6O6S [M+H]+ 475.1394, found 475.1391 (error 0.6 ppm).
(1′R, 4′S)-9-[4-((N-(2-Hydroxybenzoyl)sulfamoyl)oxymethyl)cyclopent-2-en-l-yl]adenine triethylammonium salt (11)
Compound 33 (50 mg, 0.086 mmol, 1.0 equiv) was stirred in 80% aq TFA (2.0 mL) for 3 h then concentrated in vacua. Purification by flash chromatography (25:75:1 MeOH/EtOAc/Et3N) afforded the title compound (36 mg, 80%) as a viscous oil: Rf = 0.4 (4:1 EtOAc/MeOH); [α]20D +22 (c 0.21, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.80–1.95 (m, 1H), 2.80–2.95 (m, 1H), 3.18 (q, J = 7sss.2 Hz, 6H), 3.23 (br s, 1H), 4.15–4.30 (m, 2H), 5.69 (t, J = 6.0 Hz, 1H), 5.90–6.00 (m, 1H), 6.21 (d, J = 5.4 Hz, 1H), 6.70–6.80 (m, 2H), 7.26 (t, J = 7.8 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), 8.18 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 34.3, 46.7, 59.9, 71.2, 116.7, 118.1, 119.0, 119.5, 129.9, 130.0, 133.1, 137.6, 139.9, 149.1, 151.8, 155.7, 160.7, 161.9, 173.3; HRMS (ESI+) calcd for C18H19N6O5S [M+H]+ 431.1132, found 431.1102 (error 7.0 ppm).
(1′S, 4′R)-9-[3-((N-(2-hydroxybenzoyl)sulfamoyl)oxymethyl)cyclopent-1-yl]adenine triethylammonium salt (12)
To compound 11 (25 mg, 0.047 mmol, 1.0 equiv) in MeOH (5 mL) was added 10% Pd/C (20 mg) and the reaction stirred for 6 h under an H2 atm. The reaction mixture was filtered thru a plug of Celite and the solids washed with MeOH (4×10 mL). The filtrate was concentrated under reduced pressure. Purification by flash chromatography (30:70:1 MeOH/EtOAc/Et3N) afforded the title compound (9.0 mg, 36%) as a viscous oil: Rf = 0.4 (4:1 EtOAc/MeOH); [α]20D +1.0 (c 0.44, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.82–1.92 (m, 1H), 1.94–2.04 (m, 2H), 2.06–2.16 (m, 1H), 2.22–2.32 (m, 1H), 2.44–2.50 (m, 1H), 2.50–2.60 (m, 1H), 3.18 (q, J = 7.2 Hz, 6H), 4.17 (dd, J = 10.2, 6.6 Hz, 1H), 4.21 (dd, J = 10.2, 6.6 Hz, 1H), 4.86–4.96 (m, 1H), 6.72–6.82 (m, 2H), 7.27 (t, J = 7.8 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 8.16 (s, 1H), 8.27 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.6, 32.7, 36.4, 38.5, 47.9, 57.1, 73.6, 117.9, 119.2, 120.3, 120.8, 131.3, 134.3, 140.9, 150.7, 153.4, 157.2, 161.9, 174.6; HRMS (ESI+) calcd for C18H21N6O5S [M+H]+ 433.1289, found 433.1327 (error 8.8 ppm).
9-[(((N-(2-Hydroxybenzoyl)sulfamoyl)oxy)ethoxy)methyl]adenine triethylammonium salt (13)
This was prepared from 3649 (100 mg, 0.346 mmol, 1.0 equiv) and 26 (338 mg, 1.04 mmol, 3.0 equiv) using the general salicylation procedure A. Purification by flash chromatography (85:15:1 EtOAc/MeOH/Et3N) afforded 37 (80 mg, 44%).
Compound 37 prepared above was dissolved in MeOH (15.0 mL) and stirred under an H2 atm in presence of 10% Pd/C (20 mg). After 4 h the reaction mixture was filtered through a plug of Celite. Purification by flash chromatography afforded the title compound as a white solid (65 mg, 98%): mp = 93–95 °C; Rf = 0.35 (3:1 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 3.17 (q, J = 7.2 Hz, 6H), 3.82 (t, J = 4.2 Hz, 2H), 4.25 (t, J = 4.2 Hz, 2H), 5.67 (s, 2H), 6.72–6.84 (m, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.89 (d, J = 7.2 Hz, 1H), 8.19 (s, 1H), 8.24 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 46.7, 67.5, 68.3, 72.9, 116.6, 118.0, 118.8, 119.5, 130.1, 133.1, 141.9, 149.7, 152.9, 156.2, 160.8, 173.6; HRMS (ESI+) calcd for C15H17N6O6S [M+H]+ 409.0925, found 409.0904 (error 5.1 ppm).
Cloning, Overexpression, and Purification of MbtA
The mbtA gene was amplified by PCR from M. tuberculosis BAG RV143 using the following primers 5′CCAGCCCCCCTGTTAAGGAG3′ and 5′CCGAACGTGCCCATGAGTC3′ and cloned into pCR2.1-TOPO (Invitrogen) using the manufacturers instructions creating pCDD001. This plasmid served as template for a second round of PCR using the primers 5′ATGCCACCTAAAGCGGCAGATGGCCGCCGA3′ and 5′GGAAGCTTAATGGCAGCGCTGGGTCGTCACGGGA3′. The resulting mbtA gene fragment was cloned into pET SUMO (Invitrogen) according to the manufacturers instructions to create pCDD003. After successfully cloning mbtA the gene cassette was confirmed by sequencing. pCDD003 was then electroporated into E. coli BL21(DE3) containing the groEL groES chaperone plasmid pGRO7 (Takara). LB (500 mL) supplemented with kanamycin (50μg/mL), chloramphenicol (25μg/ml), MgCl2 (10 mM), and arabinose (0.5 mg/ml) was inoculated with 5 mL of overnight culture. After growing to an OD600 at 37 °C the cultures were induced with 0.4 mM IPTG and grown for an additional 4 h at 30 °C. The cultures were then centrifuged and the pellet was frozen at −20 °C overnight. To remove GroEL from MbtA during the purification process, a modified ATP-dependent removal protocol was employed.52 Frozen pellets were resuspended in 22.5 mL GroEL stripping buffer A (100 mM TEA-HC1, 170 mM NaCl, pH 7.4) and sonicated. The lysate was centrifuged 10 min at 30,000×g and the pellet was discarded. Subsequently 2.5 mL GroEL stripping buffer B (100 mM TEA-HC1, 200 mM MgCl2, 100 mM ATP, pH 7.4) was added and the lysate was incubated for 30 min at 4 °C. To assure the removal of GroEL 140 μl of denatured E. coli proteins prepared as previously described52 was added and the lysate was allowed to incubate for another 1 h at 4 °C. After this second incubation, 2 mL of 50% Ni-NTA (Qiagen) was added to the lysate and allowed to incubate on an end-over-end mixer for 1 h at 4 °C. The mixture was poured into a column and the flow through was collected. The column was washed with 16 mL of wash buffer (50 mM sodium phosphate, 300 mM NaCl, 20 mM imidazole, pH 8.0) and MbtA was eluted with 3 mL of elution buffer (50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole, pH 8.0) of which the first 0.5 mL was discarded and the final 2.5mL collected. The protein fraction was desalted on a PD-10 column (GE Healthcare) into Sumo digestion buffer without DTT (50 mM Tris-HCl, 0.2% Igepal CA-630 (Sigma), 150 mM NaCl). The protein concentration was measured by the Bradford Assay (Bio-Rad), after which DTT was added to 1 mM and SUMO protease was added to 15 U/mg. The reaction was incubated 15 h at 4 °C. After digestion 0.5 mL of 50% Ni-NTA was added back to the sample to remove the SUMO tag, SUMO protease, and other E. coli proteins carried through purification which have some affinity for Ni-NTA. The mixture was incubated at 4 °C for 1 h and then loaded on a column to separate the flow through from the resin. The flow through was collected and desalted on a PD-10 column into MbtA storage buffer (10 mM Tris-HCl, 1 mM EDTA, 5% glycerol, pH 8.0) and stored at −80 °C (2–5 mg protein/L culture).
Enzyme Kinetic Studies. ATP/PPi Exchange Assay54
Reactions were performed under initial velocity conditions in a total volume of 101 μL. The reaction was set up in a volume of 90 μL and contained 250 μM salicylic acid, 10 mM ATP, 1 mM PPi and 7 nM MbtA in assay buffer (75 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM DTT). In reactions designed to measure inhibition of 8–13, the inhibitors (1 μL) in DMSO or DMSO only as a control were added. For experiments investigating the steady-state kinetic parameters of salicylic acid and ATP, reactions were set up as above using 14 nM MbtA and either holding the ATP concentration constant at 10 mM and varying the salicylic acid concentration (2.25 μM, 4.5 μM, 9.0 μM, 18.0 μM, 36.0 μM) or holding the salicylic acid concentration constant at 250 μM and varying the ATP concentration (37.5 μM, 75 μM, 150 μM, 300 μM, 600 μM, l0mM). Titration of MbtA against compound 8 to attain the accurate enzyme concentration used 7 nM enzyme in assay buffer along with 250 μM salicylic acid and 200μM ATP (to attain the desired [E]/Ki ratio of 200). The reaction components were allowed to equilibrate for 10 min at 23 °C. Reactions were initiated by the addition of 10 μL (0.5 μCi 32PPi, Perkin Elmer 84.12Ci/mmol) in 50 mM sodium phosphate buffer pH 7.8 and placed at 37 °C for 20 min. Reactions were quenched by the addition of 200 μl quenching buffer (350 mM HC1O4, 100 mM PPi, 1.8% w/v activated charcoal). The charcoal was pelleted by centrifugation and washed once with 500 μl H2O. The washed pellet was resuspended in 200 μL H2O and transferred to a scintillation vial then mixed with 15 mL scintillation fluid (RPI) and counted on a Beckman LS6500. The counts from the bound γ-[32P]-ATP were directly proportional to initial velocity of the reaction.
Data Analysis
For inhibitors that displayed tight-binding inhibition (KIapp < 200·[E]) the fractional activity (vi/v0) where vi is the reaction velocity at a given [I] and v0 is the reaction velocity of the DMSO control were fit by non-linear regression analysis to the Morrison equation (eq 1) constraining [E] to 7.0 nM using GraphPad prism version 4.0 to obtain KIapp values.40 The enzyme concentration in turn was determined by active-site titration employing inhibitor 8.40 The dose response curves of the fractional activity versus [I] for inhibitor 8 is shown in figure 3 and 9–13 are shown in figures S5–S9.
| (1) |
| (2) |
| (3) |
M. tuberculosis H37Rv MIC Assay
Minimum inhibitory concentrations (MICs) were determined in quadruplicate in iron-deficient GAST according to the broth microdilution method using drugs from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO.19 All measurements reported herein used an initial cell density of 104–105 cells/assay and growth monitored at 10 and at 14 days, with the untreated and DMSO-treated control cultures reaching an OD620 ~ 0.2–0.3. Plates were incubated at 37 °C (100 μl/well) and growth was recorded by measurement of optical density at 620 nm. MIC50 values were determined by fitting the dose-response curves by nonlinear regression analysis to the four parameter sigmoidal dose-response curve using GraphPad Prism version 4.0.
Y. pseudotuberculosis ATCC 6902 MIC Assay
Y. pseudotuberculois ATCC 6902 was selected as a representative strain from others in the ATCC collection based on its phenotypic similarity to other strains, nucleotide sequence confirmation of the ybtE gene, as well as siderophore production using CAS agar plates.79 Further, this strain demonstrated the ability to grow under iron-limiting conditions relative to ybtE-negative strains The MIC99 concentration of each nucleoside compound was determined in triplicate using the broth microdilution method. Y. pseudotuberculosis ATCC 6902 was cultured overnight on Difco™ Brain Heart Infusion (BHI, Becton, Dickinson and Co.) medium at 37 °C. Cells were washed twice with desferrated BHI (BHI-D, BHI treated with Chelex® 100 resin (BioRad) and supplemented with 200μM 2,2′dipyridyl). The washed cells were diluted in BHI-D to a 1.0 MacFarland standard and used as an inoculum (initial cell density of 103–104 cells/200 μL media/well.) for BHI-D or BHI-D supplemented with 150μM FeCl3-6H2O for iron rich conditions. Each compound was added to the media at varying concentrations up to 100 μM and an equivalent amount of DMSO (1%) only was used in control wells. Microtiter plate cultures were grown for 24 hrs at 37 °C and shaken using a Micromix plate shaker (program 20). Final optical density at 600 nm was measured using a SpectraMax Plus (Molecular Devices) reader.
Docking Studies
The homology model and docking runs were configured as described43, except that water molecules were held fixed during the conformational search. 10,000 search steps were performed for each compound.
Supplementary Material
1H NMR and 13C NMR spectra of 8–13, table of HPLC conditions and purities of 8–13, SDS-PAGE of purified MbtA, active-site titration plot of MbtA with 6, normalized v vs. [S] plots for the reactions of ATP and salicylic acid with MbtA, and dose-response curves of fractional MbtA activity (vi/v0) as a function of inhibitor concentrations for 9–13.
Scheme la.

aReaction conditions: (a) 2,2′-dimethoxypropane, MeSO3H, acetone, 84%; (b) NH2SO2Cl, NaH, DME, 63%; (c) DBU, DMF, 60%; (d) 80% aq TFA, 66%.
Acknowledgments
We thank Dr. Robert Vince for invaluable advice and for supplying aristeromycin and acycloadenosine. We thank the Minnesota Supercomputing Institute VWL lab for computer time and the Pasteur Research Institute (Paris) for suppyling a bacterial artificial chromosome library of M. tuberculosis H37Rv. This research was supported by grants from NIH (R01AI070219) and the Center for Drug Design in the Academic Health Center of the University of Minnesota to C.C.A.
NON STANDARD ABBREVIATIONS
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- MBP
maltose binding protein
- MIC
minimum inhibitory concentration
- NHS
N-hydroxysuccinimide
- NRPS
nonribosomal peptide synthetase
- PAS
para-aminosalicylic acid
- PDB
protein data bank
- PKS
polyketide synthase
- SAR
structure activity relationships
- SUMO
small ubiquitin modifying protein
- TB
tuberculosis
- TBAF
tetrabutylammonium fluoride
- TBS
tert-butyldimethylsilyl
- TFA
trifluoroacetic acid
- XDR
extensively-drug-resistant
References
- 1.World Health Organization. Fact sheet on tuberculosis. 2005 http://www.who.int/mediacentre/factsheets/fsl04/en/print.html. [PubMed]
- 2.CDC. Worldwide emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs. MMWR. 55(10):TK-TK. [PubMed] [Google Scholar]
- 3.Raymond KN, Dertz EA, Kim SS. Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci USA. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 5.Quadri LE. Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol Microbiol. 2000;37:1–12. doi: 10.1046/j.1365-2958.2000.01941.x. [DOI] [PubMed] [Google Scholar]
- 6.Clarke TE, Tari LW, Vogel HJ. Structural biology of bacterial iron uptake systems. Curr Top Med Chem. 2001;1:7–30. doi: 10.2174/1568026013395623. [DOI] [PubMed] [Google Scholar]
- 7.Snow GA. Isolation and structure of mycobactin-T, a growth factor of Mycobacterium tuberculosis. Biochem J. 1965;97:166–175. doi: 10.1042/bj0970166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gobin J, Moore CH, Reeve JR, Jr, Wong DK, Gibson BW, Horwitz MA. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc Natl Acad Sci USA. 1995;92:5189–5193. doi: 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snow GA. Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vergne AF, Walz AJ, Miller MJ. Iron chelators from mycobacteria (1954–1999) and potential therapeutic applications. Nat Prod Rep. 2000;17:99–116. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 11.Twort FW, Ingram GLY. A method for isolating and cultivating the Mycobacterium enteritidis chronicae pseudotuberculosis bovis, Jöhne, and some experiments on the preparation of a diagnostic vaccine for pseudo-tuberculosis enteritis of bovines. Proc Roy Soc Ser B. 1912;84:517–530. [Google Scholar]
- 12.Hu J, Miller MJ. Total synthesis of a mycobactin S, a siderophore growth promoter of Mycobacterium smegmatis, and determination of its growth inhibitory activity against Mycobacterium tuberculosis. J Am Chem Soc. 1997;119:3462–3468. [Google Scholar]
- 13.Xu Y, Miller MJ. Total synthesis of mycobactin analogues as potent antimycobacterial agents using a minimal protecting group strategy. J Org Chem. 1998;63:4314–4322. [Google Scholar]
- 14.Miller MJ, Xu Y. Antimycobacterial Agents. U. S. Patent 6,310,058. 2001 Oct. 30;
- 15.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 16.Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 17.Krithika R, Marathe U, Saxena P, Ansari MZ, Mohanty D, Gokhale RS. A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2069–2074. doi: 10.1073/pnas.0507924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitt MP, Predich M, Doukhan L, Smith I, Holmes RK. Characterization of an iron-dependent regulatory protein (IdeR) of Mycobacterium tuberculosis as a functional homolog of the diphtheria toxin repressor (DtxR) from Corynebacterium diphtheriae. Infect Immun. 1995;63:4284–4289. doi: 10.1128/iai.63.11.4284-4289.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner D, Maser J, Lai B, Cai Z, Barry CE, 3rd, Honer Zu Bentrup K, Russell DG, Bermudez LE. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J Immunol. 2005;174:1491–1500. doi: 10.4049/jimmunol.174.3.1491. [DOI] [PubMed] [Google Scholar]
- 21.Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol. 2005;1:149–153. doi: 10.1038/nchembio717. [DOI] [PubMed] [Google Scholar]
- 22.Ratledge C, Brown KA. Inhibition of mycobactin formation in Mycobacterium smegmatis by p-aminosalicylate. A new proposal for the mode of action of p-aminosalicylate. Am Rev Respir Dis. 1972;106:774–776. doi: 10.1164/arrd.1972.106.5.774. [DOI] [PubMed] [Google Scholar]
- 23.Mandell GL, Petri WAJ. Goodman and Oilman’s the Pharmacological Basis of Therapeutics. 9. McGraw-Hill; New York: 1996. Antimicrobial agents: Drugs used in the chemotherapy of tuberculosis, Mycobacterium avium complex disease, and leprosy; p. 1164. [Google Scholar]
- 24.Murray MJ, Murray AB, Murray MB, Murray CJ. The adverse effect of iron repletion on the course of certain infections. Br Med J. 1978;2:1113–1115. doi: 10.1136/bmj.2.6145.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manabe YC, Saviola BJ, Sun L, Murphy JR, Bishai WR. Attenuation of virulence in Mycobacterium tuberculosis expressing a constitutively active iron repressor. Proc Natl Acad Sci USA. 1999;96:12844–12848. doi: 10.1073/pnas.96.22.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marahiel MA, Stachelhaus T, Mootz HD. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 27.Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 28.Crosa JH, Walsh CT. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerbarh O, Ciulli A, Howard NL, Abell C. Salicylate biosynthesis: overexpression, purification, and characterization of Irp9, a bifunctional salicylate synthase from Yersinia enterocolitica. J Bacterial. 2005;187:5061–5066. doi: 10.1128/JB.187.15.5061-5066.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison AJ, Ramsay RJ, Baker EN, Lott JS. Crystallization and preliminary X-ray crystallographic analysis of Mbtl, a protein essential for siderophore biosynthesis in Mycobacterium tuberculosis. Acta Crystallograph Sect F. 2005;61:121–123. doi: 10.1107/S1744309104031215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison AJ, Yu M, Gardenborg T, Middleditch M, Ramsay RJ, Baker EN, Lott JS. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J Bacteriol. 2006;188:6081–6091. doi: 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moody DB, Young DC, Cheng TY, Rosat JP, Roura-Mir C, O’Connor PB, Zajonc DM, Walz A, Miller MJ, Levery SB, et al. Science. 2004;303:527–531. doi: 10.1126/science.1089353. [DOI] [PubMed] [Google Scholar]
- 33.Card GL, Peterson NA, Smith CA, Rupp B, Schick BM, Baker EN. The crystal structure of Rvl347c, a putative antibiotic resistance protein from Mycobacterium tuberculosis, reveals a GCN5-related fold and suggests an alternative function in siderophore biosynthesis. J Biol Chem. 2005;280:13978–13986. doi: 10.1074/jbc.M413904200. [DOI] [PubMed] [Google Scholar]
- 34.Schimmel P, Tao J, Hill J. Aminoacyl t-RNA synthetases as targets for new anti-infectives. FASEB J. 1998;12:1599–1609. [PubMed] [Google Scholar]
- 35.Kim S, Lee SW, Choi E-C, Choi SY. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl Microbiol Biotechnol. 2003;61:278–288. doi: 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- 36.May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci USA. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semret M, Zhai G, Mostowy S, Cleto C, Alexander D, Cangelosi G, Cousins D, Collins DM, van Soolingen D, Behr MA. Extensive genomic polymorphism within Mycobacterium avium. JBacterial. 2004;186:6332–6334. doi: 10.1128/JB.186.18.6332-6334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gulick AM, Lu X, Dunaway-Mariano D. Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry. 2004;43:8670–8679. doi: 10.1021/bi049384m. [DOI] [PubMed] [Google Scholar]
- 39.Finking R, Neumuller A, Solsbacher J, Konz D, Kretzschmar G, Schweitzer M, Krumm T, Marahiel MA. Aminoacyl adenylate substrate analogues for the inhibition of adenylation domains of nonribosomal peptide synthetases. Chembiochem. 2003;4:903–906. doi: 10.1002/cbic.200300666. [DOI] [PubMed] [Google Scholar]
- 40.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. Wiley; Hoboken, NJ: 2005. [PubMed] [Google Scholar]
- 41.May JJ, Finking R, Wiegeshoff F, Weber TT, Bandur N, Koert U, Marahiel MA. Inhibition of the D-alanine:D-alanyl carrier protein ligase from Bacillus subtilis increases the bacterium’s susceptibility to antibiotics that target the cell wall. FEBS J. 2005;272:2993–3003. doi: 10.1111/j.1742-4658.2005.04700.x. [DOI] [PubMed] [Google Scholar]
- 42.Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 43.Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006;273:409–419. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- 44.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 45.Vannada J, Bennett EM, Wilson D, Boshoff HI, Barry CEI, Aldrich CC. Design, synthesis and biological evaluation of beta-ketosulfonamide adenylation inhibitors as antitubercular agents. Org Lett. 2006;8:4707–710. doi: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vince R, Hua M. Synthesis and anti-HIV activity of carbocyclic 2′,3′-didehydro-2′,3′-dideoxy 2,6-disubstituted purine nucleosides. J Med Chem. 1990;33:17–21. doi: 10.1021/jm00163a004. [DOI] [PubMed] [Google Scholar]
- 47.Parry RJ, Bornemann V, Subramanian R. Biosynthesis of the Nucleoside Antibiotic Aristeromycin. J Am Chem Soc. 1989;111:5819–5824. [Google Scholar]
- 48.Picherit C, Wagner F, Uguen D. The sequel to a carbocyclic nucleoside synthesis: a divergent access to both arenediazonium ions and aryl triflates. Tetrahedron Lett. 2004;45:2579–2583. [Google Scholar]
- 49.Peterson EM, Brownell J, Vince R. Synthesis and biological evaluation of 5′-sulfamoylated purinyl carbocyclic nucleosides. J Med Chem. 1992;35:3991–4000. doi: 10.1021/jm00100a003. [DOI] [PubMed] [Google Scholar]
- 50.Saksena AK, Girijavallabhan VM, Ganguly AK. Process for preparing cyclopentyl purine derivatives. U.S. Patent 4, 999, 428. 1989 Apr 14;
- 51.Schaeffer HJ, Gurwana S, Vince R, Bittner S. Novel substrate of adenosine deaminase. J Med Chem. 1971;14:367–369. doi: 10.1021/jm00286a024. [DOI] [PubMed] [Google Scholar]
- 52.Rohman M, Harrison-Lavoie KJ. Separation of copurifying GroEL from glutathione-S-transferase fusion proteins. Protein Expr Purif. 2000;20:45–47. doi: 10.1006/prep.2000.1271. [DOI] [PubMed] [Google Scholar]
- 53.Copeland R. A practical introduction to structure, mechanism, and data analysis. 2. Wiley-VCH; New York, NY: 2000. Chapter 9 Tight-binding inhibitors; p. 313. [Google Scholar]
- 54.Linne U, Marahiel MA. Reactions catalyzed by mature and recombinant nonribosomal peptide synthetases. Methods Enzymol. 2004;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- 55.Pope AJ, Lapointe J, Mensah L, Benson N, Brown MJ, Moore KJ. Characterization of isoleucyl-tRNA synthetase from Staphylococcus aureus. I: Kinetic mechanism of the substrate activation reaction studied by transient and steady-state techniques. J Biol Chem. 1998;273:31680–31690. doi: 10.1074/jbc.273.48.31680. [DOI] [PubMed] [Google Scholar]
- 56.Pope AJ, Moore KJ, McVey M, Mensah L, Benson N, Osbourne N, Broom N, Brown MJ, O’Hanlon P. Characterization of isoleucyl-tRNA synthetase from Staphylococcus aureus. II. Mechanism of inhibition by reaction intermediate and pseudomonic acid analogues studied using transient and steady-state kinetics. J Biol Chem. 1998;273:31691–31701. doi: 10.1074/jbc.273.48.31691. [DOI] [PubMed] [Google Scholar]
- 57.Williams JW, Morrison JF. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 58.Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 59.Ikeda S, Chakravarty R, Ives DH. Multisubstrate analogs for deoxynucleoside kinases. J Biol Chem. 1986;261:15836–15843. [PubMed] [Google Scholar]
- 60.Fromm HJ. Reversible enzyme inhibitors as mechanistic probes. Methods Enzymology. 1995;249:123–143. doi: 10.1016/0076-6879(95)49033-7. [DOI] [PubMed] [Google Scholar]
- 61.Sun G, Voigt JH, Filippov IV, Marquez VE, Nicklaus MC. PROSIT: Pseudo-Rotational Online Service and Interactive Tool, Applied to a Conformational Survey of Nucleosides and Nucleotides. J Chem Inf Comput Sci. 2004;44:1752–1762. doi: 10.1021/ci049881+. [DOI] [PubMed] [Google Scholar]
- 62.Perry RD, Balbo PB, Jones HA, Fetherston JD, DeMoll E. Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology. 1999;145:1181–1190. doi: 10.1099/13500872-145-5-1181. [DOI] [PubMed] [Google Scholar]
- 63.Payne RJ, Kerbarh O, Miguel RN, Abell AD, Abell C. Inhibition studies on salicylate synthase. Org Biomol Chem. 2005;3:1825–1827. doi: 10.1039/b503800f. [DOI] [PubMed] [Google Scholar]
- 64.Lin H, Fischbach MA, Gatto GJJ, Liu DR, Walsh CT. Bromoenterobactins as potent inhibitors of a pathogen-associated, siderophore-modifying C-glycosyltransferase. J Am Chem Soc. 2006;128:9324–9325. doi: 10.1021/ja063236x. [DOI] [PubMed] [Google Scholar]
- 65.Callahan BP, Lomino JV, Wolfenden R. Nanomolar inhibition of the enterobactin biosynthesis enzyme EntE: synthesis, substituent effects, and additivity. Bioorg Med Chem Lett. 2006;16:3802–3805. doi: 10.1016/j.bmcl.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 66.Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 67.Braibant M, Gilot P, Content J. The ATP-binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol Rev. 2000;24:449–467. doi: 10.1111/j.1574-6976.2000.tb00550.x. [DOI] [PubMed] [Google Scholar]
- 68.Rodriguez GM, Smith I. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol. 2006;188:424–430. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niederweis M. Mycobacterial porins-new channel proteins in unique outer membranes. Mol Microbiol. 2003;49:1167–1177. doi: 10.1046/j.1365-2958.2003.03662.x. [DOI] [PubMed] [Google Scholar]
- 70.Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates. Proc Natl Acad Sci USA. 2000;97:2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 72.Challis GL, Ravel J, Townsend CA. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 73.Eppelmann K, Stachelhaus T, Marahiel MA. Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry. 2002;41:9718–9726. doi: 10.1021/bi0259406. [DOI] [PubMed] [Google Scholar]
- 74.Bucevic-Popovic V, Pavela-Vrancic M, Dieckmann R, Döhren HV. Relationship between activating and editing functions of the adenylation domain of apo-tyrocidin synthetase 1 (apo-TYl) Biochemie. 2006;88:265–270. doi: 10.1016/j.biochi.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 75.Conti E, Stachelhaus T, Marahiel MA, Brick P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ratledge C, Winder FG. The accumulation of salicylic acid by mycobacteria during growth on an iron-deficient medium. Biochem J. 1962;84:501–506. doi: 10.1042/bj0840501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of prolyl adenylate analogs. Bioorg Chem. 1996;24:273–289. [Google Scholar]
- 78.Dunn BM, Bruice TC. Steric and electronic effects on the neighboring general acid catalyzed hydrolysis of methyl phenyl acetals of formaldehyde. J Am Chem Soc. 1970;92:2410–2416. [Google Scholar]
- 79.Schwyn B, Neilands JB. Universal chemical assay for the detection and determination of siderophores. Anal Biochem. 1987;160:47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H NMR and 13C NMR spectra of 8–13, table of HPLC conditions and purities of 8–13, SDS-PAGE of purified MbtA, active-site titration plot of MbtA with 6, normalized v vs. [S] plots for the reactions of ATP and salicylic acid with MbtA, and dose-response curves of fractional MbtA activity (vi/v0) as a function of inhibitor concentrations for 9–13.