

Abstract
Migration of cells up the chemoattractant gradients is mediated by the binding of chemoattractants to G protein–coupled receptors and activation of a network of coordinated excitatory and inhibitory signals. Although the excitatory process has been well studied, the molecular nature of the inhibitory signals remains largely elusive. Here we report that the receptor for activated C kinase 1 (RACK1), a novel binding protein of heterotrimeric G protein βγ (Gβγ) subunits, acts as a negative regulator of directed cell migration. After chemoattractant-induced polarization of Jurkat and neutrophil-like differentiated HL60 (dHL60) cells, RACK1 interacts with Gβγ and is recruited to the leading edge. Down-regulation of RACK1 dramatically enhances chemotaxis of cells, whereas overexpression of RACK1 or a fragment of RACK1 that retains Gβγ-binding capacity inhibits cell migration. Further studies reveal that RACK1 does not modulate cell migration through binding to other known interacting proteins such as PKCβ and Src. Rather, RACK1 selectively inhibits Gβγ-stimulated phosphatidylinositol 3-kinase γ (PI3Kγ) and phospholipase C (PLC) β activity, due to the competitive binding of RACK1, PI3Kγ, and PLCβ to Gβγ. Taken together, these findings provide a novel mechanism of regulating cell migration, i.e., RACK1-mediated interference with Gβγ-dependent activation of key effectors critical for chemotaxis.
INTRODUCTION
Directed cell migration is critical for a variety of cellular processes, including cell movement during normal development, immune responses, and wound healing, as well as pathological processes such as tumor metastasis (Van Haastert and Devreotes, 2004; Charest and Firtel, 2006). Many chemoattractants, such as chemokines, act through G protein–coupled receptors (GPCRs) to promote directional cell migration (Rickert et al., 2000). These GPCRs typically activate the Gi/o family of G proteins, which in turn release Gβγ. Free Gβγ acts as a molecular switch to activate a myriad of signaling molecules, many of which are intimately involved in chemotaxis. For example, upon chemoattractant stimulation, free Gβγ elicits rapid translocation of PI3Kγ to the leading edge of cells, which in turn generates local accumulation of its lipid product phosphatidylinositol-3,4,5-trisphosphate (PIP3; Rickert et al., 2000). PIP3 recruits pleckstrin homology (PH) domain-containing proteins to the leading edge and activates small GTPases such as Rac and Cdc42, resulting in actin polymerization, cell polarization, and directional migration (Charest and Firtel, 2006). Free Gβγ also stimulates PLCβ2/3, the most abundant isoforms of PLC in leukocytes, which regulate chemotaxis of T lymphocytes but not neutrophils (Li et al., 2000; Bach et al., 2007).
Chemotaxis is a complicated phenomenon involving multiple processes including gradient sensing, polarization, and motility (Charest and Firtel, 2006; Iglesias and Devreotes, 2008). In addition to excitatory signals, it has been proposed that the binding of chemoattractants to receptors also activate inhibitory pathways to antagonize the excitation response (Charest and Firtel, 2006; Iglesias and Devreotes, 2008). This inhibitory process is necessary for chemoattractants to generate a spatially localized excitatory signal and to allow cells to adapt to a constant stimulation. Over the past several years, several proteins that are involved in this inhibitory process have been identified. For example, in Dictyostelium, phosphatase PTEN is activated and redistributed to the lateral and trailing edges of cells, where it dephosphorylates PIP3 to PIP2, leading to a sharp gradient of PIP3 between the front and rear of cells (Funamoto et al., 2002; Iijima and Devreotes, 2002). In neutrophils, a RhoA-dependent signal at the trailing edge of cells counterregulates the excitatory signals generated by Gβγ-dependent activation of PI3K and Rac at the leading edge of cells (Xu et al., 2003). However, to fully explain the chemotactic behaviors of cells, it has been proposed that there are other unknown inhibitory pathways (Xu et al., 2007; Iglesias and Devreotes, 2008).
Given its critical role in transmitting chemotactic signals, Gβγ may serve as a central point for regulation of cell migration. However, despite a wide array of binding partners having been discovered for Gβγ, little is known about how Gβγ signaling is regulated to ensure precise control of directional cell movement (Hamm, 1998). Recently, we have identified RACK1 as a novel binding partner of Gβγ (Chen et al., 2004b). RACK1 is a member of the WD40 repeat protein family that is predicted to adopt a β-propeller structure similar to that of Gβ. In addition to binding to activated protein kinase C (PKC), RACK1 has been shown to interact with a variety of proteins and may act as an adaptor/scaffold protein to orchestrate diverse cellular processes, ranging from signal transduction to cell growth (McCahill et al., 2002). However, our previous studies show that RACK1 binds to a unique region of Gβγ and blocks the activation of selective Gβγ effectors such as PLCβ (Chen et al., 2004a, 2005). Therefore, we reason that RACK1 may play a role in regulating Gβγ-stimulated cell migration.
In this article, we provide evidence that RACK1 negatively regulates chemotaxis. Down-regulation of RACK1 promotes directional migration of Jurkat and dHL60 cells, whereas overexpression of RACK1 stops cell migration in its track. Moreover, we unveil that RACK1 dose not function through other known binding proteins such as PKCβ, Src, integrin, and extracellular signal-regulated kinases (ERKs) to modulate cell migration. Rather, it acts by selectively inhibiting Gβγ-mediated PLCβ and PI3Kγ activation, due to its binding to a unique effector–contact interface of Gβγ. These findings provide a novel mechanism for RACK1 in regulating cell migration by acting on the Gβγ/effector interface.
MATERIALS AND METHODS
Cell Culture and Transfection
Jurkat T-cells and HL60 cells were cultured in RPMI-1640 supplemented with 10% fetal calf serum (FCS) and 25 mM HEPES. HEK293A cells (Invitrogen, Carlsbad, CA) were cultured in DMEM supplemented with 10% FCS. HL60 cells were induced to differentiate into human neutrophil-like cells by the addition of 1.3% DMSO for 7 d (Wang et al., 2002).
Transient transfection of HEK293A cells was achieved by using the Lipofectamine 2000 reagent (Invitrogen). Transfection of Jurkat T-cells (2–3 × 106) or dHL60 cells (1 × 107) 5 d after differentiation was performed by using Nucleofector Kit V and Necleofect II device (Amaxa Biosystems, Gaithersburg, MD) according to the manufacturer's protocol. Cells were harvested for assays 48–62 h after transfection. Jurkat cells stably expressing Flag-RACK1 or its deletion mutants Flag-BD1-2 and Flag-BD5-7 were generated after transfection and selection with increasing concentration of G418 (0.2–2 mg/ml) for 4–6 wk. Surviving cells were pooled and maintained in 1 mg/ml G418.
Small Interfering RNAs and DNA Constructs
Control small interfering RNA (siRNA) targeting green fluorescent protein (GFP) or luciferase and siRNA targeting human RACK1 (GNB2L1) gene sequence, 5′ aagctgaagaccaaccaca, were purchased from Dharmacon Research (Boulder, CO).
The expression vectors for RACK1 and its deletion mutants BD1-2, BD3-4, and BD5-7, corresponding to different blades at residues 1–102, 103–189, and 190–317 of rat RACK1 were generated by PCR. They are first cloned into the entry vector, pENTR/SD/d-TOPO and then into the destination vectors pcDNA3-DEST and pMAL-DEST for expression in mammalian cell and Escherichia coli, respectively, by using the Gateway cloning system (Invitrogen). These destination vectors contain DNA sequences for epitope tags GST, Flag, or MBP at the N-terminus. pMAL-DEST was generated by inserting a sequence cassette encoding the ccdB gene flanked by attR sites into pMAL-c2E (New England Biolab, Beverly, MA).
Immunoprecipitation and Western Blotting Analysis
Before stimulation with SDF1α (20 nM), Jurkat cells were serum-starved for 4–6 h. Cell lysates were prepared with phosphate-buffered saline containing 1% Igepal, 0.2% deoxycholate, and protease inhibitors. Immunoprecipitation of Gβγ or RACK1 was performed with 4 μg of rabbit anti-Gβ (T20) or mouse anti-RACK1 (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoprecipitates were resolved by SDS-PAGE and immunoblotted with mouse anti-RACK1 (BD Transduction Laboratories, Lexington, KY) or rabbit anti-PKC and anti-Src (Santa Cruz). To assay the interactions of Gβγ with GST-tagged RACK1 or its mutants, HEK293 cells were transiently transfected with plasmid cDNAs encoding HA-Gβ1 and Gγ2 as well as GST-RACK1 or its mutants. GST pulldown assays were then performed to determine the association of HA-Gβγ with GST-RACK1 and its mutants (Chen et al., 2004a).
Phosphorylation of ERKs and AKT was determined by Western blotting using specific antibodies against phosphor-p44/42 MAPK (Thr202/Tyr204; E10) and phosphor-AKT (Ser473; 587F11; Cell Signaling Technology). Immunoblots were analyzed by Odyssey imaging system (Li-Cor Biosciences, Lincoln, NE).
Cell Polarization Assay
Polarization of Jurkat T-cells were performed by using Sulfate latex beads (Interfacial Dynamics, Portland, OR no. 1–5000) coated with 100 nM SDF1α and 20 μg/ml fibronectin (Takesono et al., 2004). Coated beads (2 × 106) were first plated onto poly-d-lysine–coated Lab-tek chamber slides and then mixed with prechilled Jurkat T-cells (2 × 106) to form complexes at 4°C. After the unbound cells were washed off, polarization reactions were started by incubation in 37°C water bath for 2 min and stopped by the addition of 4% paraformaldehyde. Polarization of dHL60 cells to a uniform concentration of f-Met-Leu-Phe (fMLP) were performed essentially as described (Wang et al., 2002). Fixed and permeabilized cells were stained with mouse anti-RACK1 (1:400 dilution) and rabbit anti-Gβ (T20; 1:400 dilution) at 4°C overnight and then incubated with secondary antibodies Alexa 488–conjugated anti-rabbit and Cy5-conjugated anti-mouse and rhodamine-conjugated phalloidin (1:400 dilution) for 1 h at RT. Slides were visualized with a LSM510 Meta inverted confocal microscope (Carl Zeiss, Jena, Germany) with an argon/krypton laser and a Plan Apo 40× 1.3 NA oil immersion lens (Chen et al., 2004a). Images were acquired and processed with LSM5 Image software (Carl Zeiss) and Adobe Photoshop (San Jose, CA).
Chemotaxis Assays
Chemotaxis assays were performed in 96-well modified Boyden chamber (Neuro Probe, Cabin John, MD) as described with modifications (Chen et al., 2004a). Briefly, for the measurement of Jurkat cell migration, cells were first labeled with 5 μM Calcein-AM for 30 min at 37°C and washed with serum-free RPMI containing 0.1% BSA twice. Cell migration was determined by using 5 μm pore-size uncoated polycarbonate membrane filters and incubation for 3 h at 37°C in 5% CO2. Cells, 4–5 × 106, were loaded to the upper chamber. The migration of cells from the upper chamber to the lower chamber containing chemoattractants was quantified by measuring Calcein fluorescence in a fluorescent plate reader (Victor II, PerkinElmer, Waltham, MA), and converting to the number of cells based on a standard curve. The percentage of migration was calculated by the number of cells in the lower chamber divided by the sum of cells put in the upper chamber at the beginning of experiments.
To measure chemotaxis of siRNA-treated dHL60 cells, cells were cotransfected with GFP and 8 μm pore-size fibronectin-coated polycarbonate membrane filters were used in the assay. The percentage of GFP-positive cells migrating from the upper chamber to the lower chamber was calculated by counting the cells under fluorescence microscope.
Measurement of Inositol Phosphate Accumulation
Jurkat cells (2 × 106) were labeled with [3H]inositol (5 μCi/ml) in inositol-free DMEM containing 1% dialyzed FCS for 2 d. Inositol phosphate (IP) accumulation was determined as described (Chen et al., 2004a).
Rho Activation Assay
RhoGTP levels in Jurkat cells were assessed by using a Rhotekin Rho-binding domain (RBD) affinity precipitation assay (Ren and Schwartz, 2000). Briefly, after serum-starvation overnight, 1 × 107cells were stimulated with SDF1α (100 nM) for the indicated times. The reaction was stopped by adding 5× lysis buffer at 4°C (1× lysis buffer: 25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Igepal, 10 mM MgCl2, 1 mM EDTA, and 10% glycerol). Cell lysates were prepared and the level of RhoGTP in the lysates was determined by using Rhotekine-RBD bound to glutathione-Sepharose beads as described previously (Ren and Schwartz, 2000).
Purification of Gβ1γ2, PI3Kγ, and Other Proteins
PI3Kγ was expressed in Sf9 cells by infection with baculoviruses encoding 6×His-p110 and EE-p101 (kindly provided by Dr. Len Stephens, The Babraham Institute, Cambridge, United Kingdom) and sequentially purified to near homogeneity using Ni-NTG and anti-EE-Sepharose (Stephens et al., 1994). Gβ1γ2, MBP, MBP-RACK1, MBP-BD1-2, and MBP-BD5-7 were purified as described (Chen et al., 2004a, 2005). The C-terminal tail (the last 124 aa) of the rat homolog G protein–coupled receptor kinase 2 (GRK2-ct) was expressed as a His-tagged protein in E. coli and purified as described (Blackmer et al., 2001).
Measurement of PI3Kγ Activity
Gβγ-mediated PI3Kγ activation was determined essentially as described before (Kerchner et al., 2004). To determine the effect of RACK1 and its mutants, they were preincubated with Gβ1γ2 (100 nM) for 30 min before the addition of PI3Kγ (50 ng) and lipid vesicles.
Measurement of PKCζ Activity
Jurkat cells (1 × 107) were stimulated with SDF1α (50 nM) for 5 min and then lysed in the buffer (25 mM Tris, pH 7.5, 1% Triton, 0.5 mM EDTA, 150 mM NaCl, 50 mM NF, NaVO3, PMSF, and protease inhibitor cocktail). PKCζ was immunoprecipitated from the cell lysates with rabbit anti-PKCζ antibody (Santa Cruz Biotechnology). The activity of PKCζ was determined by using the PKC assay kit (Upstate Biotechnology, Lake Placid, NY) and PKC substrate peptide 2 (Upstate) as a substrate.
Mathematical Modeling
To describe RACK1 and GRK2-ct dependent inhibition of Gβγ-mediated PI3Kγ activation, we assumed that the regression curve follows the rule of competitive inhibition. Theoretical simulation of the reactions was performed by using MatLab (The Math Works, Natick, MA) on a Linux-based operating system (Ogdensburg, NY; McLaughlin et al., 2005).
Time-Lapse Experiments
dHL60 cells were stimulated with a point source of fMLP and imaged at RT using an inverted microscope (Zeiss Axiovert 200M) equipped with a cooled CCD camera (AxioCam MR3, Zeiss) driven by AxioVision Rel. 4.5 software (Wang et al., 2002). Images were taken with a Zeiss 40×, NA 1.30 Fluor DIC objective. Cell trajectories were tracked using Metamorph software (Molecular Devices, Sunnyvale, CA). The migration speed and chemotactic index were calculated as described previously (Wang et al., 2002).
Data Analysis
Unless indicated, data were representative of at least three independent experiments with similar results. Results are expressed as the mean ± 1 SEM from multiple experiments. Student's t tests were used to determine significant differences (two-tailed p < 0.05).
RESULTS
RACK1 Inhibits Chemotaxis of Jurkat Cells
To determine the function of RACK1 in directed cell migration, we first evaluated the effect of RACK1 inhibition on chemotaxis of Jurkat T-cells, which endogenously express the chemokine receptor CXCR4. As reported, the CXCR4 agonist SDF1α stimulated chemotaxis of Jurkat cells in a dose-dependent manner (Figure 1A; Curnock et al., 2003). Transfection of Jurkat cells with a siRNA against human RACK1 inhibited its expression by more than 80% (Figure 1B). The effect of the RACK1 siRNA is specific, as it did not affect the level of other proteins including Gβ and CXCR4 (Figure 1A, inset). Notably, down-regulation of RACK1 significantly enhanced SDF1α-stimulated cell migration (Figure 1A), but had no effect on general motility of cells, as the transwell migration of cells in the absence of SDF1α gradient is unaffected by decreased RACK1 expression (Figure 1A). These findings indicate that RACK1 negatively regulates chemotaxis but not random migration of Jurkat cells. Supporting this, a modestly increased expression of RACK1 (∼30%) in Jurkat cells diminished cell migration response to SDF1α stimulation (Figure 1B).
Figure 1.
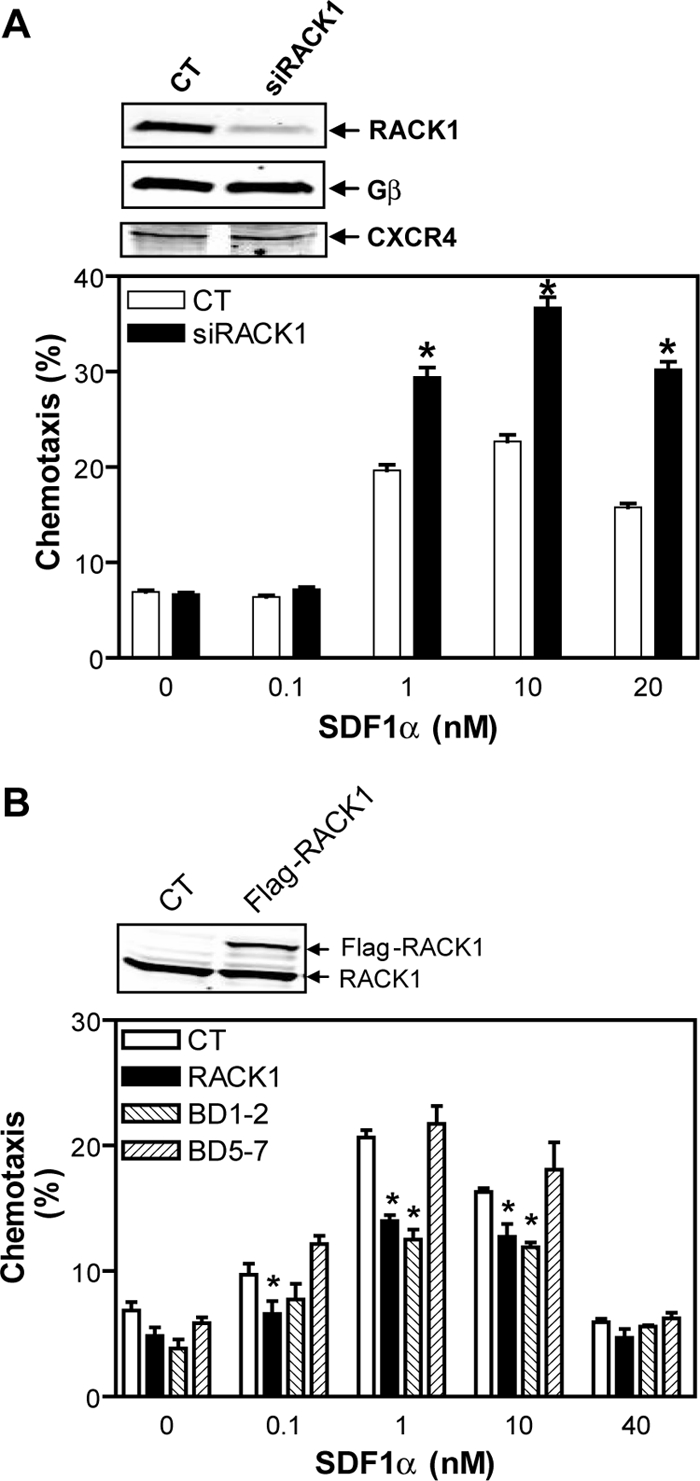
RACK1 inhibits chemotaxis of Jurkat cells. (A) Transfection of Jurkat cells with a siRNA against RACK1 (siRACK1) but not control siRNA (CT) enhances SDF1α-stimulated Jurkat cell migration. Chemotaxis was determined by the modified Boyden chamber assays. Inset, Western blots show expression of RACK1, Gβ, and CXCR4 in Jurkat cells transfected with the indicated siRNAs. (B) Jurkat cells stably expressing RACK1 and the deletion mutant BD1–2 but not BD5–7 exhibit decreased chemotactic response to SDF1α. CT, control cells transfected with vector alone. Inset, Western blot shows expression of endogenous RACK1 and Flag-RACK1 in control (CT) cells or cells stably expressing Flag-RACK1. *p < 0.05, significant difference versus control.
RACK1 Inhibits Cell Migration via Its Interaction with Gβγ
Gβγ is known to play a central role in chemotactic response of leukocytes to chemoattractants (Rickert et al., 2000). We have shown previously that RACK1 can bind Gβγ and selectively regulate its functions (Chen et al., 2004a), suggesting that RACK1 may impinge on cell migration by interaction with Gβγ. However, RACK1 is a multifunctional protein that has been shown to interact with many other proteins that are known to be involved in cell migration, such as integrin, PKC, Src, and ERKs (Schechtman and Mochly-Rosen, 2001; Chang et al., 2002; Vomastek et al., 2007). We therefore first evaluated if these RACK1-interacting proteins play a role in Jurkat cell migration. In the transwell assay, Jurkat cell migration through the filter membrane does not rely on cell adhesion to extracellular substrates via integrins because chemotaxis does not depend on whether the filter is coated with or without fibronectin or collagen (data not shown). This suggests that RACK1 is unlikely to modulate Jurkat cell migration via its interaction with integrin. Indeed, inhibition of RACK1 enhanced Jurkat cell migration equally well through the filter coated with or without fibronectin (data not shown). Studies with pharmacological inhibitors of Src kinases, PP2, a broad inhibitor of the Src family of protein tyrosine kinases, and Src inhibitor I, a selective inhibitor of Src and Lck, and an inhibitor of ERKs, PD98059, indicate that none of these proteins plays a major role in Jurkat cell migration because abrogation of their activity had little effect on chemotaxis of Jurkat cells treated with or without RACK1-siRNA (Figure 2, A–C). Similarly, inhibition of PKC with Go6976, which is known to selectively block the activity of the DAG-dependent PKCs, including the conventional and novel PKCs such as PKCβ and PKCε that are known to interact with RACK1 (Schechtman and Mochly-Rosen, 2001), did not affect chemotaxis (Figure 2A).
Figure 2.
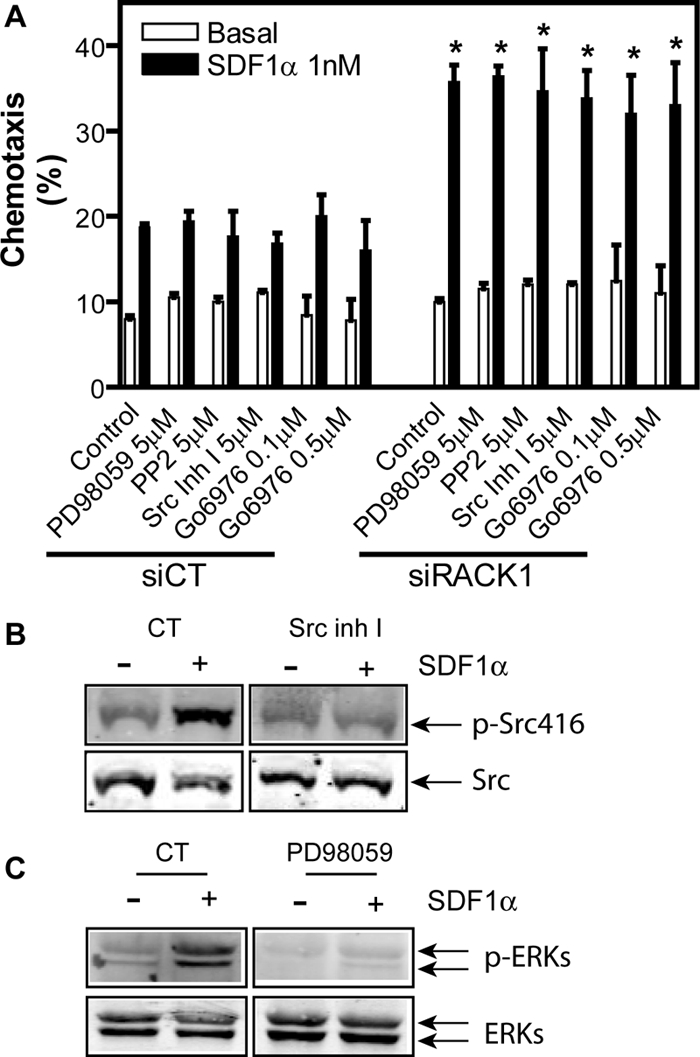
The role of Src, ERK and PKC in SDF1α-stimulated Jurkat cell migration. (A) Jurkat cells transfected with a control siRNA (siCT) or RACK1 siRNA were pretreated with or without (control) the indicated inhibitors. The transwell migration of Jurkat cells was then induced with or without (basal) 1 nM SDF1α. *p < 0.05, significance versus siCT. (B and C) Effects of the inhibitors on Src (B) and ERK (C) activation. Jurkat cells were pretreated with or without (CT) Src inhibitor I (Src Inh I) or MAPK inhibitor (PD98059) and then stimulated with SDF1α (20 nM for Src and 50 nM for ERKs) for 15 and 2 min, respectively. The activation of Src and ERKs was detected by the indicated specific antibodies against phosphorylated proteins.
To determine if RACK1 regulates Jurkat cell migration via Gβγ, we first evaluated the association of endogenous RACK1 with Gβγ in Jurkat cells by immunoprecipitation of Gβγ with an antibody that recognizes Gβ1, 2, 3, and 4 isoforms. As shown in Figure 3A, a significant amount of RACK1 was associated with Gβγ in untreated cells, and this association was significantly enhanced after SDF1α treatment for 30 min. Under the same conditions, immunoprecipitation of RACK1 did not precipitate PKCβ, Src, or the related kinase Lck (data not shown), suggesting a possibility of specific association of RACK1 with Gβγ in Jurkat cells.
Figure 3.

Interaction of RACK1 with Gβγ. (A) Association of RACK1 with Gβγ was determined after stimulation of Jurkat cells with SDF1α (20 nM) for the indicated times, and Gβγ was immunoprecipitated from the cell lysates with a control antibody (CT) or Gβ antibody that recognizes Gβ1, 2, 3, and 4 isoforms. The presence of RACK1 and Gβ in the precipitate was detected by specific antibodies. Left panel, representative Western blots; right panel, quantitative data. *p < 0.05, significant difference versus 0 min. (B) Colocalization of RACK1 and Gβγ in Jurkat cells. Cells were treated with control beads or SDF1α-conjugated beads for 2 min and then stained with specific antibodies or Alexa-568–conjugated phalloidin for F-actin. Distraction interference contrast (DIC) and fluorescent images are shown. Bar, 10 μm. The graphs on the right panel show the distribution of fluorescent intensity of Gβ, F-actin, and RACK1 along the line drawn across the cells. The images are the representatives of more than 20 cells from at least three separate experiments with similar results. (C and D) Association of RACK1 and its mutants with Gβ1γ2 (C), PKCβ or Src (D) was determined by GST pulldown assays after coexpression of HA-Gβ1γ2, PKCβ, or Src with the indicated constructs in HEK293 cells. The presence of the proteins in the GST pulldown pellets and lysates was detected with specific antibodies. A schematic representation of RACK1 structure is shown in the top panel of C.
We next assessed the cellular distribution of RACK1 and Gβγ. To induce Jurkat cell polarization, we treated cells with SDF1α-coated beads, because Jurkat cells do not polarize well in response to a uniform concentration of chemoattractants. As shown in Figure 3B, cells immobilized on control beads displayed a rounded morphology with little F-actin staining. A majority of Gβ in these cells was uniformly distributed in the plasma membrane, with a small fraction detected in the cytoplasm and nucleus. RACK1, on the other hand, was detected mostly in the cytoplasm, where it partially colocalizes with Gβγ. Stimulation of cells with SDF1α-coated beads induced cell polarization, increased F-actin accumulation and substantial relocation of Gβ at the membrane protrusion that contacts the bead (Figure 3B). Translocation of RACK1 to the polarized front end of cells was also evident in these cells and colocalization of RACK1 with Gβ was detected primarily in the membrane protrusion. Pretreatment of cells with pertussis toxin (PTx), which uncouples Gi/o classes of G proteins from their cognate receptors, inhibited cell polarization, actin accumulation and translocation of both Gβγ and RACK1 (data not shown). These findings are consistent with the previous report that free Gβγ released from activated Gi/o is primarily responsible for SDF1α-stimulated actin polymerization and cell polarization (Takesono et al., 2004).
To dissect the molecular basis of RACK1/Gβγ interaction, we determined the domains of RACK1 required for binding Gβγ. We constructed a series of RACK1 deletion mutants and expressed them as GST-conjugated proteins together with Gβγ in HEK293 cells (Figure 3C). GST pulldown assays showed that the N-terminal fragment of RACK1 consisting of the first two blades retained substantial binding to Gβγ, whereas the C-terminal fragments (BD3–4 and BD5–7) showed little binding to Gβγ, suggesting that the N-terminal domain of RACK1 is the major determinant for the interaction (Figure 3C). The Gβγ-binding sites on RACK1 are likely located on the β-propeller blades themselves and not on the short N-terminal extension, because single-blade mutants BD1 and BD2 can still interact with Gβγ (data not shown). We have shown previously that the RACK1 contact surface is also located on the β-propeller blades of Gβ (Chen et al., 2005). Because these blades are defined by the WD40 repeats, these results indicate that the RACK1/Gβγ interaction is mediated by their WD40 motifs, a possibly novel mode of protein–protein interactions. In contrast to binding to Gβγ, none of RACK1 mutants, BD1–2, BD3–4, and BD5–7, retains the ability to interact with two other known RACK1-binding proteins, PKCβ and Src, as the full-length RACK1, suggesting that unlike Gβγ, the binding of these proteins to RACK1 requires the coordinated interactions with its multiple domains (Figure 3D).
To test whether RACK1 inhibits cell migration via its interaction with Gβγ, we stably expressed Flag-BD1–2 and Flag-BD5–7 in Jurkat cells at levels comparable to the full-length RACK1. Interestingly, only cells expressing BD1–2 exhibited a diminished ability to migrate to SDF1α gradient, whereas cells expressing BD5–7 did not (Figure 1B). Because BD1–2 but not BD5–7 retains Gβγ-binding capacity, these data suggest that RACK1 regulates cell migration through its physical association with Gβγ.
RACK1 Regulates Cell Migration by Inhibiting PLC and PI3K Signaling Pathways
Gβγ regulates a variety of effectors including PLCβ, PI3Ks, and ERKs (Hamm, 1998). To test whether RACK1 regulates these pathways, we first assessed the effect of inhibiting RACK1 expression on the activity of PLCβ, PI3Ks, and ERKs. The activity of PLCβ in Jurkat cells was assessed by IP production. As shown in Figure 4A, in cells treated with a control siRNA, SDF1α stimulated little increase in IPs. However, this response was significantly enhanced by treatment of cells with a siRNA against RACK1. The increased IPs can be blocked by pretreatment of cells with either PTx or the PLC-specific inhibitor, U73122, but not by its inactive analog U73343, or the PI3K inhibitor, LY294002 (Figure 4B). Because inhibition of RACK1 did not alter the expression of CXCR4, Gβγ and PLCβ (data not shown), these findings suggest that the enhanced IP signaling is due to increased PLC activity.
Figure 4.

RACK1 regulates PLC activity. (A) Total IP production in Jurkat cells transfected with control (CT) or RACK1 siRNA (siRACK1) and stimulated with SDF1α (500 nM) for the indicated times. (B) RACK1 siRNA-transfected Jurkat cells were pretreated in the absence (CT) or presence of PTx (0.2 μg/ml) overnight, 5 μM of U73122 or U73343, or 50 μM of LY294002 for 1 h, and then stimulated with SDF1α for 10 min before the measurement of total IPs. *p < 0.05, significant difference versus basal.
To determine the effect of RACK1 on PI3K activity, we measured AKT phosphorylation. As shown in Figure 5A, RACK1 siRNA-treated cells showed significant increases in both basal and SDF1α-induced AKT phosphorylation, which was completely abolished by PTx or LY294002 (data not shown), suggesting that the activation of AKT is downstream of Gβγ and PI3K. The effect of silencing RACK1 on augmenting AKT phosphorylation is specific to GPCR-mediated signal transduction, because RACK1 depletion impaired the ability of T-cell receptor to stimulate further increase in AKT phosphorylation above the enhanced basal level (Figure 5C). Supporting the inhibitory role of RACK1 in regulating PI3K activation, overexpression of RACK1 inhibited both basal and SDF1α-stimulated AKT phosphorylation (Figure 5B). Notably, the mutant BD1–2 but not BD5–7 mimicked the inhibitory effect of the full-length RACK1 (Figure 5B and data not shown), suggesting that RACK1 likely regulates PI3K activity by binding Gβγ.
Figure 5.

RACK1 specifically regulates SDF1α-stimulated PI3K and PKCζ activities in Jurkat cells. (A–C) AKT phosphorylation at serine 473 in Jurkat cells. Cells were transiently transfected with control (CT) or RACK1 siRNA (A and C), or stably transfected with a control vector, RACK1, or BD1–2 (B) and stimulated with SDF1α (20 nM; A and B) for the indicated times or stimulated with OKT3 antibody at the indicated concentrations for 10 min (C). Representative Western blots are shown on the left panel, and quantitative analysis of data from at least three independent experiments and expressed as percentage increase over the basal in control cells is shown on the right. *p < 0.05, significant difference versus control. (D) RACK1 regulates PKCζ activity. Control (CT) or RACK1 siRNA-transfected Jurkat cells were pretreated in the absence (SDF1α and chelerythrine) or presence of LY294002 (50 μM) and stimulated with SDF1α (50 nM) for 5 min. PKCζ was then immunoprecipitated and assayed for its activity in the presence or absence of chelerythrine (10 μM). *p < 0.05, significance versus control siRNA-treated cells.
It has been shown previously that PI3Kγ simulates PKCζ to regulate human CD34+ progenitor cell migration (Petit et al., 2005). To determine if RACK1 also regulates PKCζ via PI3Kγ in Jurkat cells, we evaluated the effect of RACK1 depletion on PKCζ activity. As shown in Figure 5D, SDF1α activates PKCζ, and both the basal and SDF1α-stimulated PKCζ activities are enhanced by inhibition of RACK1. Moreover, the activity of PKCζ is sensitive to both a pan-inhibitor of PKC, chelerythrine, and PI3K inhibitor, LY 294002, suggesting that PKCζ may be regulated downstream of Gβγ-activated PI3Kγ.
In contrast to its effects on PLC, PI3K, and PKCζ activities, silencing RACK1 did not affect either basal or SDF1α-stimulated ERK activity, although the activation of ERK was completely abolished by PTx, suggesting the involvement of Gβγ (Figure 6A and data not shown). Likewise, silencing RACK1 did not affect SDF1α-induced RhoA activity, which is activated via Gα13 subunits in Jurkat cells (Figure 6B; Tan et al., 2006). Together, these findings suggest that RACK1 selectively inhibits Gβγ-mediated PLCβ and PI3K activation.
Figure 6.

RACK1 does not affect ERK and RhoA activation. (A and B) The activities of ERKs (A) and RhoA (B) in Jurkat cell transfectants stimulated with SDF1α (100 nM) for the indicated times. The level of total ERKs and RhoA, RACK1, and Gβ is also displayed.
Both PLC and PI3K signaling pathways have been shown to play a role in SDF1α-induced chemotaxis of Jurkat cells (Curnock et al., 2003; N′Diaye and Brown, 2003). Το verify that RACK1 regulates chemotaxis via these pathways, we treated cells transfected with control or RACK siRNA with specific inhibitors. As shown in Figure 7A, PTx and U73122 (but not U73343) prevented chemotaxis of both cells. The PI3K-specific inhibitors, wortmannin and LY294002, also largely abrogated cell migration (Figure 7B). Similar results were obtained with a specific PI3Kγ inhibitor, PI3Kγ inhibitor II (Camps et al., 2005), suggesting the involvement of PI3Kγ (data not shown). As expected, RACK1 siRNA-treated cells exhibited lower sensitivity to wortmannin, probably due to the enhanced PI3K activity as a result of RACK1 down-regulation (Figure 7B). Consistent with the role of PKCζ in cell migration, inhibition of its activity by chelerythrine or the palmitoylated peptide corresponding to the pseudosubstrate region of PKCζ also completely abrogated cell migration (Figure 7C). These findings indicate that RACK1-mediated inhibition of the activity of Gβγ-induced PLC, PI3K and its downstream effector, PKCζ, contributes to its abrogation of Jurkat cell migration.
Figure 7.

SDF1α-induced chemotaxis of Jurkat cells involves PLC, PI3K, and PKCζ. (A–C) Jurkat cell transfectants were pretreated with or without the indicated inhibitors. The chemotactic response of cells to buffer alone (basal) or 1 nM of SDF1α is then determined. (A) *p < 0.05, significant difference versus chemotaxis induced by SDF1α alone. (B) *p < 0.05, significant difference versus SiCT. ‡p < 0.05, significant difference versus chemotaxis induced by SDF1α alone. (C) *p < 0.05, significant difference versus chemotaxis induced by SDF1α alone. †p < 0.05, significant difference versus SiCT. Wort, wortmannin; LY, LY294002; mpPKCζ, myristoylated psudosubstrate of PKCζ.
RACK1 Inhibits Gβγ-induced PI3Kγ Activation by Steric Hindrance
We have shown previously that RACK1 inhibits Gβγ-mediated PLCβ activation by competitive binding to the region of Gβγ critical for PLCβ interaction and activation (Chen et al., 2005). In Jurkat cells, PI3Kγ is the primary PI3K responsible for SDF1α-stimulated PIP3 production and chemotaxis and is known to be directly activated by interaction with Gβγ (Curnock et al., 2003). To determine if RACK1 interferes with PI3Kγ activation via binding to Gβγ, we assessed the effect of RACK1 on Gβγ-stimulated PI3Kγ activity in vitro. As shown in Figure 8A, RACK1 itself did not stimulate PI3Kγ activity but inhibited Gβ1γ2-induced PI3Kγ activation in a dose-dependent manner as GRK2-ct, a Gβγ scavenger. However, compared with GRK-ct, RACK1 inhibition of PI3Kγ was partial. At the concentration of 20 μM, RACK1 reduced PI3Kγ activity by ∼50%, whereas at the same concentration, GRK2-ct completely abolished PI3Kγ activation (Figure 8A).
Figure 8.

RACK1 inhibits Gβ1γ2-stimulated PI3Kγ activity. (A) The activity of purified PI3Kγ stimulated by Gβ1γ2 was measured in the presence of increasing concentration of various proteins as indicated. (B) Effects of peptides on Gβ1γ2-mediated PI3Kγ activation. The activity of PI3Kγ was measured in the absence (CT) or presence of various Gβ1-derived peptides (0.5 mM). *p < 0.05 and **p < 0.01, significant difference versus the activity of PI3Kγ in the absence of peptides. (C and D) Structural determinants of Gβ1 for interactions with PI3Kγ and RACK1 (C) and GRK2 (D). Based on the crystal coordinates of Gβ1γ1 (Sondek et al., 1996), the molecular surface of Gβ1γ1 was generated using the UCSF chimera package (Pettersen et al., 2004). The surface that interacts with PI3Kγ was generated based on the data from B, whereas those for RACK1 and GRK2 were from previous reports (Lodowski et al., 2003; Chen et al., 2005).
To determine if the lower potency of RACK1 in inhibiting PI3Kγ reflects the relatively lower binding affinity of Gβγ to RACK1 (EC50 ∼ 520 nM) than PI3Kγ (EC50 ∼ 5.6 nM) and GRK2-ct (EC50 ∼ 100 nM; Pumiglia et al., 1995; Kerchner et al., 2004; Chen et al., 2005), we have generated a mathematical model to describe the effect of RACK1 and GRK2-ct on PI3Kγ activation (Supplementary Figure S1, A–C). We assumed that both RACK1 and GRK2-ct competitively inhibit Gβγ-dependent PI3Kγ activity. Initial simulations predicted regression curves for both GRK2ct- and RACK1-mediated inhibition of PI3Kγ qualitatively matching those observed experimentally but slightly left-shifted (Supplementary Figure S2A), suggesting that the empirically determined potencies of RACK1 and GRK2-ct in inhibiting PI3Kγ may be slightly lower than the theoretical predictions. However, an almost perfect fit between the simulations and empirical data for both GRK2-ct and RACK1 can be achieved when either the affinities of Gβ1γ2 for RACK1 and GRK2-ct are set to be twofold lower (200 and 1040 nM, respectively), or the affinity of Gβ1γ2 for PI3Kγ is set to be twofold higher (2.8 nM; Supplementary Figure S2, B and C). Because these values are well within the range of variations reported for each parameter in the literature (Pumiglia et al., 1995; Maier et al., 1999; Maier et al., 2000; Kerchner et al., 2004; Chen et al., 2005), this indicates that the experimental data we obtained for both GRK2-ct and RACK1 are comparable to the theoretical predictions. Taken together, these results indicate that like GRK2-ct, RACK1 regulates PI3Kγ activity by competitive binding to Gβγ. Interestingly, the inhibitory effect of RACK1 on PI3Kγ can be mimicked by the mutant BD1–2 but not BD5–7 or MBP alone, further underscoring the notion that RACK1 regulates PI3Kγ activity through its direct interaction with Gβγ (Figure 8A).
RACK1 was shown previously to bind to a noncanonical region of Gβγ involved in activation of select effectors (Chen et al., 2005). To determine if this region is also involved in PI3Kγ stimulation, we further deciphered the domains on Gβγ that are critical for PI3Kγ activation. We evaluated effects of a series of peptides derived from the β-propeller regions of Gβ1 on the activity of PI3Kγ (Chen et al., 2005). To ensure maximal effects, we used a saturating concentration (0.5 mM) of the peptides. As shown in Figure 8B, of 15 peptides tested, only peptides 86–105, 113–122, 113–135, 136–147, 159–167, 201–209, and 309–316 inhibited 15–40% of Gβγ-stimulated PI3Kγ activity. These findings suggest that activation of PI3Kγ involves multiple domains of Gβ, which contribute differentially to PI3Kγ activation. When mapped to the crystal structure of Gβ1γ1, the corresponding residues of the six inhibitory peptides occupy an extended surface of Gβ1. Two inhibitory peptides, p86–105 and 309–316, fall within the RACK1 contact surface, whereas five inhibitory peptides, p86–105, p113–122, p136–147, p201–209, p309–316, and p328–337, overlap with the GRK2 binding sites in Gβ (as determined from the cocrystal structure of the GRK2/Gβ1γ2 complex (Lodowski et al., 2003); Figure 8, C and D). The fact that RACK1 and GRK2 binding sites on Gβγ overlap with the PI3Kγ contact surface indicates that RACK1 and GRK2 inhibit PI3Kγ by preventing its access to the region of Gβγ critical for its activation.
RACK1 Regulates Chemotaxis of Neutrophil-like dHL60 Cells
Having established that RACK1 inhibits chemotaxis of Jurkat cells, a transformed T-cell line, we sought to explore whether RACK1 also plays a role in migratory response of dHL60 cells, a human neutrophil-like cell line. Like Jurkat cells, dHL60 cells also express RACK1 but its level is significantly lower than that in Jurkat cells, although the level of total Gβ is comparable between these two cell lines (Figure 9A). As seen in Jurkat cells, RACK1 was located mostly in the cytoplasm, whereas Gβ was detected mainly in the plasma membrane of unstimulated cells (Figure 9B). However, in cells that polarized in response to a uniformly increased concentration of chemoattractant, fMLP, both Gβ and RACK1 were distributed in an anterior-to-posterior gradient. As in Jurkat cells, Gβ and RACK1 were mainly colocalized to the leading edge of cells (Figure 9B). Interestingly, a substantial amount of Gβ also accumulated in the cytoplasm, a phenomenon that has been noticed previously in other cell types (Kino et al., 2005; Saini et al., 2007). Moreover, the anterior-to-posterior gradient of Gβ was shallower than that of RACK1. This shallow distribution of Gβ is consistent with that was reported in polarized Dictyostelium discoideum amoebas (Jin et al., 2000). We showed previously that free Gβγ can promote the translocation of RACK1 from the cytoplasm to the plasma membrane (Chen et al., 2004a). The fact that the gradient of RACK1 in polarized dHL60 cells exceeds that of Gβγ suggests that in addition to Gβγ-induced translocation, other mechanisms must amplify and maintain the gradient of RACK1 in these cells.
Figure 9.

Expression, cellular distribution and functions of RACK1 in dHL60 cells. (A) Immunoblot showing expression of RACK1 and Gβ in Jurkat and dHL60 cells. An equal number of cells (2 × 106) was loaded. (B) Cellular distribution of RACK1 and Gβ in dHL60 cells treated with or without (control) a uniform concentration of fMLP (0.5 μM) for 3 min. Cells were stained with specific antibodies against Gβ and RACK1. F-actin was labeled with Alexa-568–conjugated phalloidin. DIC and fluorescent images are shown. Bar, 10 μm. The graphs on the right show the distribution of fluorescent intensity of Gβ, F-actin, and RACK1 along the line drawn across the cells. The images are the representatives of more than 20 cells from at least three separate experiments with similar results. (C) Down-regulation of RACK1 enhances chemotactic response of dHL60 cells. dHL60 cells were transfected with GFP, together with a control siRNA (CT) or a siRNA against RACK1. Chemotaxis of GFP-positive cells was determined by the modified Boyden chamber assays. *p < 0.05, significance versus control.
To determine if RACK1 plays a role in regulating dHL60 migration, we first examined the effect of inhibiting RACK1 expression on the transwell migration of dHL60 cells. To our surprise, despite the level of RACK1 being significantly lower in dHL60 cells than in Jurkat cells, dHL60 cells transfected with RACK1 siRNA still showed a significant increase in chemotaxis toward fMLP (Figure 9C). These findings indicate that as in Jurkat cells, the local accumulation of RACK1 at the leading edge is sufficient to exert a negative regulation on the chemotaxis of dHL60 cells.
To further understand the role of RACK1 in the dynamic process of cell migration, we cotransfected dHL60 cells with RACK1 or RACK1 siRNA and PH-AKT-GFP, a bioprobe for PIP3, by using an optimized procedure that yields more than 80% cotransfection efficiency (Srinivasan et al., 2003). In addition to being a transfection marker, PH-AKT-GFP also serves as a probe for examining the effect of altering RACK1 expression on the spatiotemporal kinetics of PI3K activation. As reported, PH-AKT-GFP was distributed mostly in the cytoplasm of unstimulated dHL60 cells (Figure 10A; Servant et al., 2000). In responding to a gradient of fMLP delivered by a micropipette, control dHL60 cells rapidly developed a pseudopod at the part of cells that received the strongest stimulation. Moreover, fMLP stimulated translocation of PH-AKT-GFP from the cytoplasm to the pseudopod at the leading edge, resulting in a steep anterior to posterior gradient, which was stable over the entire course of cell migration (Figure 10A and Supplementary Video 1). These cells migrated linearly toward the source of stimuli with a speed of ∼5 μm/min and a chemotaxis index of 0.8, which reflects an almost straight line migration of cells from the starting point to the fMLP-containing micropipette (Figure 10A). Cotransfection of dHL60 cells with a siRNA against RACK1 did not cause significant changes either in their migration speed, directionality or the spatial and temporal kinetics of PH-AKT-GFP translocation, when exposed to a point source of fMLP (Figure 10, B and F, and Supplementary Video 2). The failure to modulate dHL60 cell migration by RACK1 depletion may be due to the fact that the micropipette system delivers a much steeper chemoattractant gradient than the modified Boyden chamber used in the transwell assays (Servant et al., 2000), such that the change in dHL60 cell migration due to the down-regulation of RACK1 could not be detected.
Figure 10.

RACK1 regulates migration of dHL60 cells toward a point source of fMLP. (A–E) Time-lapse images of dHL60 cell migration toward a point source of fMLP. dHL60 cells were transfected with PH-AKT-GFP together with a control (A), RACK1 siRNA (B), RACK1 (C), BD1–2 (D), or BD5–7 (E). The migration of cells imaged by Nomarski microscopy (top panel) and the spatial localization of PH-AKT-GFP (bottom panel) at the indicated times before and after fMLP stimulation are shown. Time-lapse videos of these cells are shown in the Supplemental Material. Bar, 10 μm. (F) Outlines of dHL60 cells responding to stimulation of fMLP (10 μM) delivered by a micropipette. Each set of outlines represents the migration path of a single cell from each transfection condition at the indicated time intervals after exposure to fMLP. Asterisks (*) indicate the location of stimuli. The chemotactic index and migration speed of the corresponding transfected cells calculated from multiple experiments are also shown. For cells expressing RACK1 and BD1–2, the chemotactic index and migration speed were calculated after the beginning of directional migration. Data are expressed as the mean ± SEM.
Overexpression of RACK1 did not cause significant changes in cell morphology and the cytoplasmic localization of PH-AKT-GFP in unstimulated cells. However, a majority of RACK1-overexpressing cells showed a significant delay in generating a pseudopod and asymmetric accumulation of PH-AKT-GFP. After exposure to a gradient of fMLP, these cells were still able to translocate PH-AKT-GFP to the plasma membrane but they failed to accumulate PH-AKT-GFP asymmetrically at the face of cells exposed to the strongest stimulation for the first several minutes (Figure 10C and Supplementary Video 3-1). During this period, RACK1-overexpressing cells did not generate a persistent pseudopod toward the source of stimuli as seen in the control cells, but rather generated lamellae that extended in multiple directions. Consequently, these cells did not demonstrate directional migration during the first several minutes of stimulation. However, once these cells began to migrate toward fMLP, they behaved similarly as the control cells in terms of PH-AKT-GFP accumulation, pseudopod formation, migration speed, and chemotactic index (Figure 10, C and F, and Supplementary Video 3-1). These results suggest that in order to promote pseudopod formation and directed cell migration, the accumulation of PIP3 needs to reach a threshold, which may be postponed by RACK1 overexpression because of its inhibition of local PI3K activation and PIP3 production. These findings are consistent with the fact that RACK1 only partially inhibits Gβγ-mediated PI3Kγ activation (Figure 8A). However, we did observe that some cells expressing RACK1 completely lost the ability to migrate toward the chemoattractant (Supplementary Video 3-2). This may reflect higher levels of RACK1 expression in these cells, resulting in a more severe inhibition of PI3K activation and PIP3 generation, thereby disrupting the positive feedback exerted by PIP3 on PI3K activation, which has been shown to be required for the formation of persistent pseudopod and directed cell migration (Wang et al., 2002; Weiner et al., 2002).
Interestingly, cells expressing BD1–2 showed chemotactic defects similar to those of RACK1-overexpressing cells, whereas cells expressing BD5–7 retained the wild-type-like chemotactic responses (Figure 10, D–F, and Supplementary Videos 4 and 5). These findings suggest that as in Jurkat cells, RACK1 regulates chemotaxis of dHL60 cells by its interaction with Gβγ, thereby competing with PI3Kγ binding and inhibiting its activation.
DISCUSSION
In the present study, we provided evidence that RACK1 regulates directed cell migration, and it does so through a novel mechanism that works on the Gβγ and effector interface. Our data indicate that RACK1 acts as a negative regulator of chemotaxis. Thus, silencing RACK1 enhances SDF1α- and fMLP-stimulated chemotaxis of Jurkat and dHL60 cells, whereas overexpressing RACK1 abrogates cell migration. The fact that RACK1 inhibits chemotaxis of both Jurkat and dHL60 cells induced via two different GPCRs suggests that RACK1 may have a general role in regulating GPCR-mediated leukocyte migration.
Several lines of evidence indicate that RACK1 regulates GPCR-directed cell migration by acting on Gβγ to intervene selectively in the activation of its effectors PLC and PI3K. First, the activity of PLC and PI3K can be significantly augmented by down-regulation of RACK1, whereas overexpression of RACK1 has an opposite effect. Second, inhibiting RACK1 neither affects Gα13-mediated RhoA activation, nor has an effect on Gβγ-mediated ERK activation, indicating that RACK1 selectively regulates Gβγ effectors. Third, the inhibitory effect of RACK1 on effector activation and chemotaxis can only be mimicked by its mutant BD1-2, which retains Gβγ-binding capacity but does not bind other RACK1-interacting proteins such as Src and PKCβ that are known to be involved in cell migration, suggesting that RACK1 exerts its function by physical association with Gβγ. Supporting this, endogenous RACK1 was found to form a complex with Gβγ, and these proteins are colocalized at the leading edge of both polarized Jurkat and dHL60 cells. Finally, our in vitro data provided direct evidence that RACK1 inhibits Gβγ-stimulated PLCβ and PI3Kγ activity by binding to Gβγ. We showed previously that RACK1 inhibits PLCβ activation by competing for its binding to the signal transfer region of Gβγ (Chen et al., 2005). In the present studies, we demonstrate that RACK1 uses a similar mechanism to regulate Gβγ-stimulate PI3Kγ activation. Thus, as with PLCβ2, RACK1 and its mutant BD1–2 inhibit PI3Kγ in a dose-dependent manner. Compared with GRK2ct, RACK1 displays a lower potency in inhibiting PI3Kγ (IC50 ∼ 20 μM vs. 4 μM for GRK2). Mathematical simulation indicates that the lower potency of RACK1 in inhibiting PI3Kγ reflects the relatively lower binding affinity of Gβγ to RACK1 (EC50 ∼ 500 nM) than GRK2ct (EC50 ∼ 100 nM; data not shown; Pumiglia et al., 1995; Chen et al., 2005). It is worth noting that we do not know the exact binding affinity of RACK1 to Gβγ in intact cells because RACK1 may undergo posttranslational modification, which could significantly alter its binding ability. However, the relatively lower potency of RACK1 in inhibiting Gβγ signaling may facilitate its ability to regulate Gβγ effectors in a spatiotemporal-specific manner because the inhibitory effect of RACK1 will critically depend on its relative local concentration in cells to the effectors.
Using a peptide-based approach, we also identified residues on Gβγ that are critical for PI3Kγ activation. Although in this assay the concentration of these peptides required to inhibit PI3K is relatively high and the effect of inhibition (15–40%) is modest, we believe that this reflects the relative contribution of the corresponding residues of the peptides to Gβγ-mediated PI3K activation rather than nonspecific effects. First, the inhibitory effect of the peptides is effector-specific. For example, in our previous studies (Chen et al., 2005), the peptide p44–54 inhibits the binding of Gβγ to PLCβ2 by 60% and directly activates PLCβ2, but it does not affect either the basal or Gβγ-stimulated PI3K activity in the current studies. Similarly, although the peptide p177–189 caused more than 70% inhibition of Gβγ-dependent PLCβ2 activity (Chen et al., 2005), it does not affect PI3K activation. Conversely, the peptide 309–316, which inhibited PI3K activation by 20%, had no effects on PLCβ2 activation (Chen et al., 2005). Second, some of the Gβγ effector-binding sites identified by the peptide-based approaches have been corroborated by site-directed mutagenesis studies. For example, by substituting the charged amino acid within residues 44–54 of Gβ with alanines, we confirmed the results from the peptide-based studies that these residues play a critical role in PLCβ2 activation (Chen et al., 2005). Although we have not yet evaluated the contribution of different amino acids of Gβ to PI3K activation, previous studies from Garrison's group demonstrated that mutation of residues H311, R314, and V315 impaired the ability of Gβγ to activate PI3Kγ (Kerchner et al., 2004). These residues are contained in the peptide 309–316, which inhibited ∼20% of Gβγ-dependent PI3K activity in our studies. Though our data remain to be corroborated by a high-resolution crystal structure of the Gβγ/PI3Kγ complex, our findings that some of Gβ residues (residues 86–105 and 309–316) critical for PI3Kγ activation are contained in the RACK1 contact surface on Gβγ is consistent with the notion that RACK1 regulates cell migration by physical association with and steric hindrance of the access of specific effectors to Gβγ.
Our findings that RACK1 regulates chemotaxis via PLCβ and PI3Kγ are consistent with the roles of these two enzymes in leukocyte migration, as demonstrated by numerous studies from gene knockout mice of PLCβ2/3 and PI3Kγ and with pharmacological inhibitors (Rickert et al., 2000; Hawkins et al., 2006). However, the relative contribution of these two pathways in RACK1-mediated regulation of chemotaxis remains to be elucidated.
Like Gβ, RACK1 has numerous interacting proteins and is generally considered as a scaffold/adaptor protein involved in diverse of cellular processes. It has also been shown to be involved in migration of epithelia cells, partially through its interaction with PKCβ or PKCε, Src, and integrins (Buensuceso et al., 2001; McCahill et al., 2002; Buensuceso et al., 2005; Kiely et al., 2005; Doan and Huttenlocher, 2007). However, conflicting reports of RACK1 in either promoting or inhibiting cell migration are found in literature, probably reflecting the context-dependent functions of RACK1 in cells, tissues, and stimuli (Buensuceso et al., 2001; Doan and Huttenlocher, 2007). In our studies, we did not detect interaction of RACK1 with either PKCβ or Src in Jurkat cells. Moreover, in the transwell assay, Jurkat cell migration through the filter membrane does not rely on cell adhesion to extracellular substrates via integrins. These data suggest that RACK1 is unlikely to modulate leukocyte migration through its effect on the activity of PKCβ, Src, or integrins. Supporting this, the RACK1 mutant BD1–2, which does not bind PKCβ and Src, can still mimic the inhibitory effect of the full-length RACK1. Moreover, studies with pharmacological inhibitors of Src, PKCβ/ε, and ERKs indicate that they are not involved in SDF1α-stimulated Jurkat cell migration.
Our data unambiguously demonstrate that RACK1 plays a negative role in regulating leukocyte migration. Moreover, they provide further support for our previous notion that RACK1 can tune Gβγ activation of effectors to impact specific functions of Gβγ (Chen et al., 2004a). This mechanism of modulating cell migration is different from that used by other known regulators. For example, the phosphatases PTEN and SHIP impinge on directional sensing and motility of cells by dephosphorylating PIP3 (Franca-Koh et al., 2007; Nishio et al., 2007). Moreover, they are essential for the establishment of internal gradient of signaling molecules and directional sensing of Dictyostelium and neutrophils (Funamoto et al., 2002; Iijima and Devreotes, 2002; Li et al., 2005; Nishio et al., 2007). In contrast, RACK1 is not essential for gradient sensing as inhibition of RACK1 neither affects the translocation of PH-AKT-GFP to the leading edge nor the directional migration of dHL60 cells. Unlike RACK1, other regulatory proteins such as arrestins and GRKs negatively modulate chemotactic responses of leukocytes by down-regulating functions of chemokine receptors through phosphorylation and internalization of GPCRs (Vroon et al., 2006). Regulator of G protein signaling (RGS) proteins inhibit chemotaxis by shortening the lifetime of the active Gα-GTP subunit (Kehrl, 2006). Given the fact that RACK1 modulates cell migration via competitive inhibition of Gβγ effector activation, it is conceivable that dynamic changes in the expression level of RACK1 during physiological and pathological processes of immune responses may contribute to fine tuning of leukocyte function. Additionally, because RACK1 is ubiquitously expressed, and chemokine receptors are involved in the migration of diverse cell types, including fibroblasts and endothelial and tumor cells, RACK1 may also function in other cellular processes, such as wound healing, angiogenesis, and tumor metastasis (Gillitzer and Goebeler, 2001; Balkwill, 2004; Strieter et al., 2005).
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Anita Preininger, Corey Fowler, and Christopher Wells for critical reading of the manuscript. This work was supported in part by National Institutes of Health Grant EY010291 (H.E.H.).
Abbreviations used:
- dHL60
differentiated HL60
- ERK
extracellular signal-regulated kinase
- fMLP
f-Met-Leu-Phe
- GPCR
G protein–coupled receptor
- Gβγ
G protein βγ
- GRK2-ct
the C-terminal tail of G protein–coupled receptor kinase 2
- IP
inositol phosphate
- PH
pleckstrin homology
- PI3K
phosphatidylinositol 3-kinase
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PKC
protein kinase C
- PLC
phospholipase C
- PTx
pertussis toxin
- RACK1
receptor for activated C kinase 1.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-04-0433) on July 2, 2008.
REFERENCES
- Bach T. L., et al. Phospholipase cbeta is critical for T cell chemotaxis. J. Immunol. 2007;179:2223–2227. doi: 10.4049/jimmunol.179.4.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat. Rev. Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Blackmer T., Larsen E. C., Takahashi M., Martin T. F., Alford S., Hamm H. E. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- Buensuceso C. S., Obergfell A., Soriani A., Eto K., Kiosses W. B., Arias-Salgado E. G., Kawakami T., Shattil S. J. Regulation of outside-in signaling in platelets by integrin-associated protein kinase C beta. J. Biol. Chem. 2005;280:644–653. doi: 10.1074/jbc.M410229200. [DOI] [PubMed] [Google Scholar]
- Buensuceso C. S., Woodside D., Huff J. L., Plopper G. E., O'Toole T. E. The WD protein Rack1 mediates protein kinase C and integrin-dependent cell migration. J. Cell Sci. 2001;114:1691–1698. doi: 10.1242/jcs.114.9.1691. [DOI] [PubMed] [Google Scholar]
- Camps M., et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Chang B. Y., Harte R. A., Cartwright C. A. RACK1: a novel substrate for the Src protein-tyrosine kinase. Oncogene. 2002;21:7619–7629. doi: 10.1038/sj.onc.1206002. [DOI] [PubMed] [Google Scholar]
- Charest P. G., Firtel R. A. Feedback signaling controls leading-edge formation during chemotaxis. Curr. Opin. Genet. Dev. 2006;16:339–347. doi: 10.1016/j.gde.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Chen S., Dell E. J., Lin F., Sai J., Hamm H. E. RACK1 regulates specific functions of Gbetagamma. J. Biol. Chem. 2004a;279:17861–17868. doi: 10.1074/jbc.M313727200. [DOI] [PubMed] [Google Scholar]
- Chen S., Lin F., Hamm H. E. RACK1 binds to a signal transfer region of G betagamma and inhibits phospholipase C beta2 activation. J. Biol. Chem. 2005;280:33445–33452. doi: 10.1074/jbc.M505422200. [DOI] [PubMed] [Google Scholar]
- Chen S., Spiegelberg B. D., Lin F., Dell E. J., Hamm H. E. Interaction of Gbetagamma with RACK1 and other WD40 repeat proteins. J. Mol. Cell Cardiol. 2004b;37:399–406. doi: 10.1016/j.yjmcc.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Curnock A. P., Sotsios Y., Wright K. L., Ward S. G. Optimal chemotactic responses of leukemic T cells to stromal cell-derived factor-1 requires the activation of both class IA and IB phosphoinositide 3-kinases. J. Immunol. 2003;170:4021–4030. doi: 10.4049/jimmunol.170.8.4021. [DOI] [PubMed] [Google Scholar]
- Doan A. T., Huttenlocher A. RACK1 regulates Src activity and modulates paxillin dynamics during cell migration. Exp. Cell Res. 2007;313:2667–2679. doi: 10.1016/j.yexcr.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franca-Koh J., Kamimura Y., Devreotes P. N. Leading-edge research: PtdIns(3,4,5)P3 and directed migration. Nat. Cell Biol. 2007;9:15–17. doi: 10.1038/ncb0107-15. [DOI] [PubMed] [Google Scholar]
- Funamoto S., Meili R., Lee S., Parry L., Firtel R. A. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002;109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- Gillitzer R., Goebeler M. Chemokines in cutaneous wound healing. J. Leukoc. Biol. 2001;69:513–521. [PubMed] [Google Scholar]
- Hamm H. E. The many faces of G protein Signaling. J. Biol. Chem. 1998;273:669–672. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- Hawkins P. T., Anderson K. E., Davidson K., Stephens L. R. Signalling through Class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Iglesias P. A., Devreotes P. N. Navigating through models of chemotaxis. Curr. Opin. Cell Biol. 2008;20:35–40. doi: 10.1016/j.ceb.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Iijima M., Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- Jin T., Zhang N., Long Y., Parent C. A., Devreotes P. N. Localization of the G protein betagamma complex in living cells during chemotaxis. Science. 2000;287:1034–1036. doi: 10.1126/science.287.5455.1034. [DOI] [PubMed] [Google Scholar]
- Kehrl J. H. Chemoattractant receptor signaling and the control of lymphocyte migration. Immunol. Res. 2006;34:211–227. doi: 10.1385/IR:34:3:211. [DOI] [PubMed] [Google Scholar]
- Kerchner K. R., Clay R. L., McCleery G., Watson N., McIntire W. E., Myung C. S., Garrison J. C. Differential sensitivity of phosphatidylinositol 3-kinase p110gamma to isoforms of G protein betagamma dimers. J. Biol. Chem. 2004;279:44554–44562. doi: 10.1074/jbc.M406071200. [DOI] [PubMed] [Google Scholar]
- Kiely P. A., Leahy M., O'Gorman D., O'Connor R. RACK1-mediated integration of adhesion and insulin-like growth factor I (IGF-I) signaling and cell migration are defective in cells expressing an IGF-I receptor mutated at tyrosines 1250 and 1251. J. Biol. Chem. 2005;280:7624–7633. doi: 10.1074/jbc.M412889200. [DOI] [PubMed] [Google Scholar]
- Kino T., Tiulpakov A., Ichijo T., Chheng L., Kozasa T., Chrousos G. P. G protein beta interacts with the glucocorticoid receptor and suppresses its transcriptional activity in the nucleus. J. Cell Biol. 2005;169:885–896. doi: 10.1083/jcb.200409150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., et al. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- Li Z., Jiang H., Xie W., Zhang Z., Smrcka A. V., Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Lodowski D. T., Pitcher J. A., Capel W. D., Lefkowitz R. J., Tesmer J. J. Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gbetagamma. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- Maier U., Babich A., Macrez N., Leopoldt D., Gierschik P., Illenberger D., Nurnberg B. Gbeta 5gamma 2 is a highly selective activator of phospholipid-dependent enzymes. J. Biol. Chem. 2000;275:13746–13754. doi: 10.1074/jbc.275.18.13746. [DOI] [PubMed] [Google Scholar]
- Maier U., Babich A., Nurnberg B. Roles of non-catalytic subunits in gbetagamma-induced activation of class I phosphoinositide 3-kinase isoforms beta and gamma. J. Biol. Chem. 1999;274:29311–29317. doi: 10.1074/jbc.274.41.29311. [DOI] [PubMed] [Google Scholar]
- McCahill A., Warwicker J., Bolger G. B., Houslay M. D., Yarwood S. J. The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol. Pharmacol. 2002;62:1261–1273. doi: 10.1124/mol.62.6.1261. [DOI] [PubMed] [Google Scholar]
- McLaughlin J. N., Shen L., Holinstat M., Brooks J. D., Dibenedetto E., Hamm H. E. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J. Biol. Chem. 2005;280:25048–25059. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- N′Diaye E. N., Brown E. J. The ubiquitin-related protein PLIC-1 regulates heterotrimeric G protein function through association with Gbetagamma. J. Cell Biol. 2003;163:1157–1165. doi: 10.1083/jcb.200307155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M., et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat. Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- Petit I., Goichberg P., Spiegel A., Peled A., Brodie C., Seger R., Nagler A., Alon R., Lapidot T. Atypical PKC-zeta regulates SDF-1-mediated migration and development of human CD34+ progenitor cells. J. Clin. Invest. 2005;115:168–176. doi: 10.1172/JCI21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pumiglia K. M., LeVine H., Haske T., Habib T., Jove R., Decker S. J. A direct interaction between G-protein beta gamma subunits and the Raf-1 protein kinase. J. Biol. Chem. 1995;270:14251–14254. doi: 10.1074/jbc.270.24.14251. [DOI] [PubMed] [Google Scholar]
- Ren X. D., Schwartz M. A. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- Rickert P., Weiner O. D., Wang F., Bourne H. R., Servant G. Leukocytes navigate by compass: roles of PI3Kgamma and its lipid products. Trends Cell Biol. 2000;10:466–473. doi: 10.1016/s0962-8924(00)01841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini D. K., Kalyanaraman V., Chisari M., Gautam N. A family of G protein betagamma subunits translocate reversibly from the plasma membrane to endomembranes on receptor activation. J. Biol. Chem. 2007;282:24099–24108. doi: 10.1074/jbc.M701191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechtman D., Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene. 2001;20:6339–6347. doi: 10.1038/sj.onc.1204778. [DOI] [PubMed] [Google Scholar]
- Servant G., Weiner O. D., Herzmark P., Balla T., Sedat J. W., Bourne H. R. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondek J., Bohm A., Lambright D. G., Hamm H. E., Sigler P. B. Crystal structure of a G-protein beta gamma dimer at 2.1A resolution [see comments] [corrected] [published erratum appears in Nature 1996 Feb 29; 379(6568), 847] Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- Srinivasan S., Wang F., Glavas S., Ott A., Hofmann F., Aktories K., Kalman D., Bourne H. R. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L., Smrcka A., Cooke F. T., Jackson T. R., Sternweis P. C., Hawkins P. T. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- Strieter R. M., Burdick M. D., Gomperts B. N., Belperio J. A., Keane M. P. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005;16:593–609. doi: 10.1016/j.cytogfr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Takesono A., Horai R., Mandai M., Dombroski D., Schwartzberg P. L. Requirement for Tec kinases in chemokine-induced migration and activation of Cdc42 and Rac. Curr. Biol. 2004;14:917–922. doi: 10.1016/j.cub.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Tan W., Martin D., Gutkind J. S. The Galpha13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J. Biol. Chem. 2006;281:39542–39549. doi: 10.1074/jbc.M609062200. [DOI] [PubMed] [Google Scholar]
- Van Haastert P. J., Devreotes P. N. Chemotaxis: signalling the way forward. Nat. Rev. Mol. Cell Biol. 2004;5:626–634. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- Vomastek T., Iwanicki M. P., Schaeffer H. J., Tarcsafalvi A., Parsons J. T., Weber M. J. RACK1 targets the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol. Cell. Biol. 2007;27:8296–8305. doi: 10.1128/MCB.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroon A., Heijnen C. J., Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J. Leukoc. Biol. 2006;80:1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- Wang F., Herzmark P., Weiner O. D., Srinivasan S., Servant G., Bourne H. R. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- Weiner O. D., Neilsen P. O., Prestwich G. D., Kirschner M. W., Cantley L. C., Bourne H. R. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Wang F., Van Keymeulen A., Herzmark P., Straight A., Kelly K., Takuwa Y., Sugimoto N., Mitchison T., Bourne H. R. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114:201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- Xu X., Meier-Schellersheim M., Yan J., Jin T. Locally controlled inhibitory mechanisms are involved in eukaryotic GPCR-mediated chemosensing. J. Cell Biol. 2007;178:141–153. doi: 10.1083/jcb.200611096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.