

Abstract
Werner syndrome (WS) is a human genetic disorder characterized by extensive clinical features of premature aging. Ataxia-telengiectasia (A-T) is a multisystem human genomic instability syndrome that includes premature aging in some of the patients. WRN and ATM, the proteins defective in WS and A-T, respectively, play significant roles in the maintenance of genomic stability and are involved in several DNA metabolic pathways. A role for WRN in DNA repair has been proposed; however, this study provides evidence that WRN is also involved in ATM pathway activation and in a S-phase checkpoint in cells exposed to DNA interstrand cross-link–induced double-strand breaks. Depletion of WRN in such cells by RNA interference results in an intra-S checkpoint defect, and interferes with activation of ATM as well as downstream phosphorylation of ATM target proteins. Treatment of cells under replication stress with the ATM kinase inhibitor KU 55933 results in a S-phase checkpoint defect similar to that observed in WRN shRNA cells. Moreover, γH2AX levels are higher in WRN shRNA cells than in control cells 6 and 16 h after exposure to psoralen DNA cross-links. These results suggest that WRN and ATM participate in a replication checkpoint response, in which WRN facilitates ATM activation in cells with psoralen DNA cross-link–induced collapsed replication forks.
INTRODUCTION
Proliferating cells are continuously exposed to agents that cause DNA damage and/or interfere with the progression of DNA replication (i.e., induce DNA replication fork arrest). Because such events can adversely affect genomic stability, cells have evolved S-phase signaling cascades, including DNA damage checkpoint responses and DNA repair mechanisms, to remove or repair DNA damage. In cells with DNA damage, the S-phase checkpoint inhibits the firing of late origins of replication (intra-S checkpoint) and prevents cells from entering mitosis with persistent DNA lesions (S-M checkpoint; Bartek et al., 2004). Together, these mechanisms promote accurate completion of DNA replication before cell division. Cells with defective S-phase cell cycle checkpoints are characterized by high levels of genomic instability.
Many commonly used cancer therapeutic agents sensitize cancer cells to replication-blocking DNA damage (Bryant et al., 2005; Farmer et al., 2005). For example, psoralen plus UVA (PUVA) induces interstrand cross-links (ICLs), which covalently bind the complementary strands of the DNA double helix, causing DNA replication fork arrest, and inducing cytotoxic DSBs during S-phase (Rothfuss and Grompe, 2004). Similarly, camptothecin (CPT) induces and traps topoisomerase I-DNA covalent complexes, stabilizing otherwise transient DNA single-strand breaks and ultimately producing cytotoxic DNA double-strand breaks (DSBs) during S-phase (Pommier et al., 2003). Exposure to hydroxyurea (HU), which depletes dNTP pools, has similar effects on cycling cells (Lundin et al., 2002). Although the mechanisms by which these clastogens act are distinct, they all indirectly induce DSBs in the vicinity of stalled DNA replication forks, a process known as replication fork collapse. It is generally thought that the Mre11-Rad50-Nbs1 (MRN) complex senses DSBs induced by ionizing irradiation and radiomimetic chemicals (Uziel et al., 2003; Stracker et al., 2004; Lee and Paull, 2005); however, less is known about the mechanisms involved in the cellular response to DSBs associated with replication fork collapse.
DSB-induced replication arrest is usually transduced through the nuclear protein kinases ATM and ATR, which are members of a family of proteins characterized by a PI3-like domain (Abraham, 2004). ATM, a primary inducer of the cellular response to DSBs, activates the DSB response network by phosphorylating key players in its numerous branches (Shiloh, 2003, 2006). Loss or inactivation of ATM is associated with the genomic instability syndrome ataxia-telangiectasia (A-T; Chun and Gatti, 2004), which is characterized by neuronal degeneration, immunodeficiency, radiation sensitivity, and cancer predisposition. ATR responds primarily to UV damage and stalled replication forks, but it is also involved in the response to DSBs and shares some protein targets with ATM (Shechter et al., 2004b). ATM and ATR are also involved in initiation and progression of DNA replication in unstressed cells (Abraham, 2001; Shechter et al., 2004a). When ATM or ATR is depleted in Xenopus cell free extracts, restart of damaged replication forks can induce DSBs (Trenz et al., 2006). Recent reports suggest that ATM acts upstream of ATR in the S-phase checkpoint response to γ-irradiation; however, this may not be true for cells exposed to UV light or HU, agents that do not directly induce DNA DSBs (Cuadrado et al., 2006; Jazayeri et al., 2006; Stiff et al., 2006). Therefore, it appears that ATM and ATR cooperate in activating the S-phase checkpoint, but their exact roles in this process may differ depending on the nature of the DNA damage and the type of replication-associated stress.
Werner syndrome (WS) is a rare autosomal recessive disorder characterized by premature onset of age-related pathologies and genomic instability. The gene mutated in WS, WRN, encodes a conserved DNA helicase of the RecQ family (Yu et al., 1996). WRN possesses 3′ → 5′ helicase, 3′ → 5′ exonuclease, and DNA-dependent ATPase activities (Gray et al., 1997; Huang et al., 1998). Biochemical and cellular studies in mice provide evidence that WRN copurifies with the 17S multiprotein DNA replication complex via direct binding to proliferating cell nuclear antigen (PCNA; Lebel et al., 1999). In addition, the Xenopus orthologue of WRN, FFA-1, is required for the formation of replication foci (Yan et al., 1998). Consistent with these observations, WS fibroblasts show a reduced number of replication initiation sites, a prolonged S-phase, and defective sister chromatid exchange at telomeres in unstressed cells (Takeuchi et al., 1982; Poot et al., 1992; Crabbe et al., 2004; Laud et al., 2005). In addition to a role for WRN in normal DNA replication, RecQ helicases have been implicated in replication checkpoint(s). Deletion of the yeast homolog of WRN, SGS1, leads to defective S-phase checkpoint activation in the stressed cells (Frei and Gasser, 2000). Furthermore, one report showed that activation of ATM is dependent on another RecQ helicase, BLM, after prolonged exposure to HU but not after exposure to the radiomimetic agent bleomycin (Davalos et al., 2004). Interestingly, WS cells are hypersensitve to DNA ICLs, but only mildly sensitive to γ-irradiation–induced DSBs (Poot et al., 2001; Yannone et al., 2001; Bohr et al., 2001). WS cells with persistent DNA ICLs undergo apoptosis in S-phase, which is likely due to accumulation of cytotoxic DSBs during DNA replication (Poot et al., 2002; Rothfuss and Grompe, 2004). Nonetheless, the exact role of WRN in the response to replication fork collapse is not understood.
Although ample evidence suggests that WRN resolves recombinational intermediates to facilitate DSB repair (Prince et al., 2001; Saintigny et al., 2002), relatively little is known about the role of WRN in the initial response to DSBs. Recent reports indicated that recruitment of WRN to sites of chromosome structural changes including DSBs is a rapid response (Lan et al., 2005; Turaga et al., 2007). Although ATR is thought to play a key role in the response to many types of replication stress including high doses of PUVA (Pichierri and Rosselli, 2004; Alderton et al., 2006; Jazayeri et al., 2006), ATM and WRN were independently identified as regulators of chromosomal replication (Lebel et al., 1999; Shechter et al., 2004a). These results led us to propose and test whether WRN and ATM might cooperate with each other during the response to replication-dependent DSBs. Indeed, the results presented here demonstrate that WRN and ATM cooperatively participate in an intra-S checkpoint in cells with collapsed replication forks.
MATERIALS AND METHODS
Cells and Reagents
The generation and maintenance of WRN short-hairpin RNA (shRNA), ATM shRNA and control cells using the U-2 OS osteosarcoma cell line have been described previously (Cheng et al., 2006; Ziv et al., 2006). The pBSCA-6XHis WRN expression vector was transfected into cells using PolyFect reagent (Qiagen, Chatsworth, CA) as previously described (Cheng et al., 2003). The ATM inhibitor, KU55933, was kindly donated by Dr. Graeme Smith (Kudos Pharmaceuticals). Nocodazole, 8-methoxypsoralen, campotothecin, HU, and angelicin were purchased from Sigma (St. Louis, MO), and aphidicolin from Calbiochem (La Jolla, CA). All were dissolved in DMSO except for HU, which was dissolved in water.
Flow Cytometric Analysis
Cell monolayers were treated with trypsin, and single cells were fixed overnight in 70% ethanol at −20°C. After centrifugation, RNase A and propidium iodide were added and incubated in the dark using the Cellular DNA Flow Cytometric Analysis Reagent Set according to the manufacturer's suggestion (Roche, Indianapolis, IN). Samples were analyzed using a FACScan (Becton Dickinson Immunocytometry Systems, Mountain View, CA). Cell cycle analysis on histograms from propidium iodide–stained cells was performed using Multicycle (Phoenix Flow Systems, San Diego, CA), where doublets and debris were modeled by software algorithms.
Checkpoint Assays
For intra-S checkpoint assay that measures DNA synthesis after DNA damage (Jaspers and Zdzienicka 2006), cells were preincubated with [14C]thymidine for 24 h. After extensive washing, cells were treated with 8-methoxypsoralen (0.1 μg/ml, 8-MOP, Sigma) for 10 min and 365-nm UV light at 1.8 J/cm2 for 5 min (PUVA; Majumdar et al., 1998). Cells were then pulsed with [3H]thymidine after the indicated times and collected 2 h later. Alternatively, cells were γ-irradiated (0–20 Gy), immediately pulsed with [3H]thymidine, and collected 2 h later. For Figure 4C, ethanol-fixed WRN shRNA and control cells were permeabilized with 0.25% Triton X-100 in PBS on ice for 15 min. After centrifugation, cells were stained with phospho-histone H3 antibody (Upstate Biotechnology, Lake Placid, NY) in PBS containing 1% BSA, followed by FITC-conjugated secondary antibody and propidium iodide staining. Mitotic cells were examined by flow cytometry.
Figure 4.

WRN-dependent ATM activation. (A) ATM autophosphorylation on Ser-1981 was quantified in control and WRN shRNA cells treated with PUVA (0.1 μg 8-methoxypsoralen/ml), HU (0.5 mM), or γ-irradiation. Cell lysates were prepared at the indicated time points as described for Western analyses (see Materials and Methods). (B) As in A, except Western blot was probed with antibodies to ATM substrates. (C) Phosphorylation of ATM substrates in control and WRN shRNA U-2 OS cells 16 h after treatment with PUVA (0.1 μg 8-methoxypsoralen/ml). Representative images were shown. (D) Fluorescence intensity after staining with the anti-phospho-(SQ/TQ) antibody was averaged over fields of cells using the softwares Axio Vision 3.1 and AIM Rel 3.2 (Zeiss).
Western Blotting Analysis
Whole cell lysates and immunoblot analysis were described previously (Cheng et al., 2003). The following antibodies were used: pS1981 ATM (clone 10H11.E12) and pS139 H2AX (clone JBW301) were from Upstate Biotechnology; ATM (Epitomics, alone Y170); pS966 SMC1 (clone BL311) from Bethyl (Montgomery, TX); pS1423 BRCA1 and H2AX (Novus, Littleton, CO); BRCA1 and WRN (Transduction Laboratories, Lexington, KY); SMC1, pS317 CHK1, and CHK1 (Cell Signaling Technology, Beverly, MA); and lamin B (Santa Cruz Biotechnology, Santa Cruz, CA).
Immunofluorescence
Cells were grown on Lab-Tek glass chamber slides (Nunc, Rochester, NY). For colocalization studies, cells were treated with bromodeoxyuridine (BrdU, 10 μM) and incubated at 37°C for 30 min in normal culture medium before PUVA treatment. The slides were washed with PBS between every indicated step. After treatment with a hypotonic lysis solution (10 mM Tris-HCl, pH 7.4, 2.5 mM MgCl2, 1 mM PMSF, and 0.5% Nonidet P-40) for 8 min on ice, cells were fixed in 4% paraformaldehyde for 10 min at room temperature (Balajee and Geard, 2001). Fixed cells were permeabilized with PBS + 0.2% Triton X-100 at room temperature for 5 min, blocked in PBS + 2% fetal bovine serum for 1 h at room temperature, and incubated with rabbit anti-ATM mAb (Epitomics, 1:500) previously found to be ATM-specific in immunostaining (Biton et al., 2006) and mouse anti-WRN mAb (clone 2D06) at 37°C for 1 h. Subsequently, cells were incubated with Alexa 633–conjugated anti-mouse IgG and Alexa 568–conjugated anti-rabbit IgG antibodies (Molecular Probes, Eugene, OR, 1:1000) at 37°C for 1 h in the dark. The chamber slides were then fixed again with 4% paraformaldehyde in PBS for 5 min at room temperature, and the DNA was denatured by incubating in 2 N HCl at 37°C for 30 min. After blocking as described above, the slides were immunostained with sheep anti-BrdU antibody (GeneTex, 2 μg/ml) at 37°C for 1 h, followed by incubation with Alexa 488 anti-sheep IgG antibody. DNA was stained with 1 μM DAPI in PBS for 20 min at room temperature, and the slides were mounted with Prolong Gold antifade reagent (Molecular Probes). Immunofluorescence was analyzed by a confocal microscope (Zeiss 510, Thornwood, NY).
To determine general ATM/ATR phosphorylation, we used the pS/TQ antibody (Cell Signaling), which was raised against a collection of peptides containing the p-SQ or p-TQ motifs. Cells were grown in the glass chamber slides and fixed in 4% paraformaldehyde for 10 min at room temperature and then permeabilized with PBS + 0.2% Triton X-100. Fixed cells were then blocked in 10% goat serum overnight at 4°C, followed by immunostaining with the pS/TQ antibody (1:1500) at 37°C for 2 h. Subsequently, slides were incubated with Alexa 568–conjugated anti-rabbit IgG antibody (1:1000, Molecular Probes). DNA was stained with 1 μM DAPI in PBS for 20 min at room temperature, and the slide chambers were mounted with Prolong Gold antifade reagent (Molecular Probes). Immunofluorescence signals were visualized by a fluorescence microscope (Zeiss Axiovert 200 M) and quantitated using the software AxioVision 4.5.
Pulse-Field Gel Electrophoresis
U-2 OS cells were treated with the indicated clastogens. After trypsinization, 106 cells were melted into 0.7% agarose insert. These inserts were incubated in 0.5 M EDTA (pH 8.0), 1% N-laurylsarcosyl, and proteinase K (1 mg/ml) at 50°C for 48 h and then washed four times in Tris-EDTA buffer before loading onto a 1% agarose (chromosomal grade) gel. Chromosomes were separated by pulsed-field gel electrophoresis for 24 h (CHEF Mapper, Bio-Rad, Richmond, CA; 120° angle, 60–240-s switch time, 4 V/cm). The gel was subsequently stained with ethidium bromide for analysis.
RESULTS
WRN Participates in a S-Phase Checkpoint Activated by Clastogens That Induce Replication-dependent DSBs
The role of WRN in the S-phase checkpoint was examined by treating WRN-deficient and control U-2 OS cells with DNA-damaging agents and examining cell cycle progression. In these experiments, we used WRN-deficient cells that stably expressed a WRN-specific shRNA (WRN shRNA cells) and control cells stably express a nonspecific shRNA (control shRNA cells). Cells were treated with clastogens that cause collapsed replication forks (PUVA, CPT, and HU), DSBs (γ-irradiation), DNA monoadducts (angelicin) or that inhibit replication (aphidicolin) and then were analyzed for cell cycle progression by flow cytometry. The majority of control and WRN shRNA cells treated with PUVA (0.1 μg 8-methoxy-psoralen/ml, Figures 1A and 2A) arrested in S-phase 16 h after treatment. However, by 24 h after treatment, significantly more control cells were arrested in S-phase and relatively more WRN shRNA cells appeared at G2/M phase. Similar results were obtained in cells treated with CPT or HU. Both cell types arrested in S-phase 8 h after removal of CPT from the culture media (1 μM for 3 h), but WRN shRNA cells progressed through S-phase at a faster rate than control cells (Figures 1B and 2B; 16-h time point). As such, the population of WRN shRNA cells contained more G2/M and fewer S-phase cells 16 h after CPT removal. For cells treated with HU, the majority of control and WRN shRNA cells arrested in S-phase 4 h after removal of HU from the culture media (0.5 mM for 24 h), but fewer WRN shRNA cells remained in S-phase 8 h after HU removal (Figures 1C and 2C). Because PUVA, CPT, and HU all induce DSBs during DNA replication, these data suggest that WRN may play a role in the S-phase checkpoint induced by collapsed replication forks. Lack of such replication checkpoint in WRN shRNA cells may compromise cell proliferation, as we have shown previously that WRN shRNA cells are more sensitive than control cells to PUVA-induced cell death (Cheng et al., 2006). The cell doubling times for unstressed U-2 OS cells are ∼20 h, consistent with the time frame of the observed S-phase checkpoint defect in WRN shRNA cells after replication stress.
Figure 1.

Role of WRN in S-phase checkpoint. Control and WRN shRNA U-2 OS cells were treated with (A) PUVA (0.1 μg 8-methoxypsoralen/ml), (B) CPT (1 μM, 3 h), (C) HU (0.5 mM, 24 h), (D) ionizing radiation (6 Gy), (E) aphidicolin (1 μg/ml, 24 h), and (F) angelicin (0.1 μg/ml)+UVA. Cells were harvested at the indicated time points and analyzed by flow cytometry (n = 3–4).
Figure 2.

Quantification of cell cycle profiles as described in Figure 1. (A) PUVA; (B) CPT; (C) HU.
The ability of replication-independent γ-irradiation induced DSBs to induce an S-phase checkpoint was also tested in control and WRN-deficient cells. Cells were treated with 6 Gy γ-irradiation and analyzed by flow cytometry. The number of control and WRN shRNA cells in S-phase was higher at 3 and 6 h after γ-irradiation, and most of these cells arrested in G1 or at G2/M by 16 h after irradiation (Figure 1D and Supplemental Figure S1). However, during the first 48 h after γ-irradiation, there was no apparent difference in cell cycle profiles in control and WRN shRNA cells. Aphidicolin is a known replication inhibitor that does introduce significant DSBs (Saleh-Gohari et al., 2005). After treatment with aphidicolin (1 μg/ml, 24 h), control and WRN shRNA cells accumulated in S-phase by 4 h and at G2/M by 8 h after removal of aphidicolin; both cell types progressed through the cell cycle with similar kinetics (Figure 1E and Supplemental Figure S2). In addition, angelicin (0.1 μg/ml), an agent that forms DNA monoadducts but not DNA ICLs in the presence of UVA, did not stimulate a WRN-dependent S-phase checkpoint (Figures 1F and Supplemental Figure S3). Taken together, the WRN-dependent S-phase checkpoint regulation likely requires formation of sufficient amount of replication-dependent DSBs.
Pulse-field gel electrophoresis (PFGE) was performed to confirm the presence or absence of DSBs in treated cells (Figure 3; see also Saleh-Gohari et al., 2005). The results confirmed that DSBs were generated under the following conditions: PUVA (0.1 μg 8-methoxy-psoralen/ml, 24 h), CPT (1 μM for 3 h, followed by 16-h recovery), HU (0.5 mM for 24 h, followed by 8 h recovery), and γ-irradiation (6 Gy, 30 min recovery; Figure 3). In contrast, DSB induction by exposure to aphidicolin (1 μg/ml for 24 h, followed by 6-h recovery) is only marginal, the level being much lower than those of γ-irradiation, PUVA, CPT, or HU.
Figure 3.
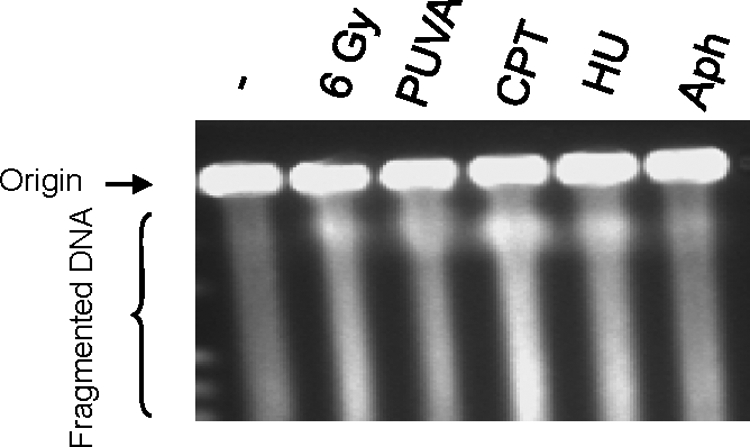
Pulse-field gel electrophoresis of control shRNA U-2 OS cells. DNA from U-2 OS cells was analyzed by PFGE after exposure to DNA damaging agents as follows: 30 min after 6 Gy γ-irradiation, 24 h after PUVA treatment (0.1 μg 8-methoxypsoralen/ml), 16-h recovery after CPT treatment (1 μM for 3 h), 8-h recovery after HU treatment (0.5 mM for 24 h), and 6-h recovery after Aph treatment (1 μg/ml for 24 h).
We have previously shown that U-2 OS cells exhibit increased endogenous chromosome breaks as evidenced by PFGE (Bartkova et al., 2006). This is consistent with the background DNA fragments observed in unstressed U-2 OS cells (Figure 3). The specificity of WRN knockdown was confirmed by complementing WRN shRNA cells with ectopic His-tagged WRN. It is our experience that Western analysis allows us to detect exogenously expressed WRN in WRN shRNA cells, suggesting that the system of WRN overexpression outpaces that of WRN knockdown. Twenty-four hours after treatment with PUVA, 26 ± 1% of WRN-knockdown cells arrested in S-phase, whereas 60 ± 9% of WRN knockdown cells expressing ectopic WRN arrested in S-phase; thus, complementation with wild-type WRN restored near wild-type response to PUVA (69 ± 11% in S-phase, Supplemental Figure S4). These results strongly support the observation that WRN plays a significant role in the S-phase checkpoint in the PUVA-treated U-2 OS cells.
PUVA-induced Activation of the ATM Pathway Requires WRN
There are at least two possible roles that WRN might play in the S-phase checkpoint: WRN might regulate early S-phase checkpoint kinases, such as ATM, or it might act downstream of checkpoint signaling cascades. The effect of WRN on ATM activation was examined by determining ATM autophosphorylation on Ser-1981. Human ATM activation involves autophosphorylation on Ser-1981, Ser-367, and Ser-1893 (Bakkenist and Kastan, 2003; Kozlov et al., 2006). Interestingly, ATM autophosphorylation on Ser-1981 was deficient in WRN shRNA cells (Figure 4A, top panels, left and right) 3–16 h after PUVA treatment using Western blotting analysis. However, ATM autophosphorylation on Ser-1981 was proficient in control shRNA cells from 3 to 16 h (Figure 4A) and gradually returned to the control level during the period from 24 to 72 h after PUVA treatment (not shown). Similarly, ATM autophosphorylation on Ser-1981 was severely repressed in WRN shRNA cells, but was proficient in control shRNA cells 0–24 h after recovery from HU treatment (Figure 4A, middle panels). ATM autophosphorylation on Ser-1981 was not detectable in WRN shRNA cells 0–24 h after recovery from HU treatment, whereas ATM autophosphorylation subsided in control shRNA cells at 24 h, a time point when cells have been released from S-phase arrest (Figure 1C). Thus, our results suggest that replication stress-induced activation of the ATM pathway is diminished, but not delayed, in WRN shRNA cells. Altogether, the results suggest that wild-type WRN is required for optimal ATM autophosphorylation in cells after PUVA treatment, and this might be a general event in response to collapsed replication forks based on the HU treatment data. In contrast, ATM autophosphorylation does not appear to be WRN-dependent in cells treated with 6 Gy of γ-irradiation (Figure 4A, bottom panels).
The results shown in Figure 4A predicted that downstream events in the ATM signaling pathway might also be defective in WRN shRNA cells treated with PUVA. To test this possibility, we determined phosphorylation of SMC1 on Ser-966, BRCA1 on Ser-1423, and CHK1 on Ser-317, all of which have been reported as ATM phosphorylation targets during activation of the S-phase checkpoint (Gatei et al., 2000; Xu et al., 2001; Kitagawa et al., 2004). Indeed, WRN shRNA cells were deficient in PUVA-induced phosphorylation of SMC1, BRCA1, and CHK1 at these phosphorylation sites (Figure 4B). In contrast, ATM substrates such as SMC1 were phosphorylated at normal levels in WRN shRNA cells exposed to γ-irradiation (Figure 4B, bottom panels). In addition, the amount of total SMC1, BRCA1, CHK1, and lamin B was similar in control and WRN shRNA cells under all conditions tested. Remarkably, it appears that WRN affects ATM activation and regulates S-phase checkpoint with similar kinetics in PUVA- or HU-treated cells (Figures 1 and 4). Thus, WRN is required for the ATM pathway activation in the response to PUVA, but not to γ-irradiation.
These results were confirmed using immunostaining for phosphorylated substrates of ATM; the experiment utilized an antibody that recognizes the ATM phosphorylation motif phosphoserine/phosphothreonine followed by glutamine (pS/TQ; Uziel et al., 2003). We found that there were significantly more pS/TQ-positive control shRNA cells than WRN shRNA cells 16 h after PUVA treatment (19 vs. 10%, p <0.05; Figure 4, C and D). There are comparable results as evidenced by phosphorylation of ATM target motifs (Figures 4, C and D) and specific pS/TQ-containing proteins (Figure 4A), further supporting the conclusion that WRN regulates PUVA-induced ATM pathway activation.
WRN and ATM Function in a PUVA-induced Intra-S-Phase Checkpoint
The above studies implicate that the ATM-mediated downstream events such as intra-S checkpoint were also impaired. To address this hypothesis, we tested whether WRN and ATM might inhibit DNA synthesis in replicating cells treated with PUVA (0.1 μg 8-MOP/ml). The results showed an initial decrease in the rate of DNA synthesis similarly in all the three cells, but the rates were higher in WRN and ATM shRNA cells than in control shRNA cells 8 and 12 h after PUVA treatment (Figure 5A). In contrast, the kinetics of DNA synthesis was similar in control and WRN shRNA cells exposed to IR (Figure 5B). Previous studies showed that A-T and Nijmegen breakage syndrome (NBS) cells are characterized by a defect in inhibition of DNA synthesis, known as intra-S checkpoint, after exposure to γ-irradiation (Jaspers and Zdzienicka, 2006). Similarly, ATM shRNA cells failed to inhibit DNA replication after γ-irradiation (Figure 5B). Collectively, our results here expand the role of ATM in the intra-S checkpoint to the more general response to collapsed replication forks. However, the extent of PUVA-resistant DNA synthesis is greater in WRN than in ATM shRNA cells, suggesting a central and upstream role of WRN, in relative to ATM, in PUVA-induced intra-S checkpoint.
Figure 5.

Role of WRN and ATM in intra-S-phase checkpoint in cells treated with PUVA and γ-irradiation. (A) Rate of DNA synthesis in control, WRN and ATM shRNA U-2 OS cells after exposure to PUVA (0.1 μg 8-methoxypsoralen/ml). Cells were harvested at the indicated time points (n = 3). DNA synthesis was measured as described in Materials and Methods. (B) Rate of DNA synthesis in control, WRN and ATM shRNA U-2 OS cells 2 h after exposure to the indicated doses of ionizing radiation (n = 3).
The presence of an incomplete replication fork can inhibit cells from entering mitosis (Bartek et al., 2004). We next examined DNA content and the extent of histone H3 Ser-10 phosphorylation (H3-Ser10P) after exposure to PUVA. The relative amount of H3-Ser10P was used as an indicator of cells that have completed DNA replication and entered mitosis (Ajiro et al., 1996). The results showed that the fraction of H3-Ser10P–positive cells was higher in WRN shRNA cells than in control cells 12 h after treatment with PUVA (Supplemental Figure S5). This result is consistent with the presence of a defective S-phase checkpoint in WRN shRNA cells.
Enhanced Level of PUVA-induced γH2AX in WRN shRNA Cells
It is known that PUVA can induce replication-dependent DSBs (Rothfuss and Grompe, 2004), a well-established trigger of ATM activation. Because we found a deficiency in PUVA-induced ATM activation in WRN shRNA cells (Figure 4), we asked whether PUVA-induced DSB formation is inhibited in WRN shRNA cells. Immunofluorescence (Figure 6A) and Western analysis (Figure 6B) results show that γH2AX levels increase in a time-dependent manner, and the extent of induction is greater in WRN shRNA than in control shRNA cells 16 h after PUVA treatment. In contrast, γ-irradiation (6 Gy) induces a fast mode of γH2AX formation, the level of which is similar in WRN and control shRNA cells (Figure 6). The amount of total H2AX protein is similar in WRN and control shRNA cells under all conditions tested (Figure 6B). Taken together, the deficiency in PUVA-induced ATM activation in WRN shRNA cells is unlikely to be attributed to the extent of PUVA-induced DSB formation. Possibly, kinases other than ATM are responsible for H2AX phosphorylation in PUVA-treated WRN shRNA cells. Thus, the results support the view that WRN functionally regulates ATM pathway activation.
Figure 6.

Role of WRN in γH2AX formation in cells treated with PUVA and γ-irradiation. Control and WRN shRNA U-2 OS cells were treated with PUVA (0.1 μg 8-methoxypsoralen/ml), and ionizing radiation. (A) Cells were fixed and stained with mouse antibodies against γH2AX, followed by detection using confocal microscope. (B) Cell lysates were prepared at the indicated time points and were analyzed by Western analyses using antibodies against γH2AX and H2AX (see Materials and Methods).
WRN and ATM Colocalize at Sites of DNA Replication in the Absence of PUVA
Next, we tested if WRN and ATM colocalize to replication foci by performing coimmunofluorescence for ATM and WRN in U-2 OS cells (Balajee and Geard, 2001). Cells were pulse-labeled with BrdU to identify replication foci. Soluble nucleoplasm was extracted, and cells were fixed before immunostaining. In unstressed cells (no PUVA treatment), ATM colocalizes with WRN at most BrdU-positive replication foci (Supplemental Figure S6A). Interestingly, WRN and ATM colocalize less frequently in non-S-phase cells, which are BrdU-negative (arrow, Supplemental Figure S6B). This result indicates that ATM and WRN constitutively associate with replication foci in S-phase.
WRN and ATM Function in a Common Process of Cellular Response to PUVA
The above data suggest that ATM and WRN may be epistatic in the cellular response to collapsed replication forks. This hypothesis was tested by treating control and WRN shRNA cells sequentially with the ATM kinase inhibitor KU55933 and PUVA. The results showed that PUVA-treated control cells underwent S-phase arrest 24 h after treatment and that this S-phase checkpoint was inhibited to a similar extent by WRN shRNA, KU55933, or both (Figure 7A). However, the fraction of G1 cells increased in cells treated with KU 55933 (open arrows, Figure 7A). The latter result may reflect the fact that ATM may also play a role in G2/M checkpoints (Shiloh, 2003). Thus, cells were cotreated with PUVA and the microtubule blocker nocodazole for 24 h, to trap cells in G2/M phase. Cells arrested in S-phase by PUVA were not affected by nocodazole until they progress into the G2/M phase, whereby they were trapped. As expected, trapped cells that progressed beyond S-phase arrested at G2/M phase. Although most control cells arrested in S-phase, the distribution of cells through the cell cycle was similar in WRN shRNA cells treated with or without KU 55933 (Figure 7B). These results are consistent with the hypothesis that WRN and ATM kinase function in the same pathway during the S-phase checkpoint response to PUVA interstrand cross-link–induced DNA DSBs.
Figure 7.

Convergence between WRN and ATM in the cellular response to PUVA. (A) Control and WRN shRNA U-2 OS cells were treated with PUVA (0.1 μg 8-methoxypsoralen/ml), ATM inhibitor (ATMi, KU55933, 10 μM), or both and harvested at 24 h. (B) Control and WRN shRNA U-2 OS cells were cotreated with nocodazole, PUVA, and ATMi as described in A.
DISCUSSION
This study describes a novel upstream role of WRN in the response to PUVA-induced DNA DSBs. Based on flow cytometric, DNA synthesis, immunofluorescence, Western and PFGE results, we conclude that WRN is required for an intra-S-phase checkpoint in the response to replication-dependent DSBs, but not for the response to acutely formed DSBs induced by γ-irradiation. Furthermore, WRN appears to modulate ATM activation during the S-phase checkpoint, facilitating autophosphorylation of ATM on Ser-1981 and phosphorylation of downstream ATM protein substrates. Lack of WRN increases PUVA-induced γH2AX formation, suggesting that lack of the WRN-dependent ATM pathway activation leads to increased genomic instability. These findings demonstrate that WRN cooperates with ATM to implement an intra-S-phase checkpoint in cells with collapsed replication forks. Because ATM is known to act upstream of DNA damage response, WRN and ATM may be poised for a rapid cellular response to collapsed replication forks.
Previous studies of the intrinsic catalytic properties of WRN and functional protein–protein interactions involving WRN suggested that WRN plays an important role in homologous recombination and base excision repair subpathways (Opresko et al., 2004a). For example, WRN helicase activity is stimulated by replication protein A (RPA; Brosh et al., 1999), a ssDNA-binding protein that play a role in DSB repair. WRN helicase activity is stimulated by Nbs1, a component of the MRN complex that is required for DSB-induced S-phase checkpoint and homologous recombination (Tauchi et al., 2002; Cheng et al., 2004). These previously published reports document a downstream role for WRN in DSB repair; this study reports that WRN also acts upstream in cellular signaling pathways, facilitating an intra-S-phase checkpoint in cells with collapsed replication forks. It is of future interests to determine whether WRN′s catalytic activities are required for ATM pathway activation after replication fork collapse.
Recent studies demonstrate that the ATM and ATR pathways interact during the response to DNA damage (Cuadrado et al., 2006; Jazayeri et al., 2006; Stiff et al., 2006). In most cells in S-phase, ATM acts upstream of ATR during the response to DSBs (Cuadrado et al., 2006; Jazayeri et al., 2006); in contrast, ATR acts upstream of ATM in response to stalled replication forks or UV treatment (Stiff et al., 2006). Our past work suggests that WRN is not significantly involved in the UV response pathway (Karmakar and Bohr, 2005). Consistent with this, we observed that WRN appears to play only an ancillary role in UV-induced S-phase arrest (Supplemental Figure S7). A previous report suggests that ATR, but not ATM, is required for inhibition of DNA synthesis after cellular exposure to PUVA (Pichierri and Rosselli, 2004). However, they used a time course up to only 9 h, and most likely the majority of cells are not yet arrested in S-phase under these conditions, and it is too early for the accumulation of collapsed forks. DNA cross-links per ser do not induce DNA breaks; they are converted into DNA DSBs during DNA replication in S-phase (Rothfuss and Grompe 2004).
It is known that ATM and ATR share some phosphorylation targets. Although CHK1 Ser-317 is generally thought to be a ATR phosphorylation target, CHK1 phosphorylation by ATM in response to γ-irradiation has also been reported (Gatei et al., 2003). In cells treated with PUVA, CHK1 Ser-317 is phosphorylated before ATM and H2AX are phosphorylated (Figures 4, A and B, and 6B), suggesting that at an early stage ATR may phosphorylate CHK1 in response to PUVA-induced DNA ICLs without the involvement of replication-dependent DSBs. Because CHK1 phosphorylation at 1 h occurs in both types of cells, this event appears to be WRN-independent. Furthermore, we earlier showed that collapsed replication forks after inhibition of PARP-1 clearly activates ATM, but does not activate ATR (Bryant and Helleday, 2006). Therefore, DNA breaks may be a major determinant for ATM and/or ATR involvement in replication stress. Although this report focuses on WRN-dependent ATM pathway activation involving replication-dependent DSBs, additional studies are required to define the exact sequence of events and molecular mechanism of pathways that may involve additional protein partners such as ATR.
This study shows that WRN is required for the ATM pathway activation in cells with PUVA-induced DSBs during replication. It is worth noting that WRN binds DNA substrates that resemble replication intermediates (von Kobbe et al., 2003), whereas ATM does not. In fact, ATM has no intrinsic DNA binding capability (Lee and Paull, 2005). In this context, it is possible that WRN increases the accessibility of ATM to collapsed replication forks and that a functional interaction exists between these two proteins. Here, we also show that WRN and ATM colocalize to DNA replication foci in unstressed cells, which is in accordance with previous studies showing that WRN colocalizes with PCNA at replication foci (Opresko et al., 2004b). DNA DSB mediators, such as MDC1 and 53BP1, contain the BRCT phosphoprotein interaction domain that facilitate transient binding to γH2AX (Schultz et al., 2000; Goldberg et al., 2003; Lou et al., 2006). 53BP1 also contains the tudor domain that binds Lys-79 methylated histone H3 when the site is exposed by DNA DSBs (Huyen et al., 2004). Moreover, chromatin relaxation is triggered in the vicinity of a DSB after DNA damage induction (Kruhlak et al., 2006), likely resulting in increased accessibility of DSB mediators and ATM to damaged nucleosomes. In comparison, before the fork encounters a nick, chromatin structures 5′ to collapsed replication forks are already relaxed as a result of the progression of the replication machinery. Because WRN does not contain BRCT or tudor domains and does not bind directly to γH2AX (Cheng et al., 2005), WRN may not function as a DNA damage mediator. Rather, WRN has a HRDC domain that is required for binding to DSBs (Lan et al., 2005). Taken together, this suggests that under conditions of replication-dependent DSBs, WRN is required for ATM pathway activation.
Previous studies show that ATM is required for the cellular response to replication stress (Bolderson et al., 2004). This study shows that WRN can play an important role in activating ATM and suggests that WRN may increase the accessibility of ATM to damaged/collapsed replication forks. The exact mechanism by which this might occur is not yet known; however, it may involve direct interaction or shared interaction partners between WRN and ATM. One candidate is the MRN complex that also directly interacts with and activates ATM (Lee and Paull, 2005). Importantly, the MRN complex binds directly to both WRN and ATM (Cheng et al., 2004; Lee and Paull, 2005). Thus, it is possible that WRN and the MRN complex function together to signal the presence of collapsed replication forks, resulting in ATM activation. Future studies are needed to determine whether WRN directly stimulates ATM activation independent of MRN or whether MRN plays a direct role in WRN-dependent activation of ATM.
Studies in yeast suggest that WRN participates in the early response to replication stress. Rgh1 and Sgs1 are the sole members of the RecQ helicase family in fission and budding yeasts, respectively. Rqh1 is not essential for a HU-induced S-phase checkpoint, but prevents aberrant recombination events during checkpoint recovery (Stewart et al., 1997). In contrast, Sgs1 has been implicated in checkpoint activation and in preventing the accumulation of aberrant recombinational intermediates in response to replication stress (Frei and Gasser, 2000; Liberi et al., 2005). In humans, earlier studies suggested that WRN and RPA prevent aberrant recombination intermediates in HU-arrested S-phase cells (Constantinou et al., 2000; Saintigny et al., 2002). Therefore, WRN likely plays dual roles in the response to replication-dependent DSBs: first to stimulate activation of ATM and second to resolve recombination intermediates. It is not yet understood what factors might regulate these two functions of WRN. However, it is possible that posttranslational modifications of WRN regulate WRN functions and activity. For example, phosphorylation of WRN in stressed cells is ATM- and ATR-dependent (Pichierri et al., 2003) and phosphorylation of WRN alters its DNA binding properties (J. Lee and V. Bohr, unpublished data).
The findings presented here may be related to the observation that WRN-deficient cells are much more sensitive to CPT-, HU-, and ICL-induced cytotoxicity than to γ-irradiation (Poot et al., 2001; Yannone et al., 2001; Cheng et al., 2004). This is consistent with the observation that the WRN-dependent ATM-dependent response pathway is not activated by γ-irradiation. This early function of WRN after DNA damage can potentially impact on many ATM-dependent genome surveillance pathways. Indeed, WRN shRNA cells exhibit decreased replication stress-induced phosphorylation of SMC1 on Ser-966, BRCA1 on Ser-1423, and CHK1 on Ser-317, as well as defective S-phase checkpoint pathways; these proteins are important downstream effectors of the ATM-dependent S-phase checkpoint (Xu et al., 2001; Kim et al., 2002; Gatei et al., 2003). WRN could also act at a later step during resolution of intermediates in recombinational DNA repair. Loss of these two WRN functions would increase genomic instability as well as the cytotoxicity associated with unrepaired DSBs in replicating cells. This is supported by the observation that there is increased γH2AX formation in WRN shRNA cells. Other kinases are apparently responsible for H2AX phosphorylation in PUVA-treated WRN shRNA cells when ATM activation is defective. It should be noted that although WRN plays a role in ATM pathway activation at the later time point after PUVA or HU treatment, this should not be interpreted as a late response. Considering the nature of PUVA and HU in inducing DNA breaks during DNA replication, this supports a specific role of WRN in S-phase where replication-dependent DNA breaks occur. It is also tempting to speculate that WRN could ensure that homologous recombination is only initiated when S-phase arrest has been achieved.
In summary, this study provides evidence that WRN and ATM function cooperatively at the interface between cell cycle checkpoints and DNA replication. We propose a scenario (Figure 8) in which the WRN-ATM complex may play a role at damaged replication forks. If replication-dependent DSBs are induced, WRN facilitates ATM activation, which in turn activates downstream effectors and intra-S-phase arrest. In contrast, γ-irradiation–induced ATM activation does not require WRN. Although there is a difference in the premature aging phenotypes between WS and A-T, our results suggest that a deficiency in the WRN-ATM pathway could increase DNA damage-related symptoms of aging. Mutations in either WRN or ATM cause several common cellular features, including senescence, chromosome breaks, and telomere instability. Thus, the novel WRN-dependent ATM activation and its substrate phosphorylation described here may have significant implications for our understanding of aging as well as the complex relationship between cellular checkpoint mechanisms and genomic stability.
Figure 8.

Model of the Role of WRN and ATM in response to collapsed replication forks or DSBs.
Supplementary Material
ACKNOWLEDGMENTS
We thank G. Smith and A. May for materials and technical support; F. Indig and R. Earley for confocal microscope; and S. A. Martomo, A. Balajan, R. Wersto, and Y. Liu for comments. This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging, and Lilly Lawski Foundation (D.M.).
Abbreviations used:
- A-T
ataxia-telengiectasia A-T
- CPT
camptothecin
- DSBs
double-strand breaks
- HU
hydroxyurea
- ICLs
interstrand cross-links
- MRN
Mre11-Rad50-Nbs1
- PUVA
psoralen plus UVA
- PFGE
pulse-field gel electrophoresis
- WS
Werner syndrome
- WRN
Werner protein.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E07-07-0698) on July 2, 2008.
REFERENCES
- Abraham R. T. PI3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair. 2004;3:883–887. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Abraham R. T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Ajiro K., Yoda K., Utsumi K., Nishikawa Y. Alteration of cell cycle-dependent histone phosphorylations by okadaic acid. Induction of mitosis-specific H3 phosphorylation and chromatin condensation in mammalian interphase cells. J. Biol. Chem. 1996;271:13197–13201. doi: 10.1074/jbc.271.22.13197. [DOI] [PubMed] [Google Scholar]
- Alderton G. K., Galbiati L., Griffith E., Surinya K. H., Neitzel H., Jackson A. P., Jeggo P. A., O'Driscoll M. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat. Cell Biol. 2006;8:725–733. doi: 10.1038/ncb1431. [DOI] [PubMed] [Google Scholar]
- Bakkenist C. J., Kastan M. B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Balajee A. S., Geard C. R. Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nucleic Acids Res. 2001;29:1341–1351. doi: 10.1093/nar/29.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J., Lukas C., Lukas J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Bartkova J., et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Biton S., Dar I., Mittelman L., Pereg Y., Barzilai A., Shiloh Y. Nuclear ataxia-telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells. J. Biol. Chem. 2006;281:17482–17491. doi: 10.1074/jbc.M601895200. [DOI] [PubMed] [Google Scholar]
- Bohr V. A., Souza P. N., Nyaga S. G., Dianov G., Kraemer K., Seidman M. M., Brosh R. M., Jr. DNA repair and mutagenesis in Werner syndrome. Environ. Mol. Mutagen. 2001;38:227–234. doi: 10.1002/em.1076. [DOI] [PubMed] [Google Scholar]
- Bolderson E., Scorah J., Helleday T., Smythe C., Meuth M. ATM is required for the cellular response to thymidine induced replication fork stress. Hum. Mol. Genet. 2004;13:2937–2945. doi: 10.1093/hmg/ddh316. [DOI] [PubMed] [Google Scholar]
- Brosh R. M., Jr., Orren D. K., Nehlin J. O., Ravn P. H., Kenny M. K., Machwe A., Bohr V. A. Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem. 1999;274:18341–18350. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- Bryant H. E., Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34:1685–1691. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Cheng W. H., Kusumoto R., Opresko P. L., Sui X., Huang S., Nicolette M. L., Paull T. T., Campisi J., Seidman M., Bohr V. A. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34:2751–2760. doi: 10.1093/nar/gkl362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W. H., Sakamoto S., Fox J. T., Komatsu K., Carney J., Bohr V. A. Werner syndrome protein associates with gamma H2AX in a manner that depends upon Nbs1. FEBS Lett. 2005;579:1350–1356. doi: 10.1016/j.febslet.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Cheng W. H., von Kobbe C., Opresko P. L., Arthur L. M., Komatsu K., Seidman M. M., Carney J. P., Bohr V. A. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- Cheng W. H., von Kobbe C., Opresko P. L., Fields K. M., Ren J., Kufe D., Bohr V. A. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol. Cell. Biol. 2003;23:6385–6395. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun H. H., Gatti R. A. Ataxia-telangiectasia, an evolving phenotype. DNA Repair. 2004;3:1187–1196. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Constantinou A., Tarsounas M., Karow J. K., Brosh R. M., Bohr V. A., Hickson I. D., West S. C. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L., Verdun R. E., Haggblom C. I., Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- Cuadrado M., Martinez-Pastor B., Murga M., Toledo L. I., Gutierrez-Martinez P., Lopez E., Fernandez-Capetillo O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos A. R., Kaminker P., Hansen R. K., Campisi J. ATR and ATM-dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle. 2004;3:1579–1586. doi: 10.4161/cc.3.12.1286. [DOI] [PubMed] [Google Scholar]
- Farmer H., et al. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Frei C., Gasser S. M. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Gatei M., Scott S. P., Filippovitch I., Soronika N., Lavin M. F., Weber B., Khanna K. K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000;60:3299–3304. [PubMed] [Google Scholar]
- Gatei M., et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- Goldberg M., Stucki M., Falck J., D'Amours D., Rahman D., Pappin D., Bartek J., Jackson S. P. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Gray M. D., Shen J. C., Kamath-Loeb A. S., Blank A., Sopher B. L., Martin G. M., Oshima J., Loeb L. A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- Huang S., Li B., Gray M. D., Oshima J., Mian I. S., Campisi J. The premature ageing syndrome protein, WRN, is a 3′ → 5′ exonuclease. Nat. Genet. 1998;20:114–116. doi: 10.1038/2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y., Zgheib O., Ditullio R. A., Jr., Gorgoulis V. G., Zacharatos P., Petty T. J., Sheston E. A., Mellert H. S., Stavridi E. S., Halazonetis T. D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- Jaspers N. G., Zdzienicka M. Z. Inhibition of DNA synthesis by ionizing radiation: a marker for an S-phase checkpoint. Methods Mol. Biol. 2006;314:51–59. doi: 10.1385/1-59259-973-7:051. [DOI] [PubMed] [Google Scholar]
- Jazayeri A., Falck J., Lukas C., Bartek J., Smith G. C., Lukas J., Jackson S. P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Karmakar P., Bohr V. A. Cellular dynamics and modulation of WRN protein is DNA damage specific. Mech. Ageing Dev. 2005;126:1146–1158. doi: 10.1016/j.mad.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Kim S. T., Xu B., Kastan M. B. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa R., Bakkenist C. J., McKinnon P. J., Kastan M. B. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov S. V., Graham M. E., Peng C., Chen P., Robinson P. J., Lavin M. F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–3514. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak M. J., Celeste A., Dellaire G., Fernandez-Capetillo O., Muller W. G., McNally J. G., Bazett-Jones D. P., Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L., Nakajima S., Komatsu K., Nussenzweig A., Shimamoto A., Oshima J., Yasui A. Accumulation of Werner protein at DNA double-strand breaks in human cells. J. Cell Sci. 2005;118:4153–4162. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- Laud P. R., Multani A. S., Bailey S. M., Wu L., Ma J., Kingsley C., Lebel M., Pathak S., DePinho R. A., Chang S. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005;19:2560–2570. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel M., Spillare E. A., Harris C. C., Leder P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J. Biol. Chem. 1999;274:37795–37799. doi: 10.1074/jbc.274.53.37795. [DOI] [PubMed] [Google Scholar]
- Lee J. H., Paull T. T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Liberi G., Maffioletti G., Lucca C., Chiolo I., Baryshnikova A., Cotta-Ramusino C., Lopes M., Pellicioli A., Haber J. E., Foiani M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005;19:339–350. doi: 10.1101/gad.322605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Lundin C., Erixon K., Arnaudeau C., Schultz N., Jenssen D., Meuth M., Helleday T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 2002;22:5869–5878. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A., et al. Targeted gene knockout mediated by triple helix forming oligonucleotides. Nat. Genet. 1998;20:212–214. doi: 10.1038/2530. [DOI] [PubMed] [Google Scholar]
- Opresko P. L., Cheng W. H., Bohr V. A. Junction of RecQ helicase biochemistry and human disease. J. Biol. Chem. 2004a;279:18099–18102. doi: 10.1074/jbc.R300034200. [DOI] [PubMed] [Google Scholar]
- Opresko P. L., Otterlei M., Graakjaer J., Bruheim P., Dawut L., Kolvraa S., May A., Seidman M. M., Bohr V. A. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell. 2004b;14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Pichierri P., Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004;23:1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P., Rosselli F., Franchitto A. Werner's syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–1500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- Pommier Y., et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. 2003;532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Poot M., Gollahon K. A., Emond M. J., Silber J. R., Rabinovitch P. S. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB J. 2002;16:757–758. doi: 10.1096/fj.01-0906fje. [DOI] [PubMed] [Google Scholar]
- Poot M., Hoehn H., Runger T. M., Martin G. M. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp. Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- Poot M., Yom J. S., Whang S. H., Kato J. T., Gollahon K. A., Rabinovitch P. S. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J. 2001;15:1224–1226. doi: 10.1096/fj.00-0611fje. [DOI] [PubMed] [Google Scholar]
- Prince P. R., Emond M. J., Monnat R. J., Jr. Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001;15:933–938. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuss A., Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 2004;24:123–134. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny Y., Makienko K., Swanson C., Emond M. J., Monnat R. J., Jr. Homologous recombination resolution defect in werner syndrome. Mol. Cell. Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh-Gohari N., Bryant H. E., Schultz N., Parker K. M., Cassel T. N., Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz L. B., Chehab N. H., Malikzay A., Halazonetis T. D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D., Costanzo V., Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004a;6:648–655. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- Shechter D., Costanzo V., Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair. 2004b;3:901–908. doi: 10.1016/j.dnarep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem. Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Stewart E., Chapman C. R., Al-Khodairy F., Carr A. M., Enoch T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 1997;16:2682–2692. doi: 10.1093/emboj/16.10.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T., Walker S. A., Cerosaletti K., Goodarzi A. A., Petermann E., Concannon P., O'Driscoll M., Jeggo P. A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006;25:5775–5782. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker T. H., Theunissen J. W., Morales M., Petrini J. H. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair. 2004;3:845–854. doi: 10.1016/j.dnarep.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Takeuchi F., Hanaoka F., Goto M., Akaoka I., Hori T., Yamada M., Miyamoto T. Altered frequency of initiation sites of DNA replication in Werner's syndrome cells. Hum. Genet. 1982;60:365–368. doi: 10.1007/BF00569220. [DOI] [PubMed] [Google Scholar]
- Turaga R., Massip L., Chavez A., Johnson B., Lebel M. Werner syndrome protein prevents DNA breaks upon chromatin structure alternation. Aging Cell. 2007;6:471–481. doi: 10.1111/j.1474-9726.2007.00301.x. [DOI] [PubMed] [Google Scholar]
- Tauchi H., et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature. 2002;420:93–98. doi: 10.1038/nature01125. [DOI] [PubMed] [Google Scholar]
- Trenz K., Smith E., Smith S., Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C., Thoma N. H., Czyzewski B. K., Pavletich N. P., Bohr V. A. Werner syndrome protein contains three structure-specific DNA binding domains. J. Biol. Chem. 2003;278:52997–53006. doi: 10.1074/jbc.M308338200. [DOI] [PubMed] [Google Scholar]
- Xu B., Kim S., Kastan M. B. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol. Cell. Biol. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Chen C. Y., Kobayashi R., Newport J. Replication focus-forming activity 1 and the Werner syndrome gene product. Nat. Genet. 1998;19:375–378. doi: 10.1038/1263. [DOI] [PubMed] [Google Scholar]
- Yannone S. M., Roy S., Chan D. W., Murphy M. B., Huang S., Campisi J., Chen D. J. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J. Biol. Chem. 2001;276:38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- Yu C. E., et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- Ziv Y., Bielopolski D., Galanty Y., Lukas C., Taya Y., Schultz D. C., Lukas J., Bekker-Jensen S., Bartek J., Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.