

Abstract
We investigated the functional and structural implications of SNAP25 having two SNARE motifs (SN1 and SN2). A membrane-bound, intramolecular FRET probe was constructed to report on the folding of N-terminal SN1 and C-terminal SN2 in living cells. Membrane-bound constructs containing either or both SNARE motifs were also singly labeled with donor or acceptor fluorophores. Interaction of probes with other SNAREs was monitored by the formation of SDS-resistant complexes and by changes in FRET measured in vitro using spectroscopy and in the plasma membrane of living cells using TIRF microscopy. The probes formed the predicted SDS-resistant SNARE complexes. FRET measurements revealed that syntaxin induced a close association of the N-termini of SN1 and SN2. This association required that the SNARE motifs reside in the same molecule. Unexpectedly, the syntaxin-induced FRET was prevented by VAMP. Both full-length SNAP25 constructs and the combination of its separated, membrane-bound constituent chains supported secretion in permeabilized chromaffin cells that had been allowed to rundown. However, only full-length SNAP25 constructs enabled robust secretion from intact cells or permeabilized cells before rundown. The experiments suggest that the bidentate structure permits specific conformations in complexes with syntaxin and VAMP and facilitates the function of SN1 and SN2 in exocytosis.
INTRODUCTION
The exocytotic release of hormones and neurotransmitters is mediated by the interaction of three membrane-associated soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein (SNAP) receptors or SNAREs (Sollner et al., 1993): syntaxin, SNAP-25, and VAMP (synaptobrevin). The importance of these proteins for secretion was originally suggested when they were identified as substrates of the various botulinum neurotoxins and tetanus toxin, although their precise role in the fusion event was unclear.
In solution, the cytosolic domains of these SNARE proteins associate to form a four-helix bundle that is resistant to dissociation by SDS (Hayashi et al., 1994). X-ray crystallographic studies reveal that the N-termini of the helices lie in parallel, with two of the four helices contributed by SNAP-25 and one each by syntaxin and VAMP (Sutton et al., 1998). Studies of the proteins in solution are consistent with this structure (Fasshauer et al., 1997; Lin and Scheller, 1997), which may reflect a configuration of the SNAREs during or after fusion. Physiological interactions of SNAP-25 and syntaxin in the plasma membrane probably occur with a 1:1 ratio (Fasshauer, 2003;Rickman et al., 2004), although in vitro studies of soluble SNARE motifs also demonstrate a 2:1 binding stoichiometry (Fasshauer et al., 1997;Xiao et al., 2001). Little is known about the interactions of membrane-bound SNAREs in living cells and the relationships of the interactions to secretion. However, there is strong evidence that the membrane influences SNARE properties, thereby influencing exocytosis (Fasshauer and Margittai, 2004;De Haro et al., 2004;Zhang et al., 2005).
It has been suggested that in situ SNARE complex assembly begins at the cytosolic N-termini and continues toward the membrane-anchored C-termini, thus bringing the secretory vesicle membrane and the plasma membrane in close proximity. In vitro studies with soluble domains suggest that the N-terminal domains of syntaxin and SN1 [SNAP25 (1-80)] and SN2 [SNAP25(142-206)] interact to provide a nucleation site for VAMP (Fasshauer and Margittai, 2004). Experiments in PC12 cells suggest that syntaxin first interacts with SN1 of SNAP-25 before VAMP interacts with SN2 (An and Almers, 2004). However, other in situ experiments are consistent with SNAP-25 first interacting with VAMP before a strong interaction with syntaxin (Chen et al., 2001). Capacitance measurements of exocytosis in chromaffin cells are consistent with partially zippered SNARE complexes being a prelude to full zippering of the complex and exocytosis (Xu et al., 1999;Rettig and Neher, 2002).
The present study was motivated by our desire to investigate SNARE interactions in living cells. Most previous structural studies of SNARES investigated complex formation of soluble peptides in vitro, using a variety of techniques including PAGE, circular dichroism, and x-ray crystallography. We have exploited the presence of two SNARE motifs in SNAP25 to construct an intramolecular FRET probe with donor fluorophore (cerulean) on the N-terminus of SN1 and acceptor fluorophore (citrine) on the N-terminus of SN2. This probe becomes palmitoylated and attaches to the plasma membrane similarly to endogenous SNAP25. In addition, individual membrane-bound SN1 and SN2 chains were labeled with donor and acceptor fluorophores, respectively. We determined the behavior of these probes as they interacted with other SNAREs in a membrane environment by monitoring their ability to form SDS-resistant complexes, by measuring changes in FRET, and by determining their ability to function in exocytosis. Comparison of the function of full-length SNAP25 with that of its constituent chains SN1 and SN2 revealed that the bidentate molecule interacts with syntaxin by drawing the N-termini of the linked SN1 and SN2 together in a way that separated SN1 and SN2 do not and enhances the ability of SN1 and SN2 to support secretion.
MATERIALS AND METHODS
Molecular Biology
The cDNA of mouse SNAP25b (a gift from Prof. T.J.F Martin, University of Wisconsin, Madison) with or without a D179K mutation (to render the constructs resistant to botulinum neurotoxin type E [BoNT E]) was used for all probes. SNAP-25 plasmids labeled with green fluorescent protein (GFP) variants were constructed by PCR and inserted into pEGFP-C1 based plasmids (Clontech, Palo Alto, CA). Citrine (cit) is a YFP variant that is insensitive to Cl− and is more photostable (Griesbeck et al., 2001). Cerulean (cer) is a cyan fluorescent protein (CFP) variant that is brighter and more photostable than its parent (Rizzo et al., 2004). All GFP variants had a A206K mutation to prevent dimerization (Zacharias et al., 2002). Specific details of their construction are given in Supplemental Materials.
Cell Preparation
Preparation of bovine chromaffin cells and transient transfection were performed as described previously (Wick et al., 1993). For human growth hormone (hGH) secretion experiments, cells were plated in 12-well plates (22.6-mm well diameter). HEK293 cells were plated at a density of 1.5 ∼ 2 × 106 cells/well in six-well plates and transfected in 2 ml of antibiotic-free DMEM/well with 7.5 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and 2.8 μg of DNA. Cells were twice rinsed with DMEM after 6 h, and medium containing antibiotics was added. One day later, cells were subjected to spectrophotometry or optical FRET analysis. For optical experiments, cells were plated on glass coverslips (no. 1.5, Warner Instruments, Hamden, CT) lying at the bottom of six-well plates. Coverslips were coated with poly-d-lysine to promote cell adhesion.
Secretion Experiments
Secretion experiments were generally performed 4–5 d after transfection of bovine chromaffin cells by CaPO4 precipitates. Intact cell experiments were performed in a physiological salt solution (CaPSS) containing 145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 15 mM HEPES, pH 7.4. Cells were incubated with or without 20 μM DMPP (1,1-dimethyl-4-phenylpiperazinium, a nicotinic agonist) for 2 min at 30°C. Secretion experiments in permeabilized cells were performed in potassium glutamate solution (KGEP) containing 139 mM potassium glutamate, 20 mM PIPES (pH 6.6), 2 mM MgATP, 20 μM digitonin, and 5 mM EGTA with either no added Ca2+ or sufficient Ca2+ to yield 30 μM buffered Ca2+. In some experiments, the cells were first permeabilized in the absence of Ca2+ and then were stimulated by 30 μM buffered Ca2+ in KGEP without digitonin. hGH was measured with a highly sensitive ELISA from Roche Applied Science (Indianapolis, IN). Stimulated secretion was expressed as the difference between the percentage of the total cellular hGH that was released into the medium in the presence of nicotinic agonist or Ca2+ and the percentage of hGH released in the absence of stimulation (typically 1%). Fractional recovery after BoNT E was calculated according to the following formula using the percentage of stimulated hGH secretion for each condition: [(construct + BoNT E) − (neo + BoNT E)]/[(neo alone) − (neo + BoNT E)], where neo represents the control condition without exogenous SNAP25 constructs. There was usually 0.5–2.0 ng of hGH and 60 nmol of catecholamine/22.6-mm diameter well, and n = four wells per group. Data are expressed as mean ± SEM unless otherwise indicated.
SDS-PAGE and Blots
Membrane proteins were incubated at 37° (to retain SDS-resistant complexes) or boiled for 5 min in 2× SDS sample buffer and subjected to 8% SDS-PAGE, followed by immunoblotting with monoclonal antibodies against the following epitopes: GFP (Roche Diagnostics, Indianapolis, IN, 1:3000 dilution); syntaxin (Synaptic Systems, Göttingen, Germany, 1:3000 dilution); FLAG (Sigma-Aldrich, St. Louis, MO, 1:3000 dilution); myc (BD Biosciences Clontech, Palo Alto, CA, 1:3000 dilution); SNAP25 (Sternberger Monoclonals, Lutherville, MD, SMI 81, 1:2000 dilution); and hemagglutinin (HA; Berkeley Antibody Co., Richmond, CA, 12CA5, 1:2000 dilution). Antibodies were detected by enhanced chemiluminescence (GE Healthcare-Amersham, Piscataway, NJ), and protein bands were quantitated using ImageJ (NIH; http://rsb.info.nih.gov/ij/).
Spectroscopy
Constructs were expressed in HEK293T cells transfected using Lipofectamine 2000 < (Invitrogen). Twenty-four hours after transfection, cells were harvested in 1 mM Tris (pH 7.4) containing protease inhibitors (10 mM benzamidine, 1 μg/ml aprotinin, 1 mM PMSF, 1 mM DTT) and homogenized. After a brief centrifugation at 800 × g to remove debris, membranes were pelleted at 50,000 × g for 35 min. Pellets were resuspended in TBS (20 mM Tris, 150 mM NaCl, pH 7.4) without protease inhibitors and diluted to an appropriate concentration for spectroscopy. In some experiments, membranes were solubilized by the addition of 10% Triton X-100 to a final concentration of 1% and incubated 30 min at 4°C. Samples were then cleared by centrifugation for 15 min at Vmax in an Eppendorf centrifuge at 4°C, and the supernatant was retained. Spectra were taken on a Shimadzu RF-5301PC spectrofluorimeter (Shimadzu Scientific Instruments, Columbia, MD) with excitation at 435 nm and emission scanned between 450 and 600 nm. For presentation, spectra were normalized to the peak cerulean emission (476 nm).
Total internal fluorescence microscopy–based Measurement of Plasma Membrane FRET
Donor-stimulated emission of acceptor fluorescence was measured in the plasma membrane of living HEK293T cells and chromaffin cells using total internal fluorescence microscopy (TIRFM). Objective-based TIRFM was obtained by directing a 442-nm beam from a helium/cadmium laser (HeCd laser, Melles Griot, Carlsbad, CA) through a custom side port to side-facing filter cube containing a double-notch dichroic mirror MS-442/514zpc and 514rb emission filter (514-nm notch filter, reflects 514 nm but transmits emissions <500 nm and greater than 528 nm; all filters from Chroma Technology, Brattleboro, VT) on an Olympus IX70 (inverted) microscope (Melville, NY) with the 1.5× internal magnifying lens in the emission path. An additional filter (E455lpv2) was placed in the emission path just after the filter cube to block 442-nm leakage through the dichroic mirror. The beam was focused on the periphery of the back focal plane of a 60× 1.49 NA, oil immersion objective (Olympus, Melville, NY), giving a decay constant for the evanescent field of ∼100 nm. The emission was directed through a 2.5× magnifying lens to a Dualview beam splitter (Optical Insights, Tucson, AZ) containing a 505dcxr dichroic mirror and HQ470/30 (cerulean) and E580LP (combination of cerulean and citrine) emission filters. The band widths of the emissions were 465–500 nm (cerulean alone) and 580–605 nm (citrine and cerulean). Digital images were captured on a cooled electron mobilization, back-thinned CCD camera (Ikon+, Andor Technology, South Windsor, CT) with 100–250-ms exposures and 150–250 gain (EM setting). Each pixel corresponded to 73 nm.
The images were processed using custom software written in IDL (ITT, Boulder, CO). The split images were aligned to within one pixel, backgrounds were subtracted, and the images were smoothed by 3 × 3 sliding box car binning. The cerulean spillover into the long wavelength emission (0.415 of the low wavelength intensity) was subtracted pixel-by-pixel, and the pixel-by-pixel ratio of the citrine/cerulean emission was calculated, saved as a text file, and displayed as a tif image. Citrine emission excited by 442 nm in the low-wavelength cerulean channel (465 nm–500 nm) was negligible. Relative frequencies of the pixel-by-pixel ratios were also determined in a separate program.
Chromaffin cells were stimulated to secrete by perfusion with the nicotinic agonist DMPP (20 μM). Solution was delivered to the cells using positive pressure from a computer-controlled perfusion system DAD-6VM (ALA Scientific Instruments, Westbury, NY). Images were recorded at 2 Hz.
FRET Determined by Increased Donor Emission upon Acceptor Bleaching in a Confocal Microscope
HEK293 cells were cotransfected with plasmids encoding FRET constructs of SNAP25, and syntaxin and/or FLAG-VAMP. One day later, living cells expressing various fluorescent SNAP25 constructs on the plasma membrane were visualized for FRET at 30°C in CaPSS solution using a Fluoview500 confocal microscope (Olympus). Inclusion of the cholesterol-chelator 5-methyl- β-cyclodextrin (15 mM) did not affect the results. The chelator was not present in the experiments reported here. Cerulean was excited at 405 nm, and emission was detected at 465–495 nm and 535–565 nm. Citrine was excited at 514 nm, and emission was detected at 535–565 nm. Selective photobleaching of citrine was performed by repeatedly scanning a region of the specimen at 514 nm with the laser set at maximum intensity. At least 90% of the original acceptor fluorescence was photobleached. The fluorescence emission from the donor and the acceptor were collected sequentially. The pre- and postbleach images were analyzed with ImageJ software.
Plasma membrane fluorescence was specifically measured. The Threshold function in ImageJ was used to select the brightest pixels, which were mainly on the plasma membrane. The threshold was set at 2.5 times the mode of the off-cell background. Intracellular pixels that were above threshold were subtracted to determine the plasma membrane fluorescence. The average background (off-cell) pixel fluorescence was <1% of the average pixel fluorescence of the plasma membrane. Total fluorescence intensities on the plasma membrane of both donor and acceptor were measured before and after bleaching. The percentage efficiency of bleaching FRET was calculated as follows: E = [1 − (IDA/ID)] × 100, where IDA and ID represent the donor fluorescence in the presence and the absence of acceptor.
RESULTS
Construction of FRET Probes
The goal of this study is to understand the structural basis for SNAP25 function in exocytosis. We designed intramolecular FRET probes for SNARE interactions that could both function in exocytosis and report SNARE complex formation. The FRET probes are based on SNAP25, which contains two SNARE motifs (Figure 1A). Structural studies of SNARE interactions demonstrate that the two SNARE motifs in SNAP25 fold upon SNARE complex formation so that the N-termini of the first (SN1) and second (SN2) SNARE motifs are in close proximity. The probes were constructed with the FRET pair cerulean (cer, a variant of cyan fluorescent protein) and citrine (cit, a derivative of yellow fluorescent protein), and the indicated segments of SNAP25. In the intramolecular FRET constructs, cerulean is located at the N-terminus of SN1 and citrine is located at the N-terminus of SN2. When the molecules are folded within a SNARE complex, cerulean and citrine are predicted to come together, allowing energy transfer to occur (Figure 1B). Indeed, an earlier study demonstrated the feasibility of this approach (An and Almers, 2004). All of the constructs are membrane-associated by virtue of palmitoylated cysteines in the linker region between SN1 and SN2. Constructs were named with the following conventions: C, cerulean; Y, citrine; and s, any continuous sequence in SNAP25. Three of the probes (CssYs, CssYs180, and CssY) contain a 20-amino acid duplication within the linker region (amino acids 81–100), a modification that positions the FRET partners slightly farther apart and provides eight potential palmitoylation sites rather than four. Constructs without the 20-amino acid duplication were also created. They behaved similarly in both FRET and secretion experiments, but gave somewhat smaller FRET changes and somewhat reduced function in secretion. They were not as extensively investigated, and the data are not presented. Constructs that include SN2 also contain (unless otherwise indicated) a mutation (D179K) that renders them resistant to cleavage by BoNT E, a modification that allows assessment of the functioning of the transfected protein in cells coexpressing BoNT E (Zhang et al., 2002).
Figure 1.

FRET constructs containing the D179K mutation function in secretion. (A) Schematic drawing of various SNAP25 constructs. Citrine (cit) is a YFP variant, and cerulean (cer) is a CFP variant. All GFP variants have the A206K mutation to prevent dimerization. (B) Predicted conformational change of the SNAP-25-based FRET constructs upon interaction with other SNAREs. (C and D) Cultured bovine chromaffin cells were transfected with plasmids encoding hGH and the various FRET constructs described in A, with or without a plasmid encoding the light chain of botulinum neurotoxin type E (BoNT E). Secretion was stimulated for 2 min by 20 μM DMPP. (C) Secretion in the presence of the constructs was normalized to control secretion (neo). The number of repetitions of each experiment is given in parentheses: neo (6), CssYsDK (5), and citSNAP25DK (6). (D) Fractional recovery of hGH secretion is expressed relative to the toxin-induced inhibition in the absence of construct (see Materials and Methods). The number of repetitions of each experiment were: CssYsDK (5), CssYs180 (2), CssY (2), and citSNAP25noDK (1).
FRET Constructs Function in Secretion
Before investigating the conditions under which these constructs could undergo FRET, we asked whether they retained the ability to support exocytosis. Constructs were expressed in chromaffin cells together with hGH, a marker for the regulated secretory pathway, with or without BoNT E light chain to cleave the endogenous SNAP25. All constructs were present on the plasma membrane (data not shown). If no exogenous SNAP25 was expressed, BoNT E light chain inhibited hGH secretion by 65–80%. The FRET construct containing SN2, CssYs, almost completely rescued secretion (Figure 1C). Toxin-resistant, SNAP25 labeled with citrine on its N-terminus actually enhanced secretion when expressed with or without toxin. As expected, constructs lacking an intact SN2 (CssY and CssYs180) or the D179K mutation (citSNAP25) did not rescue (Figure 1D). We conclude that the insertion of neither cerulean on the N-terminus nor citrine within the SNAP25 molecule of SNAP25 prevents function in cells in which endogenous SNAP25 is cleaved by BoNT E.
The validity of this conclusion depends on the ability of BoNT E to cleave endogenous SNAP25 in the presence of the overexpressed D179K-containing constructs. We verified that proteins with the D179K mutation do not interfere with the cleavage of endogenous SNAP25 in two ways. First, wild-type SNAP25 was equally sensitive to BoNT E whether or not citSNAP25(D179K) was present in transfected HEK293T cells (Supplemental Figure S1A). Second, equivalent amounts of endogenous SNAP25 were cleaved at all concentrations of recombinant BoNT E light chain in the presence and absence of recombinant cit-SN2(D179K) in permeabilized chromaffin cells (Supplemental Figure S1B). In addition, the degree of rescue was strongly dependent on the secretion protocol (see Figure 8), a result that would be unlikely if rescue were caused by inhibiting cleavage of endogenous SNAP25.
Figure 8.

Separated SN1 and SN2 do not FRET and do not efficiently rescue secretion. (A) CerSN1 and citSN2 were expressed in HEK293T cells alone or together, with or without syntaxin or syntaxin plus VAMP. Membranes were prepared as in Materials and Methods. The groups are listed in order of decreasing fluorescence at 526 nm (the order they appear in the figure). The spectrum of citSN2 (which contains no cerulean and thus has little emission when excited at 435 nm) was omitted from the figure. This spectrum was used to subtract the component of citrine emission due to excitation at the cerulean wavelength (435 nm), or spill-over, from each group that included citSN2. (B–E) Cultured bovine chromaffin cells were transfected with plasmids encoding hGH and cerSN1 and citSN2 alone or together, with or without BoNT E. Secretion in the presence of the constructs was normalized to control secretion (neo). Values for control secretion were 17.8% (B and C), 17.9% (D), and 22% (E). (B) Secretion was stimulated for 2 min by 20 μM DMPP. For each condition, data from two experiments were pooled, except for cerSN1 (1) and CssYs (5). (C) Secretion was stimulated for 2 min by 20 μM DMPP. (D) Cells were permeabilized for 3 min with 20 μM digitonin ± 30 μM Ca2+. (E) Cells were permeabilized for 8 min with 20 μM digitonin in the absence of Ca2+, followed by a 15-min incubation ± 30 μM Ca2+. (C–E) The data from two experiments were pooled. All proteins were coexpressed with hGH in more than 80% of the cells.
Syntaxin Alone Induces FRET in Membrane-bound SNAP25 Constructs with SN2
SNARE complex formation has been typically studied using cytosolic or soluble forms of the proteins. In the present study we investigated SNARE complex formation using fluorescent-labeled SNAP25 constructs in membranes, which more closely approximates the environment in which the SNARES function.
Membranes were prepared from HEK293 cells transiently transfected with FRET constructs with or without syntaxin 1A (Figure 2). A spectrum of cerSNAP25 (without citrine) is presented for comparison (Figure 2A). Because excitation at 435 nm results in insignificant citrine emission (data not shown), the cerSNAP25 spectrum represents the absence of FRET. Both CssY and CssYs exhibited significant FRET when expressed alone (Figure 2A), with the maximum donor-stimulated emission at 526 nm. There are likely to be several components to the FRET. One is an intramolecular interaction between the linked fluorophores. In addition there could be two types of intermolecular interactions: random collisions between the fluorophores in the membrane (Zacharias et al., 2002), which disappear when the proteins are solubilized in detergent (compare panels A and B), and specific interactions between separate SNAP25 molecules.
Figure 2.

Syntaxin alone induces FRET in membrane-bound SNAP25 constructs with SN2. Membranes were prepared from HEK293T cells expressing various FRET probes with or without syntaxin as described in Materials and Methods. Samples were excited in a spectrofluorimeter at 435 nm, and the resulting emission spectra were normalized to the peak cerulean emission (476 nm). (A) The spectrum of cerSNAP25 (without citrine) is included for comparison. (B) Aliquots of the membrane fractions were solubilized with 1% Triton X-100 as described in Materials and Methods. (C–E) FRET probes CssY and CssYs were expressed in HEK293T cells, either alone or with or without syntaxin or syntaxin plus VAMP. Membranes were prepared as in Materials and Methods. (C) CssY; CssY + syntaxin; CssY + syntaxin + VAMP. D. CssYs; CssYs + syntaxin; CssYs + syntaxin + VAMP. (E) The results in C and D are expressed as the percent change in peak citrine emission (526 nm) in the presence of syntaxin or syntaxin + VAMP.
In the absence of syntaxin, the spectra of CssY and CssYs were identical (Figure 2A), suggesting that the presence of SN2 has little effect on the degree of folding of these constructs in the absence of other SNAREs. However, SN2 had a profound effect when these probes were coexpressed with syntaxin (Figure 2, C and D). The presence of syntaxin produced a 32% increase in FRET with CssYs (Figure 2D), but caused no change in the FRET with CssY (Figure 2C). Similar effects of syntaxin were also seen in Triton X-100 extracts of these membranes (not shown).
Effects of VAMP on FRET
Because the SNARE motif of VAMP forms a stable superhelical complex with the SNARE motifs of SNAP25 and syntaxin, we expected that expression of VAMP together with CssYs and syntaxin would have no effect or might further increase FRET compared with CssYs and syntaxin alone. However, coexpression of VAMP with syntaxin and CssYs decreased FRET in membranes (Figure 2D). The results of duplicate experiments are summarized in Figure 2E. Syntaxin enhanced FRET in CssYs by 32%, and this enhancement was decreased by almost 50% upon coexpression with VAMP despite similar coexpression of syntaxin. The effect of VAMP required SN2 and syntaxin because VAMP had little effect on the emission spectrum of CssY in the presence of syntaxin (Figure 2, C and E) or of the emission spectrum of CssYs or CssY alone (not shown).
SDS-resistant Complexes Formed by FRET Probes, Syntaxin, and VAMP
The existence of SNARE complexes containing the FRET probes can be inferred from the abovementioned changes in FRET. We asked whether these complexes resemble the SDS-resistant SNARE complexes reported by others. Various combinations of FRET probes and full-length SNARE proteins were coexpressed in HEK293T cells, and cell membranes were prepared and solubilized in SDS sample buffer.
When expressed alone, both CssY (Figure 3A, lane 1) and CssYs (Figure 3B, lane 1) ran as three bands in unboiled samples, and as single bands in boiled samples (not shown). For CssY the bands ranged between 50 and 75 kDa; CssYs bands were correspondingly larger. The top band in each case corresponded to the boiled protein. Apparently, both probes exhibit folded conformations that run more quickly than the fully denatured molecule.
Figure 3.
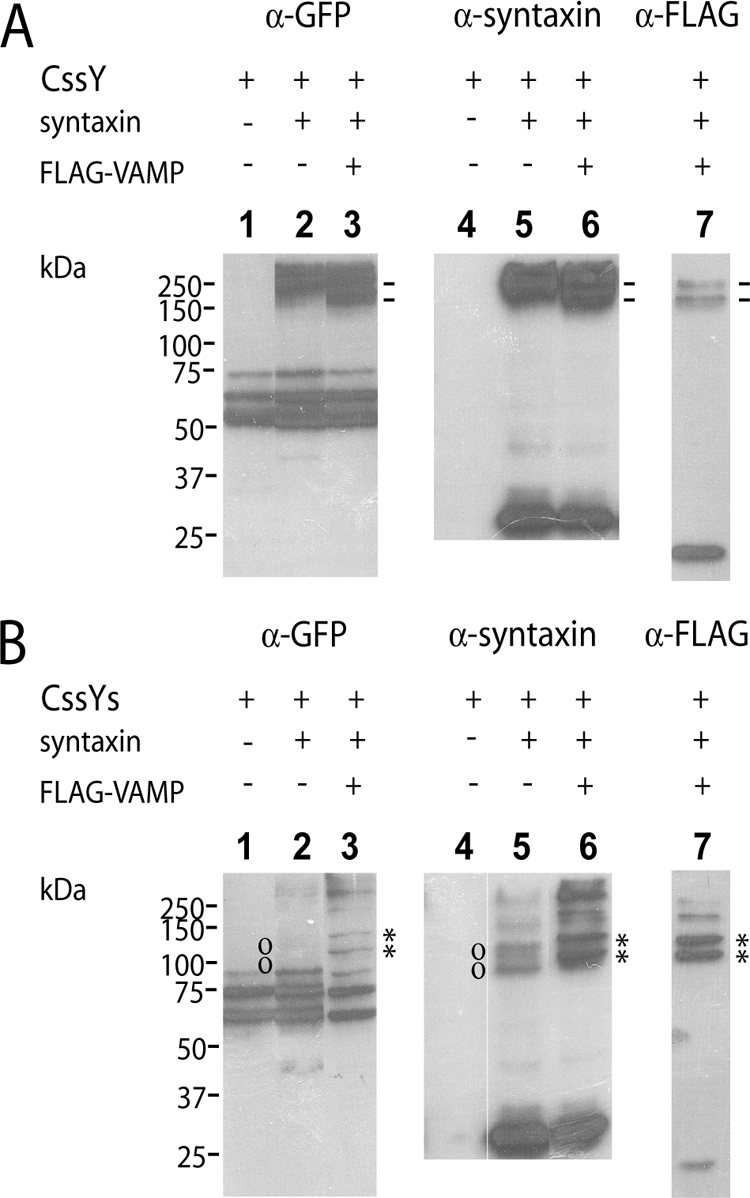
SDS-resistant complexes formed by FRET probes, syntaxin, and VAMP. Membrane samples from the experiment in Figure 2 were analyzed by SDS-PAGE followed by immunoblotting. (A) CssY. Lanes 1–3, anti-GFP. Lanes 4–6, anti-syntaxin. Lane 7, anti-FLAG-VAMP. Symbols (−) denote high-molecular-weight complexes containing CssY, syntaxin, and VAMP. (B) CssYs. Lanes 1–3, anti-GFP. Lanes 4–6, anti-syntaxin. Lane 7, anti-FLAG-VAMP. Symbols (o, *) denote complexes containing CssYs and syntaxin, or CssYs, syntaxin, and VAMP, respectively. Note that the complexes between CssYs, syntaxin, and VAMP (*) are slightly larger than those between CssYs and syntaxin (o).
Both CssY and CssYs formed SDS-resistant complexes with syntaxin. Complexes at greater than 200 kDa were prominent with CssY (Figure 3A, lanes 2 and 5) but much less evident with CssYs (Figure 3B, lanes 2 and 5). Both the apparent molecular weight and the strength of the bands suggest a complex of two CssYs and two syntaxins, analogous to the complex formed by two molecules of cytosolic SN1 and the H3 domain of syntaxin as determined by crystallography (Misura et al., 2001). The lack of SN2 probably promotes the formation of these quaternary complexes. Interestingly, the ability to form these complexes was not associated with a change in FRET in our experiments (Figure 2C).
CssYs and syntaxin also formed complexes of the correct mobility to contain one CssYs and one syntaxin (Figure 3B, lanes 2 and 5, indicated by o). The complexes are much more visible in lane 5 (anti-syntaxin) than in lane 2 (anti-GFP), suggesting that complex formation may shield the GFP epitope from the anti-GFP antibody.
In the absence of syntaxin, no stable complexes with VAMP were seen (not shown). However, membrane-bound VAMP (FLAG-VAMP) can participate in complexes with both CssY and CssYs if syntaxin is present. Most (88%) of the expressed FLAG-VAMP was in complexes with CssYs and syntaxin (Figure 3B, lane 7). The two bands marked with asterisks contained over 70% of the VAMP immunoreactivity. Conversely, when coexpressed with CssY and syntaxin, FLAG-VAMP was mainly (84%) uncomplexed (Figure 3A, lane 7), with only a small fraction of VAMP associated with the high-molecular-weight forms of CssY:syntaxin.
CssY had two complexes containing both VAMP and syntaxin at 180 and 250 kDa. CssYs had similar bands plus two much darker bands (asterisks), one of 130 kDa (the predicted molecular weight of a syntaxin:CssYs:VAMP complex) and one of ∼100 kDa, which also contains all three proteins. The 100-kDa complex may represent a more tightly folded, higher mobility trimeric complex. As expected, these bands were shifted upward by the molecular weight of FLAG-VAMP from the bands corresponding to CssYs + syntaxin in the absence of VAMP (marked by o Figure 3B, lane 5). The GFP epitope was somewhat more apparent with VAMP than in CssYs + syntaxin without VAMP (compare panel B, lanes 6 and 7), consistent with a less shielded epitope.
In summary, both CssY and CssYs form SDS-resistant complexes with syntaxin alone or together with VAMP. Complexes between CssYs and syntaxin differ in two ways from complexes between CssY and syntaxin. They represent substantially different folded configurations because the higher molecular weight CssYs forms lower molecular weight complexes. In addition, the combination of CssYs and syntaxin is much better able to interact with VAMP. The experiments do not determine whether the SDS-resistant CssYs/syntaxin complexes are responsible for FRET. However, it is likely that characteristics of CssYs that allow these complexes to form are related to the increase in FRET in spectroscopy and TIRFM (below) experiments.
A FRET Probe Lacking the C-terminal 26 Amino Acids of SNAP25 Exhibits Modest FRET with Syntaxin But Is Unable to Interact with VAMP
BoNT E cleaves SNAP25 at its C-terminus, producing a truncated protein and destroying its ability to function in secretion. We generated a probe corresponding to the BoNT E cleavage product (CssYs180) and verified that it was unable to rescue secretion in cells expressing BoNT E light chain (Figure 1D). We then asked whether it retained the ability to interact with syntaxin and VAMP. HEK293T cells were transfected with CssYs180 alone or in combination with syntaxin and VAMP, and membranes were prepared as in Figure 2. In response to syntaxin, CssYs180 exhibited a modest (15%) increase in FRET (Figure 4A). Importantly, the syntaxin-induced FRET with CssYs180 was no longer decreased by coexpression of VAMP. In the same experiment, the FRET with CssYs increased by 54% in response to syntaxin and decreased by 40% upon addition of VAMP (data not shown).
Figure 4.
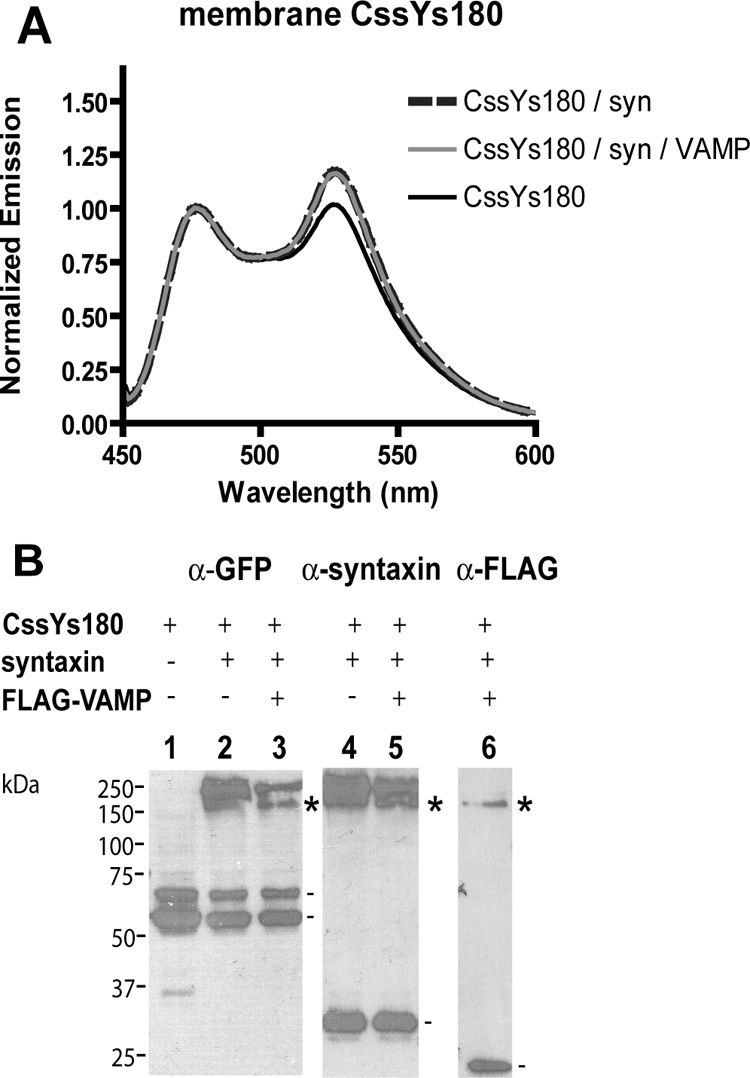
Effect of C-terminal truncation of SNAP25 on syntaxin- and VAMP-induced changes in FRET. (A) FRET constructs were transiently expressed in HEK293T cells with or without syntaxin and/or FLAG-VAMP, and membranes were prepared as in Figure 2. Membrane samples were excited in a spectrofluorimeter at 435 nm, and the resulting emission spectra were normalized to the peak cerulean emission (476 nm). (B) Aliquots of membranes containing CssYs180 from A were analyzed by SDS-PAGE followed by immunoblotting. Lanes 1–3, anti-GFP. Lanes 4 and 5, anti-syntaxin. Lane 6, anti-FLAG-VAMP. Symbols denote complexes containing CssYs180, syntaxin, and VAMP (*), or the monomeric proteins (−).
The SDS-resistant complexes formed by CssYs180, syntaxin, and VAMP were examined by SDS-PAGE and protein blotting. CssYs180 did form tight complexes with syntaxin or syntaxin and VAMP (marked by asterisks in lanes 3, 5, and 6, Figure 4B), but these complexes were of high molecular weight, and resembled the large complexes seen with CssY (Figure 3A) rather than those of CssYs (Figure 3B). Most of the VAMP was uncomplexed, similarly to experiments with CssY (Figure 3A, lane 7). There were no bands analogous to the 130- and 100-kDa bands containing CssYs, which represent a 1:1:1 association of syntaxin:CssYs:VAMP. We conclude that the truncation of the C-terminus of SN2 impairs its ability to interact fully with syntaxin and prevents its productive interaction with VAMP.
Syntaxin-dependent FRET in Plasma Membrane–bound SNAP25 Constructs Detected in Living Cells by TIRFM
The above experiments were performed in a suspension containing both intracellular and plasma membranes. Total internal fluorescence microscopy was used to measure FRET selectively in the plasma membrane in living cells. HEK293T cells expressing CssY or CssYs, alone or in combination with syntaxin or syntaxin and VAMP, were excited at 442 nm. Emission was detected at 465–500 nm (cerulean emission alone) and 580–605 nm (citrine and cerulean emissions). The citrine signal was calculated by subtracting the spillover from cerulean. FRET was measured in the images by the pixel-to-pixel ratio of the citrine/cerulean emissions. Images from cells coexpressing CssYs and syntaxin, and a histogram of the relative frequency of citrine/cerulean ratios at each pixel in the upper right cell, are shown in Supplemental Figure S2E.
Histograms similar to the one in Supplemental Figure S2E were generated for each cell (11–17 cells/group) and then averaged for each group. For CssYs (Figure 5B), syntaxin reproducibly broadened the frequency histogram and shifted the ratio to higher values (increased FRET). In contrast, the frequency peaks for CssY were narrow, and symmetrical, with no consistent difference when syntaxin was coexpressed (Figure 5A). In this experiment, CssY alone was shifted slightly to the left of the other peaks, but this was not a reproducible finding.
Figure 5.

Effect of syntaxin with or without VAMP on plasma membrane donor-stimulated FRET emission from CssY and CssYs in HEK293T cells and chromaffin cells measured by TIRFM. Cells were transfected with a plasmid encoding CssY or CssYs with or without plasmids encoding syntaxin and VAMP as indicated. Chromaffin cells were also cotransfected with BoNT E. FRET (citrine/cerulean ratios) was determined as in Supplemental Figure S2E and Materials and Methods. Syntaxin caused large increases in FRET in both HEK293T cells and chromaffin cells expressing exogenous syntaxin. Coexpression of VAMP prevented the increased FRET. There were 11–17 cells/group in each of the panels.
The effect of VAMP to reduce syntaxin-dependent FRET was also observed in the plasma membrane of living cells. VAMP coexpression prevented the shift to higher FRET ratios in CssYs + syntaxin while having no effect on the FRET ratios in CssY + syntaxin (Figure 5, A and B).
CssYs and CssY were also expressed in chromaffin cells together with BoNT E. There was no difference in donor-stimulated emission for the two proteins without cotransfected syntaxin (Figure 5, C and D). This was unexpected because CssYs should interact with endogenous syntaxin. However, the amount of transiently expressed CssYs may be much greater than that of endogenous syntaxin. Indeed, when syntaxin was transiently expressed with the probes, donor-stimulated emission from CssYs but not from CssY was increased (Figures 5, C and D, and Supplemental Figure S3). FRET changes were not detected during nicotinic stimulation either with or without cotransfected syntaxin.
FRET in HEK293T cells was also measured using confocal microscopy. FRET was measured by determining the increase in donor (cerulean) emission upon photobleaching of the acceptor (citrine). The results were similar to the TIRFM experiments. Syntaxin caused a large increase in FRET with CssYs but no increase with CssY (Supplemental Figure S4).
In summary, the syntaxin-dependent structural changes reflect a robust, intrinsic property of the syntaxin-SNAP25 interaction. It occurs in the plasma membrane of living HEK293T and chromaffin cells as well as in in vitro membrane preparations. It is evident both by measurements of donor-stimulated emission and acceptor bleaching.
Intermolecular FRET Does Not Reveal Syntaxin-dependent Conformational Changes
The FRET seen with CssY and CssYs in Figure 2 is consistent with an intramolecular interaction between the two fluorophores. However, it was also possible that the cerulean on one molecule of CssYs could interact with citrine on the second and vice versa. To test this possibility, we made full-length SNAP25 (Figure 6) with cerulean on the amino-terminus (HAcerSNAP25) and full-length SNAP25 with citrine inserted on the N-terminus of SN2, between amino acids 141 and 142 (myc-sYs-SNAP25). The two constructs were expressed alone or together, with or without syntaxin and VAMP in HEK293T cells, and membranes were prepared. Together, the two molecules produced a modest amount of FRET (Figure 6). A comparison of the ratio of citrine fluorescence and cerulean fluorescence, as well as analysis by protein blots, indicate that there was equivalent expression (∼1:1) of the citrine- and cerulean-tagged constructs in the membranes. Unlike the FRET seen with CssYs, however, FRET between HAcerSNAP25 and myc-sYs-SNAP25 was not significantly enhanced by syntaxin. Thus, the ability of syntaxin to enhance FRET requires the presence of the two fluorophores on the same molecule. The data indicate that most of the syntaxin-dependent FRET seen with CssYs is intramolecular rather than intermolecular.
Figure 6.

Intermolecular FRET does not reveal syntaxin-dependent structural changes. (A) Structure of full-length SNAP25 constructs containing single fluorophores. These constructs were also tagged with separate epitopes (HA and myc, respectively) for ease of identification of the individual species on blots. (B) SNAP25 constructs shown in A were expressed in HEK293T cells alone or together with or without syntaxin or syntaxin plus VAMP. Membranes were prepared as in Materials and Methods, and spectra were obtained using a spectrofluorimeter. The groups are listed in order of decreasing fluorescence at 526 nm (the order they appear in the figure). The spectrum of myc-sYs-SNAP25 (which contains no cerulean and thus has little emission when excited at 435 nm) was omitted from the figure. This spectrum was used to subtract the component of citrine emission due to excitation at the cerulean wavelength (435 nm) or spill-over, from each group that included myc-sYs-SNAP25. In addition, the expression of the two SNAP25 constructs was examined by SDS-PAGE and protein blotting and was determined to be ∼1:1 in all groups in which they were coexpressed.
Separated SN1 and SN2 Form SDS-resistant Complexes, But Do Not FRET
The following experiments were designed to determine whether the combination of the separated SNARE motifs interacts with syntaxin similarly to the full-length protein and whether it functions similarly in secretion.
Separated fragments of SNAP25 interact in solution with the H3 domain of syntaxin and the cytosolic portion of VAMP to form a stable, SDS-resistant complex (Hayashi et al., 1994; Chen et al., 1999). We expected to find that the membrane-anchored cerSN1 and citSN2 would form similar complexes when coexpressed with full-length syntaxin and VAMP, and this was indeed the case (Figure 7). Stable complex formation required SN1 and syntaxin. All transfections that included both these proteins resulted in higher molecular weight SDS-resistant complexes in situ (Figure 7A, lanes 2, 3, 9, and 10). Combinations of SNAREs that did not contain both SN1 and syntaxin did not form such complexes (Figure 7A, lanes 1 and 4–8). Syntaxin appeared in complexes only when expressed in combination with SN1 (Figure 7B, lanes 2, 3, 6, and 7) compared with lanes lacking SN1 (Figure 7B, lanes 1, 4, and 5). CitSN2 and/or VAMP (Figure 7C, lanes 2 and 5) form complexes only if they are expressed together with SN1 and syntaxin.
Figure 7.

Separated SN1 and SN2 form SDS-resistant complexes. (A and B) Cs (cerSN1) and sYs (citSN2) were expressed in HEK293T cells alone or together, with the indicated combinations of syntaxin and VAMP. Membranes were prepared as in Materials and Methods, and analyzed by SDS-PAGE followed by immunoblotting. In addition, FRET spectra were determined for some of these samples (see Figure 8). (A) Anti-GFP. (B) Anti-syntaxin. (C) anti-FLAG-VAMP. Complexes summarized in Table 1 are indicated by lower case letters: a and b, cerSN1 and syntaxin; c and d, cerSN1, syntaxin and VAMP; e, cerSN1, citSN2, syntaxin, and VAMP. Note that d is poorly resolved from b on these blots, but d contains VAMP (panel C, lane 2), whereas b does not (panel A, lane 2). (D) CerSN1 and mycSN2 were coexpressed with syntaxin and FLAG-VAMP in HEK293T cells. Membranes were analyzed by SDS-PAGE followed by immunoblotting. A complex containing all four proteins (designated f) was recognized by each of the indicated antibodies.
When cerSN1 and syntaxin were coexpressed, they formed high-molecular-weight species (labeled a and b). Coexpression of VAMP with cerSN1 and syntaxin resulted in two additional bands, c and d, containing VAMP (Figure 7C, lane 2). The bands b and d coincided but must represent different complexes because b occurs in the absence of VAMP (Figure 7A, lane3) and d contains VAMP (Figure 7C, lane 2). Addition of citSN2 generates a fifth complex, e (Figure 7A, lane 10; B, lane 7; and C, lane 5) containing SN1, SN2, syntaxin, and VAMP. It has the appropriate molecular weight to contain one molecule of each of the four proteins and is only found when all four proteins are expressed together. The constituents of these complexes are summarized in Table 1. Because cerSN1 and citSN2 share similar epitope tags, in a separate experiment, we verified that all four SNARE chains were in the same complex using mycSN2 rather the citrine-tagged protein used in FRET experiments (Figure 7D). The complex (f) only appeared when all four proteins were coexpressed.
Table 1.
SDS-resistant SNARE complexes detected with separated SN1 and SN2 constructs
| Complex | cerSN1/syntaxin | cerSN1/syntaxin/VAMP | cerSN1/citSN2/syntaxin/VAMP | cerSN1/mycSN2/syntaxin/VAMP |
|---|---|---|---|---|
| a | X | |||
| b | X | |||
| c | X | |||
| d | X | |||
| e | X | |||
| f | X |
Components of SDS-resistant SNARE complexes. Individual components of the various SDS-resistant SNARE complexes enumerated in Figure 8, A–D, are indicated.
The above experiments demonstrate that the combination of cerSN1 and citSN2 form complexes with syntaxin alone or with syntaxin and VAMP. Do these complexes support FRET interactions? The separated chains cerSN1 and citSN2 showed no tendency to FRET when coexpressed with syntaxin (Figure 8A). CerSN1 and citSN2 also did not FRET in combination with VAMP and syntaxin, despite the formation of the SDS-resistant SNARE complexes (Figure 7). The results suggest that the N-terminal fluorophores of the separated chains are not as closely apposed as they are in the complex with the CssYs, which has SN1 and SN2 on the same molecule.
Separated SN1 and SN2 Do Not Efficiently Rescue Secretion
The above experiments indicate that separated SN1 and SN2 interact with syntaxin differently than the linked construct (CssYs). If the changes in conformation of CssYs due to syntaxin are important for secretion, then one would predict that the separated SN1 and SN2 would be less able to rescue secretion than constructs containing both chains. We investigated the ability of the membrane-bound constructs citSN2 and cerSN1 to enable secretion in BoNT E expressing cells. CitSN2, alone or in combination with cerSN1, was not as effective as the full-length protein CssYs in restoring DMPP-induced secretion in toxin-expressing cells (Figure 8B). Although citSN2 alone rescued secretion by 22 ± 5% and the combination of cerSN1 and citSN2 rescued by 33 ± 8%, CssYs rescued secretion by 74 ± 10%. (Fractional recovery was calculated relative to the BoNT E–induced inhibition in the absence of construct; see Materials and Methods). SN1 and SN2 constructs lacking the fluorophores gave similar results (data not shown), whereas full-length SNAP25 (without an internal fluorophore) actually caused supramaximal rescue (Figure 1C). These data indicate that the full-length protein has a distinct functional advantage when compared with the separated chains of SNAP25, despite the ability of separated SN1 and SN2 to form stable complexes with other SNAREs.
The above results in intact cells differ from those in permeabilized PC12 cells, in which soluble SN2 rescues secretion (Chen et al., 1999). We therefore investigated in permeabilized cells the effectiveness of membrane-bound SN2 to support secretion. When secretion was stimulated by Ca2+ during permeabilization (Figure 8D), results were similar to those in intact cells (Figure 8C). In both cases citSN2 supported secretion poorly. However, when secretion was allowed to run down in permeabilized cells before stimulation with Ca2+, citSN2 supported an almost complete rescue (Figure 8E). The latter experiment suggests that when secretion is less robust, SN2 interacts with other SNAREs (including the membrane-bound cleavage product of BoNT E) sufficiently to enable secretion.
DISCUSSION
The present study addresses a gap in our understanding of the biochemistry and function of SNAP25: the implications of SNAP25 having two SNARE motifs on the same molecule. We created a membrane-bound, intramolecular FRET probe that reports on the folding of SN1 and SN2, thereby allowing the investigation of conformational changes in a natural membrane setting. In parallel experiments we determined the functionality in secretion of SNAP25 constructs with different folding characteristics by measuring their ability to rescue secretion in BoNT E–expressing cells. The findings indicate that linking SN1 and SN2 together in the same protein has at least two important outcomes. It facilitates the formation of complexes with syntaxin that brings the N-termini of SN1 and SN2 close together, as reflected by FRET in labeled probes. Second, it enables fast secretion in chromaffin cells. The basis for these conclusions is discussed below.
SNAP25-based FRET Probes Report on a Syntaxin-dependent Association of the N-Termini of SN1 and SN2
Unlike most previous studies using soluble SNARE domains to determine the structure of SNARE complexes in vitro, the present study investigated the structure of complexes of membrane-bound SNAREs. We used FRET to detect SNAP25 folding in membrane-bound SNARE complexes in vitro and in living cells. In both situations, syntaxin caused large FRET increases reflecting a conformational change that brings into close proximity the N-termini of SN1 and SN2. This effect of syntaxin required SN2 (in CssYs), and was reduced or abolished in truncated SNAP25 constructs (CssYs180 and CssY). The requirement for SN2 was not a result of an inability of the truncated constructs to interact with syntaxin because syntaxin formed SDS-resistant complexes with the truncated as well as the full-length constructs. Thus, the syntaxin-dependent FRET reflects specific, SN2-dependent conformations.
Membrane-bound VAMP Reverses the Syntaxin-dependent Close Association of Domains N-Terminal to SN1 and SN2
The cytosolic domains of VAMP, SNAP25 (SN1, SN2), and syntaxin (H3 domain) form a well-structured four-peptide, superhelical bundle. We expected that its formation would maintain or increase the close association of the N-terminal domains of SN1 and SN2 observed with syntaxin and CssYs alone. This was not the case. Instead, coexpression of membrane-bound VAMP together with syntaxin and CssYs prevented the FRET observed with syntaxin and CssYs alone. This effect of VAMP required an intact SN2 (Figure 4). One possible explanation is that VAMP competes with CssYs for syntaxin, thereby inhibiting complex formation with CssYs. However, 88% of the VAMP was associated with syntaxin/CssYs-containing SDS-resistant complexes (Figure 3B), strongly suggesting that a ternary complex is responsible for the decreased FRET.
The decreased FRET between complexes of CssYs, syntaxin, and VAMP compared with complexes of CssYs and syntaxin alone probably reflects a greater separation of N-termini of SN1 and SN2 in the former. If there is a 5-nm separation of the fluorophores in the presence of syntaxin (equal to the Förster distance for CFP and YFP; Tsien, 1998), then a further 2-nm separation due to interaction with VAMP would reduce the syntaxin-dependent FRET by 80%, to a level below our sensitivity. Indeed, supporting the notion of greater separation is the greater accessibility of the GFP epitope to antibody in complexes containing CssYs, syntaxin, and VAMP compared with complexes of CssYs and syntaxin (Figure 3B, compare lanes 2 and 3).
The appropriate interaction of the v-SNARE VAMP with t-SNAREs is a critical step in the secretory pathway. The FRET data suggest that part of that interaction may represent a loosening rather than a tightening of the SNAP25/syntaxin complex. If the VAMP-dependent separation of SN1 and SN2 occurs during secretion, it may take place after fusion, when VAMP diffuses into the plasma membrane (Allersma et al., 2004). This “cis” orientation corresponds to the orientation of VAMP in the present study. It is, however, also possible that the separation begins earlier, when VAMP on the granule membrane interacts with a postulated preformed syntaxin/SNAP25 complex (Fasshauer and Margittai, 2004;Pobbati et al., 2006) and thus may reflect an important conformational step in the pathway to fusion.
The Bidentate Structure of SNAP25 Increases Its Efficacy in Secretion
An important conclusion from this study is that the linkage of SN1 and SN2 facilitates their function in exocytosis. Linked constructs (citSNAP25, SNAP25, CssYs) were much better able than membrane-bound SN2 (alone or cotransfected with membrane-bound SN1) to rescue nicotinic agonist-induced secretion in BoNT E–expressing cells (Figures 1, C and D, and 8, B and C). Furthermore, expression of exogenous full-length SNAP25 (without an internal fluorophore, Figure 1C) substantially enhanced secretion, whereas cotransfected SN1 and SN2 (with or without fluorescent tags) were unable to enhance secretion (Figure 8B and data not shown). Thus, secretion from intact chromaffin cells is dependent on and limited by the amount of endogenous SNAP25, which can be compensated for by exogenous SNAP25 but not by SN2, either alone or together with SN1.
Experiments in permeabilized cells indicate that under some conditions the separated SNARE motifs of SNAP25 can interact to support secretion. When permeabilized cells were allowed to run down before stimulation with Ca2+, transfected SN2 almost completely rescued BoNT E–inhibited secretion (Figure 8E), presumably because SN2 interacted productively with the SN1-containing, BoNT E cleavage product. The enhanced ability to rescue was not simply an effect of permeabilization itself, because rescue was not as apparent in cells stimulated by incubation with Ca2+ during permeabilization (Figure 8D). The result is consistent with the ability of soluble SN2 to rescue secretion in permeabilized (cracked) PC12 cells (Chen et al., 1999).
We know that under conditions of rundown, the rates of secretion diminish as secretion becomes ATP-dependent (Holz et al., 1989; Bittner and Holz, 1992). Thus, the ability of SN2 to support secretion depends upon the requirements of the secretory pathway, with rescue possible when secretion is less robust. It is possible that relatively slow ATP- and NSF-dependent remodeling of SNARE complexes enables separated SN2 to engage the secretory machinery (Jun et al., 2007).
Unlinked SNARE motifs homologous to those in SNAP25 (Hay, 2001) are found in intracellular trafficking pathways that do not require fast responses to signaling events. What are the implications of linking the two SNARE motifs in SNAP25? Our data suggest two consequences. First, the intramolecular configuration enables unique complexes of SN1 and SN2 with syntaxin, because only intramolecular complexes undergo FRET. Second, intramolecular interactions between SN1 and SN2 are likely to be more rapid than intermolecular interactions. It is possible that both the distinct complexes and the more rapid interactions enable more rapid secretion.
FRET Changes upon Secretion
One of the reasons for developing the intramolecular FRET probe CssYs was to investigate the timing of SNARE interactions during exocytosis. On the basis of the present study, we expected to see increased FRET if stimulation increases interaction of CssYs and endogenous syntaxin, or decreased FRET when VAMP interacts with preformed complexes of CssYs and syntaxin. We detected neither an increase nor a decrease in FRET in the plasma membrane upon nicotinic stimulation. There are at least two possible explanations. The concentration of overexpressed CssYs was probably much greater than that of endogenous syntaxin, thereby causing a high fluorescence background. In addition, FRET measurements could not be made specifically at fusion sites.
A previous study using a probe similar to CssYs reported a 1–3% Ca2+-dependent increase in FRET that was not related to granule fusion (An and Almers, 2004). However, the small FRET change has been difficult to reproduce (Dr. Wolfhard Almers, personal communication). An and Almers also demonstrated an increase in FRET upon interaction with syntaxin or its H3 domain similar to the present study, but did not detect a change in FRET upon addition of the cytosolic domain of VAMP. There are methodological differences that may explain the results, including the use of soluble, truncated proteins in the previous study rather than the full-length, membrane-bound molecules used here.
Despite our inability to detect changes in FRET with a SNAP25-based probe during secretion, CssYs has promising characteristics that motivate future strategies. Future experiments will use high-sensitivity TIRF measurements, low expression of CssYs, and simultaneous optical measurement of exocytotic sites to detect the dynamics of the formation of a small number of SNARE complexes at the time and location of fusion events.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr. T.F.J. Martin for the gift of the plasmid encoding the BoNT E–resistant SNAP25 and Dr. David W. Piston (Vanderbilt University Medical Center, Nashville, TN) for the gift of the plasmid encoding cerulean. We also thank Dr. Paul F. Hollenberg (University of Michigan) for allowing us to use his spectrofluorimeter. We also acknowledge the Michigan Diabetes Research and Training Center for core laboratory support. This work was supported by National Institutes of Health Grants R01-DK050127 (R.W.H.) and R01-NS38129 (R.W.H. and D.A.).
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-04-0344) on July 2, 2008.
REFERENCES
- Allersma M. W., Wang L., Axelrod D., Holz R. W. Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection microscopy. Mol. Biol. Cell. 2004;15:4658–4668. doi: 10.1091/mbc.E04-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S. J., Almers W. Tracking SNARE complex formation in live endocrine cells. Science. 2004;306:1042–1046. doi: 10.1126/science.1102559. [DOI] [PubMed] [Google Scholar]
- Bittner M. A., Holz R. W. Kinetic analysis of secretion from permeabilized adrenal chromaffin cells reveals distinct components. J. Biol. Chem. 1992;267:16219–16225. [PubMed] [Google Scholar]
- Chen Y. A., Scales S. J., Patel S. M., Doung Y. C., Scheller R. H. SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell. 1999;97:165–174. doi: 10.1016/s0092-8674(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Chen Y. A., Scales S. J., Scheller R. H. Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron. 2001;30:161–170. doi: 10.1016/s0896-6273(01)00270-7. [DOI] [PubMed] [Google Scholar]
- De Haro L., Ferracci G., Opi S., Iborra C., Quetglas S., Miquelis R., Leveque C., Seagar M. Ca2+/calmodulin transfers the membrane-proximal lipid-binding domain of the v-SNARE synaptobrevin from cis to trans bilayers. Proc. Natl. Acad. Sci. USA. 2004;101:1578–1583. doi: 10.1073/pnas.0303274101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D., Otto H., Eliason W. K., Jahn R., Brunger A. T. Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J. Biol. Chem. 1997;272:28036–28041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- Fasshauer D. Structural insights into the SNARE mechanism. Biochim. Biophys. Acta. 2003;1641:87–97. doi: 10.1016/s0167-4889(03)00090-9. [DOI] [PubMed] [Google Scholar]
- Fasshauer D., Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J. Biol. Chem. 2004;279:7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- Griesbeck O., Baird G. S., Campbell R. E., Zacharias D. A., Tsien R. Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- Hay J. C. SNARE complex structure and function. Exp. Cell Res. 2001;15:10–21. doi: 10.1006/excr.2001.5368. [DOI] [PubMed] [Google Scholar]
- Hayashi T., McMahon H. T., Yamasaki S., Binz T., Hata Y., Sudhof T. C., Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz R. W., Bittner M. A., Peppers S. C., Senter R. A., Eberhard D. A. MgATP-independent and MgATP-dependent exocytosis. Evidence that MgATP primes adrenal chromaffin cells to undergo exocytosis. J. Biol. Chem. 1989;264:5412–5419. [PubMed] [Google Scholar]
- Jun Y., Xu H., Thorngren N., Wickner W. Sec18p and Vam7p remodel trans-SNARE complexes to permit a lipid-anchored R-SNARE to support yeast vacuole fusion. EMBO J. 2007;26:4935–4945. doi: 10.1038/sj.emboj.7601915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R. C., Scheller R. H. Structural organization of the synaptic exocytosis core complex. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- Mattheyses A., Hoppe A., Axelrod D. Polarized fluorescence resonance energy transfer microscopy. Biophys. J. 2004;87:2787–2797. doi: 10.1529/biophysj.103.036194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura K.M.S., Gonzalez L. C., Jr, May A. P., Scheller R. H., Weis W. I. Crystal structure and biophysical properties of a complex between the N-terminal SNARE region of SNAP25 and syntaxin 1a. J. Biol. Chem. 2001;276:41301–41309. doi: 10.1074/jbc.M106853200. [DOI] [PubMed] [Google Scholar]
- Pobbati A. V., Stein A., Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- Rettig J., Neher E. Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science. 2002;298:781. doi: 10.1126/science.1075375. [DOI] [PubMed] [Google Scholar]
- Rickman C., Meunier F. A., Binz T., Davletov B. High affinity interaction of syntaxin and SNAP-25 on the plasma membrane is abolished by botulinum toxin E. J. Biol. Chem. 2004;279:644–651. doi: 10.1074/jbc.M310879200. [DOI] [PubMed] [Google Scholar]
- Rizzo M. A., Springer G. H., Granada B., Piston D. W. An improved cyan fluorescent protein variant useful for FRET. Nat. Struct. Biol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Sollner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J. E. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Sutton R. B., Fasshauer D., Jahn R., Brunger A. T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 angstrom resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tsien R. Y. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Wick P. W., Senter R. A., Parsels L. A., Holz R. W. Transient transfection studies of secretion in bovine chromaffin cells and PC12 cells: generation of kainate-sensitive chromaffin cells. J. Biol. Chem. 1993;268:10983–10989. [PubMed] [Google Scholar]
- Xiao W., Poirier M. A., Bennett M. K., Shin Y. K. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat. Struct. Biol. 2001;8:308–311. doi: 10.1038/86174. [DOI] [PubMed] [Google Scholar]
- Xu T., Rammner B., Margittai M., Artalejo A. R., Neher E., Jahn R. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 1999;99:713–722. doi: 10.1016/s0092-8674(00)81669-4. [DOI] [PubMed] [Google Scholar]
- Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. Partitioning of Lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Zhang X., Kim-Miller M. J., Fukuda M., Kowalchyk J. A., Martin T. F. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/s0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Su Z., Zhang F., Chen Y., Shin Y. K. A partially zipped SNARE complex stabilized by the membrane. J. Biol. Chem. 2005;280:15595–15600. doi: 10.1074/jbc.M500736200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.