

Summary
We demonstrate that administration of a depleting antibody specific for c-kit leads to the highly efficient removal of host hematopoietic stem cells (HSCs) and high levels of donor HSC chimerism following transplantation.
Upon intravenous transplantation, hematopoietic stem cells (HSCs) can home to specialized niches, yet most HSCs fail to engraft unless recipients are subjected to toxic preconditioning. Here, we provide evidence that, aside from immune barriers, donor HSC engraftment is restricted by occupancy of appropriate niches by host HSCs. Administration of ACK2, an antibody that blocks c-kit function, led to the transient removal of >98% of endogenous HSCs in immunodeficient mice. Subsequent transplantation of these animals with donor HSCs led to chimerism levels of up to 90%. Extrapolation of these methods to humans may enable mild but effective conditioning regimens for transplantation.
Allogeneic bone marrow transplantation (BMT) generally requires conditioning of the recipient with cytoreductive treatments to prevent immunological rejection of the graft. Because these regimens can be associated with serious side effects (1), preparative treatments are often omitted for the treatment of diseases such as severe combined immunodeficiency (SCID), as these patients are incapable of rejecting donor grafts (2). Nevertheless, although large numbers of B and T lymphocytes are at least transiently generated, the levels of donor HSC engraftment are usually less than 1% following transplantation into unconditioned SCID recipients (3–6). Although several studies have concluded that transplanted HSCs can easily replace endogenous HSCs without conditioning (7, 8), earlier studies suggested that the availability of niches is a limiting factor to transplantation (9). Thus, we reasoned that donor HSC engraftment might be limited by the occupancy of appropriate niches by endogenous HSCs, and that the development of reagents that specifically remove host HSCs could lead to safer transplantation-based therapies for hematological and non-hematological disorders than those currently in use.
To address whether endogenous HSCs can be replaced by transplanted HSCs in a linear dose-dependent manner, unconditioned Rag2−/−γc−/− mice (10, 11) were transplanted with varying numbers of c-kit+lineage−Sca-1+ (KLS) CD34−CD150+ HSCs from GFP+ mice (12–16). Peripheral blood granulocyte chimerism was measured at 16 weeks post-transplant, which has been previously shown to accurately reflect donor HSC chimerism in this system (3, 15). Donor granulocyte chimerism increased measurably in doses of between 10 and 250 transplanted HSCs, but transplantation of more than 250 cells led to at most modest increases in chimerism (Fig. 1). Transplantation of 18,000 HSCs, representing approximately 70% of the total number of HSCs in an adult mouse (17, 18), led to a mean chimerism of only 3% (Fig. 1, top panel). Similar results were obtained through transplantation of unfractionated bone marrow (supporting online text and Fig. S1). These data suggest that without conditioning, HSC engraftment is limited by the number of saturable niches that are empty at the time of transplant or become empty as the transplanted cells still survive.
Figure 1. Available HSC niches can be saturated with donor HSCs.
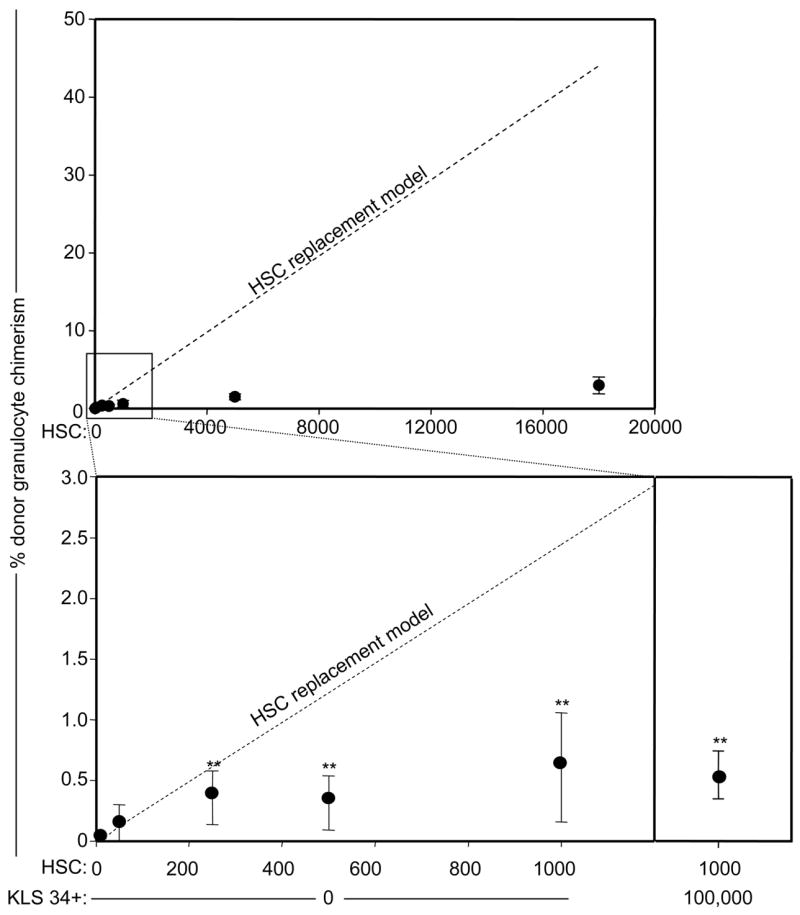
Peripheral blood of transplanted unconditioned RAG2−/−γc−/− mice was analyzed 16 weeks after HSC transplantation for GFP+ granulocytes. The bottom panel represents an expanded view of results from transplantation of 10–1000 HSCs. In the bottom right panel, mice were co-transplanted with CD45.1 1000 HSCs and 100,000 GFP+ KLS CD34+ cells. Mean values +/− SEM are shown (n = 4–5 for each dose); ** indicates p-value <0.05 relative to the chimerism arising from the 10 HSC-transplanted group. The dashed line represents the theoretical HSC chimerism if engraftment were to increase linearly with transplanted cell dose from the observed chimerism at the 50-HSC dose group.
To determine the specificity of these niches, we competitively transplanted unconditioned Rag2−/−γc−/− (CD45.2) mice with 1000 CD45.1 HSCs along with 100,000 GFP+ KLS CD34+ progenitor cells, which are the immediate progeny of HSCs (16, 19). No significant difference in donor HSC chimerism was observed in these mice relative to recipients that received 1000 HSCs alone (Fig. 1, lower right panel), suggesting that HSCs and their immediate progeny use distinct niches to maintain function.
To determine whether the specific elimination of host HSCs would allow for high levels of donor HSC engraftment, we compared a number of different candidate HSC-depleting monoclonal antibodies, including α-Sca1 (13), α-integrin α4 (20), and α-ESAM1 (21), but ultimately selected ACK2, an antibody known to recognize and antagonize c-kit (22), the receptor for stem cell factor (SCF) (23). We reasoned that if the ACK2 antibody were capable of depleting endogenous HSCs, residual antibody in the serum of mice would also deplete transplanted donor HSCs. To determine the kinetics of antibody clearance in vivo, we administered ACK2 intravenously to Rag2−/−γc−/− mice and tested the serum for the presence of antibody (12). ACK2 remained in the serum until five days post-injection; however no ACK2 was detected by 7–8 days after injection (Fig. 2A). All mice survived the ACK2 treatment with no obvious signs of distress (supporting online text, Table S1, and Fig. S2). At this time point, a ~99% decrease in the number of HSCs (Fig. 2B) was observed, and comparable levels of depletion were observed in B cell deficient and wild type mice (supplemental text and Fig. S3). To further confirm HSC depletion, at 9 days post-ACK2 treatment, 200,000 bone marrow cells were transplanted into lethally irradiated recipients (12). Transplantation of BM from ACK2-treated animals led to >90% lower engraftment relative to controls (Fig. 2C), thus confirming that ACK2 depletes functional HSCs from the BM.
Figure 2. ACK2 treatment depletes HSCs in vivo.
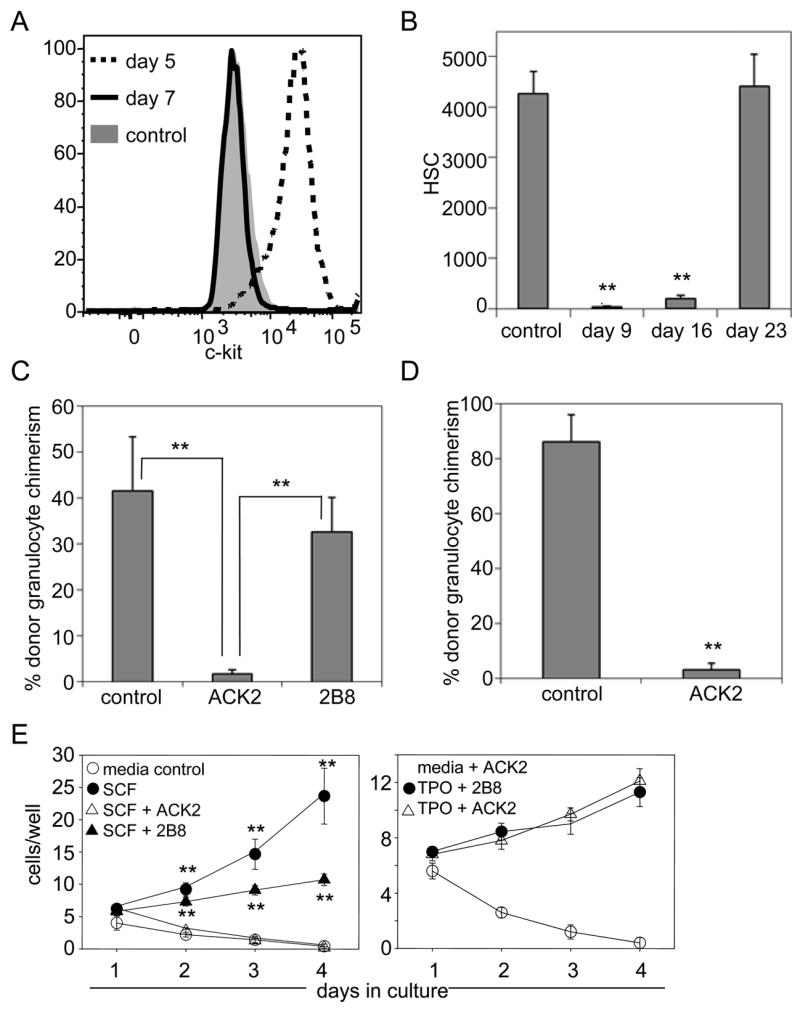
A) ACK2 is cleared from serum of RAG2−/−γc−/− mice seven days after injection. Serum of mice receiving 500μg ACK2 was analyzed every two days for ACK2 antibody by staining c-kit+ mast cells (31). B) ACK2 administration leads to depletion of BM HSCs. The number of KLS CD135− CD150+ HSCs in both femurs and tibia of ACK2-treated and control mice was determined. Mean values +/− SEM are shown (n = 3 for each time point); ** indicates p-value < 0.001. C) ACK2, but not 2B8 treatment, depletes functional HSCs from BM. 200,000 unfractionated bone marrow cells from RAG2−/−γc−/− mice treated with 500μg ACK2 or 2B8 nine days earlier were transplanted into wild type irradiated recipients alongside 200,000 untreated competitor wild type bone marrow cells. Mean values +/− SEM are shown (n = 5–8); ** indicates p-value <0.01. D) ACK2 treatment does not directly cause HSC mobilization to the spleen. Entire splenocyte populations from mice treated with 500μg ACK2 nine days earlier were transplanted alongside 200,000 competitor bone marrow cells from wild type mice. Mean values +/− SEM are shown (n = 3–9); ** indicates p-value <0.001. E) ACK2 inhibits SCF mediated HSC proliferation. HSC were isolated from wild type mice, plated at 10 cells/well cultured in the presence of SCF or TPO and ACK2 or 2B8. Proliferation was observed by light microscopy. ** indicates p-value <0.05 as compared to ACK2 treated samples.
To determine the mobilizing effects of ACK2 treatment, the entire splenocyte populations of ACK2-conditioned animals were transplanted into lethally irradiated recipients (12). The donor chimerism resulting from ACK2-treated splenocytes was reduced by >95% relative to controls (Fig. 2D). These data indicate that ACK2-treatment does not induce HSC depletion from the BM by mobilizing HSCs to the spleen. Moreover, we were unable to detect HSCs in the liver 9 days after ACK2 treatment by flow cytometry (data not shown). Mobilization was only observed during the recovery phase, well after ACK2 had already cleared from the serum (Fig. S4).
To determine the mechanism by which ACK2 depletes HSCs, we compared the effects of ACK2 treatment to that of 2B8, another c-kit monoclonal antibody of the same IgG2b isotype. 2B8 treatment did not decrease functional HSC numbers in vivo, as transplantation of bone marrow cells from mice treated with 2B8 resulted in levels of engraftment similar to those of controls (Fig. 2C). Next, we cultured purified HSCs in the presence of ACK2 and found that it completely inhibited SCF-dependent proliferation, but not thrombopoietin (TPO)-mediated proliferation (Fig. 2E) (12). These data, along with the data that distinct c-kit-expressing progenitors are depleted at differential rates (supporting online text and Fig. S5), suggest that ACK2 does not deplete through Fc-mediated functions. Rather, consistent with studies using c-kit mutant mice (24, 25) (supporting online text), the complete inhibition of c-kit signaling by ACK2 (Fig. 2E) can deplete HSCs while the partial inhibition by 2B8 cannot.
By 23 days post-treatment, HSC cell surface profiles (Fig. S4) and numbers (Fig. 2B) had returned to near normal levels. These data indicate that ACK2 causes a significant but transient depletion of host HSCs and results in a short window during which ACK2-treated animals might be receptive to donor HSC transplantation. To test whether the ablation of host HSCs could improve the efficiency of donor HSC engraftment, RAG2−/− (CD45.1) mice were conditioned with ACK2 and transplanted with 5000 wild type CD45.2 HSCs after serum clearance of ACK2. The mean donor granulocyte chimerism at 37 weeks post transplant was 16.1%, reflecting a >10-fold increase over control recipients (Fig. 3A). These engrafted HSCs also gave rise to peripheral B and T cells (Fig. 3B).
Figure 3. ACK2 treatment enhances HSC engraftment.
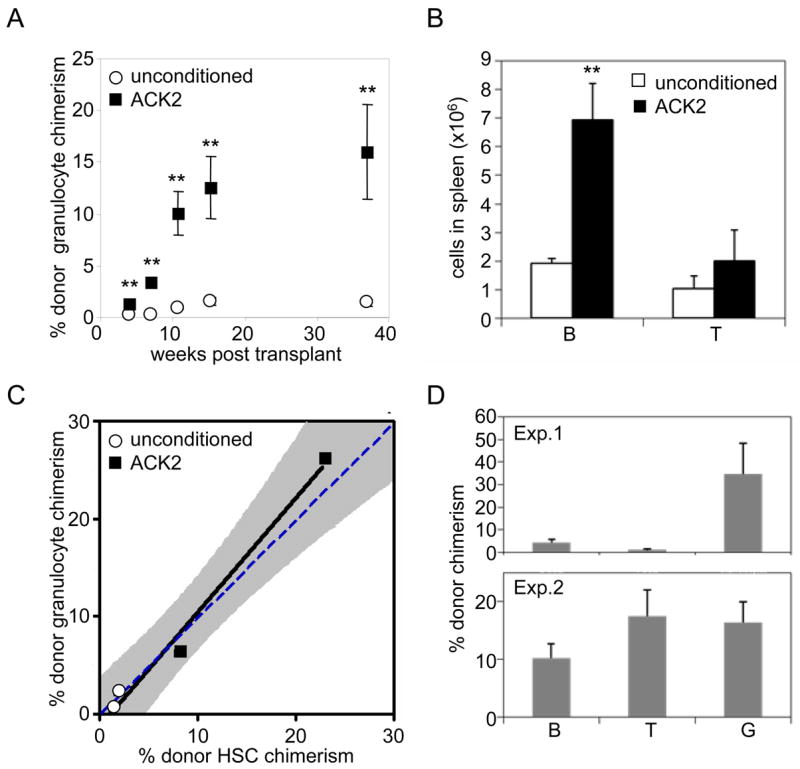
A) ACK2 conditioning leads to higher donor myeloid chimerism. Donor granulocyte chimerism was measured following transplantation of 5000 HSCs in RAG2−/− mice conditioned with ACK2 seven days prior to transplant and compared to that of unconditioned mice. Mean values +/− SEM are shown (n = 4); ** indicates p-value <0.01. B) HSC transplantation of ACK2-treated animals leads to lymphocyte reconstitution. Splenic donor-derived B and T-cells from ACK2-treated and unconditioned RAG2−/− mice were enumerated 39 weeks after transplantation with wild type HSCs. Mean values +/− SEM are shown (n = 3–5); ** indicates p-value <0.01. C) Granulocyte chimerism accurately measures BM HSC chimerism. Peripheral blood granulocyte (Ter119−CD3−B220−Mac-1highside scatterhigh) chimerism at 37 weeks post-transplantation was correlated with HSC (c-kit+ lineage− Sca-1+ CD34−CD150+) chimerism in the BM at 39 weeks post-transplantation upon sacrifice. Solid line illustrates linear regression with 95% confidence interval shaded in gray. Dashed line represents theoretical values if donor granulocyte chimerism were identical to donor HSC chimerism. D) Secondarily transplanted donor HSCs from ACK2 treated give rise to long term multilineage engraftment. Peripheral blood chimerism of B cells (B), T cells (T), and granulocytes (G) are shown 16 weeks post-secondary transplant for 2 independent experiments. Mean values +/− SEM are shown (n = 7–8 in each experiment).
We also analyzed BM HSC chimerism directly at this late time point to confirm the increase in engraftment, and found that it correlated well with peripheral blood granulocyte chimerism (Fig. 3C). Secondary transplants of donor HSCs, re-isolated from primary recipients led to multi-lineage engraftment for at least 16 weeks post-transplant (Fig. 3D), confirming that transplanted HSCs regain their normal cell surface phenotype and cell cycle status (Fig. S6) by at least 7–9 months post-transplant in ACK2-treated animals (12).
These data did not distinguish whether ACK2 treatment increased niche space or whether, as in unconditioned animals, only a small fraction of transplanted HSCs initially engrafted but then competitively expanded. If niche space had truly been freed, HSC chimerism would be expected to increase linearly with transplanted cell dose. To distinguish between these alternatives, ACK2-conditioned RAG2−/−γc−/− (CD45.2) recipient mice were transplanted with varying doses of CD45.1 LT-HSCs. Donor engraftment increased linearly with transplanted HSC dose in 2 independent experiments at both early and late timepoints (Fig. 4A), consistent with the emptying of HSC niches by ACK2 treatment. Strikingly, donor chimerism values of up to 90% were achieved at the 35,000 HSC dose (Fig. 4B), which, upon extrapolation to humans, is a clinically obtainable number (supporting online text and Fig. S7).
Figure 4. Donor chimerism increases with transplanted HSC cell number in ACK2-treated mice.

A) ACK2 treatment increases available HSC niche space. In two separate experiments, RAG2−/−γc−/− mice were treated with ACK2 and transplanted nine days later with varying doses of HSCs (CD45.1). Donor granulocyte chimerism was measured as above 24 weeks after transplantation for the first experiment, and 4 weeks for the second experiment. Mean values +/− SEM are shown. B) Flow cytometry profiles of mice transplanted with 35,000 HSCs. Chimerism of CD3−B220−Mac1high side scatterhigh peripheral blood granulocytes is shown.
Allogeneic BMT is used routinely for a number of clinical purposes, such as for the treatment of SCID (26, 27). Here, we provide evidence that in the absence of conditioning, donor HSC engraftment is limited by the occupancy of appropriate niches by host HSCs. These data offer an explanation for the poor donor HSC engraftment observed in unconditioned SCID patients (4, 5), which in turn may be responsible for the low levels of donor B lymphopoiesis and the finite duration of T cell production (6, 28).
We demonstrate above that administration of ACK2 in vivo leads to the rapid but transient depletion of host HSCs and subsequent transplantation of highly purified HSCs leads to donor chimerism levels of up to 90%. When coupled with highly specific immunosuppressive depleting antibodies, shown to be effective in both mice (29) and humans (30) as transplantation conditioners, the use of HSC-specific depleting antibodies may be an attractive alternative to conventional methods of conditioning, which carry significant health risks (1), and may thus increase the utility of allogeneic BMT for both hematological and non-hematological disorders.
Supplementary Material
References
- 1.Ferry C, Socie G. Exp Hematol. 2003 Dec;31:1182. doi: 10.1016/j.exphem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Buckley RH, et al. N Engl J Med. 1999 Feb 18;340:508. doi: 10.1056/NEJM199902183400703. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya D, Rossi DJ, Bryder D, Weissman IL. J Exp Med. 2006 Jan 23;203:73. doi: 10.1084/jem.20051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tjonnfjord GE, Steen R, Veiby OP, Friedrich W, Egeland T. Blood. 1994 Nov 15;84:3584. [PubMed] [Google Scholar]
- 5.Muller SM, Kohn T, Schulz AS, Debatin KM, Friedrich W. Blood. 2000 Dec 15;96:4344. [PubMed] [Google Scholar]
- 6.Cavazzana-Calvo M, et al. Blood. 2007 May 15;109:4575. doi: 10.1182/blood-2006-07-029090. [DOI] [PubMed] [Google Scholar]
- 7.Brecher G, Ansell JD, Micklem HS, Tjio JH, Cronkite EP. Proc Natl Acad Sci U S A. 1982 Aug;79:5085. doi: 10.1073/pnas.79.16.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart FM, Crittenden RB, Lowry PA, Pearson-White S, Quesenberry PJ. Blood. 1993 May 15;81:2566. [PubMed] [Google Scholar]
- 9.Micklem HS, Clarke CM, Evans EP, Ford CE. Transplantation. 1968 Mar;6:299. [PubMed] [Google Scholar]
- 10.Shinkai Y, et al. Cell. 1992 Mar 6;68:855. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 11.Goldman JP, et al. Br J Haematol. 1998 Nov;103:335. doi: 10.1046/j.1365-2141.1998.00980.x. [DOI] [PubMed] [Google Scholar]
- 12.Materials and methods and supplementary figures are available as supporting material on Science Online.
- 13.Spangrude GJ, Heimfeld S, Weissman IL. Science. 1988 Jul 1;241:58. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 14.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. Cell. 2005 Jul 1;121:1109. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 15.Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Science. 2001 Nov 30;294:1933. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- 16.Osawa M, Hanada K, Hamada H, Nakauchi H. Science. 1996 Jul 12;273:242. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 17.Colvin GA, et al. Leukemia. 2004 Mar;18:575. doi: 10.1038/sj.leu.2403268. [DOI] [PubMed] [Google Scholar]
- 18.Sudo K, Ema H, Morita Y, Nakauchi H. J Exp Med. 2000 Nov 6;192:1273. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi DJ, et al. Proc Natl Acad Sci U S A. 2005 Jun 28;102:9194. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams DA, Rios M, Stephens C, Patel VP. Nature. 1991 Aug 1;352:438. doi: 10.1038/352438a0. [DOI] [PubMed] [Google Scholar]
- 21.Forsberg EC, et al. PLoS Genet. 2005 Sep;1:e28. doi: 10.1371/journal.pgen.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogawa M, et al. J Exp Med. 1991 Jul 1;174:63. doi: 10.1084/jem.174.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witte ON. Cell. 1990 Oct 5;63:5. doi: 10.1016/0092-8674(90)90280-r. [DOI] [PubMed] [Google Scholar]
- 24.Fleischman RA, Mintz B. Proc Natl Acad Sci U S A. 1979 Nov;76:5736. doi: 10.1073/pnas.76.11.5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller CL, et al. Exp Hematol. 1996 Feb;24:185. [PubMed] [Google Scholar]
- 26.Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Lancet. 1968 Dec 28;2:1364. doi: 10.1016/s0140-6736(68)92672-x. [DOI] [PubMed] [Google Scholar]
- 27.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Lancet. 1968 Dec 28;2:1366. doi: 10.1016/s0140-6736(68)92673-1. [DOI] [PubMed] [Google Scholar]
- 28.Sarzotti M, et al. J Immunol. 2003 Mar 1;170:2711. doi: 10.4049/jimmunol.170.5.2711. [DOI] [PubMed] [Google Scholar]
- 29.Gambel P, Francescutti LH, Wegmann TG. Transplantation. 1984 Aug;38:152. [PubMed] [Google Scholar]
- 30.Cosimi AB, et al. N Engl J Med. 1981 Aug 6;305:308. doi: 10.1056/NEJM198108063050603. [DOI] [PubMed] [Google Scholar]
- 31.Nakano T, et al. J Exp Med. 1985 Sep 1;162:1025. doi: 10.1084/jem.162.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.We thank D. Bryder for helpful discussions and technical assistance, L. Jerabek for laboratory management, C. Richter for antibody production, L. Hidalgo, D. Escoto, 9 and J. Dollaga for animal care, C. Park for histological expertise, and M. Longaker and D. Rossi for critical reading of the manuscript. None of the material presented here has been published or is under consideration for publication elsewhere. This work was supported by National Institutes of Health grants 5R01HL058770 and 5R01CA086065 (to I.L.W.). A.C. was supported by a fellowship from the Medical Scholars Program at Stanford University School of Medicine, D.B. was supported by fellowships from the Cancer Research Institute and from the National Institutes of Health (T32AI0729022 and 5K01DK078318), and D.K. was supported by National Institutes of Health grant 5K08HL076335. Affiliations that might be perceived to have biased this work are as follows: I.L.W. owns significant Amgen stock, cofounded and consulted for Systemix, is a cofounder and director of Stem Cells, Inc., and cofounded and is a director of Cellerant, Inc. All other authors have no conflicting financial interests.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.