

Abstract
FXYD1 is a major regulatory subunit of the Na,K-ATPase, and the principal substrate of hormone-regulated phosphorylation by c-AMP dependent protein kinases A and C in heart and skeletal muscle sarcolemma. It is a member of an evolutionarily conserved family of membrane proteins that regulate the function of the enzyme complex in a tissue-specific and physiological-state-specific manner. Here we present the three-dimensional structure of FXYD1 determined in micelles by NMR spectroscopy. Structure determination was made possible by measuring residual dipolar couplings in weakly oriented micelle samples of the protein. This allowed us to obtain the relative orientations of the helical segments of the protein, and also provided information about the protein dynamics. The structural analysis was further facilitated by the inclusion of distance restraints, obtained from paramagnetic spin label relaxation enhancements, and by refinement with a micelle depth restraint, derived from paramagnetic Mn line broadening effects. The structure of FXYD1 provides the foundation for understanding its intra-membrane association with the Na,K-ATPase α subunit, and suggests a mechanism whereby the phosphorylation of conserved Ser residues, by protein kinases A and C, could induce a conformational change in the cytoplasmic domain of the protein, to modulate its interaction with the α subunit.
Keywords: FXYD1, Na, K-ATPase, structure, membrane protein, micelles, NMR, RDC
The Na,K-ATPase is the principal enzyme responsible for maintaining the gradient of Na and K ion concentrations across cell membranes. The energy required to pump ions into and out of cells is generated by the hydrolysis of ATP in the cytosol, through a process that involves distinct phosphorylated intermediates of the enzyme with different affinities for Na and K ions. The enzyme complex is formed by a catalytic α subunit (110 kD), formed by ten membrane spanning helices and two large cytoplasmic loops that fold into four distinct functional domains, an auxiliary β subunit (31 kD), with one membrane spanning helix and a highly glycosylated extracellular domain, and a third regulatory subunit (7–17 kD) which modulates the enzyme kinetics, with one membrane spanning helix and variable extra- and intra-cellular domains. The tissue-specific homologs of this third subunit belong to the FXYD protein family, which derives its name from a highly conserved signature sequence of amino acids (Phe-X-Tyr-Asp) preceding the transmembrane domain. The FXYD proteins, first discovered associated with the Na,K-ATPase in kidney membranes and in heart sarcolemma, have been the focus of recent attention due to their ability to finely regulate the activity of the enzyme complex in various physiological settings (1–4).
The FXYD genes are expressed predominantly in tissues that are electrically excitable, or that specialize in transport. Six of the seven mammalian FXYD family members, and the shark homolog, have been shown to interact specifically with the Na,K-ATPase and to modulate its functional properties in a tissue-specific and physiological-state-specific manner, and their effects on the enzyme’s rate constant, and its affinity for intracellular Na or extracellular K ions have been described (1–4). While some FXYD proteins increase the Na affinity, thereby activating the enzyme’s function at lower Na concentrations, others have the opposite effect, and these contrasting activities have been ascribed, in part, to specific differences in amino acids of the FXYD transmembrane domains, which are otherwise highly homologous throughout the protein family across species. The FXYD functions are further modulated by their cytoplasmic domains, which may associate with different regions of the α subunit to regulate enzymatic activity, and by post-translational modifications including phosphorylation (FXYD1) and glycosylation (FXYD5).
In heart and skeletal muscle sarcolemma, FXYD1 (PLM; phospholemman) is an integral part of the Na,K-ATPase enzyme complex (5, 6) where it plays a role in regulating muscle contractility. It is also the principal substrate of hormone-stimulated phosphorylation by c-AMP dependent protein kinases A and C (PKA and PKC) and other kinases (6–8), and its activity in different physiological settings is affected by its phosphorylation state (9–12). In addition, FXYD1, as well as at least three other FXYD family members, can induce ionic currents in Xenopus oocytes and in phospholipid bilayers (13, 14), however, the direct formation of ion channels has not been demonstrated in vivo, and recent evidence indicates that the role of FXYD proteins in ion transport regulation is solely related to their association with the Na,K-ATPase and other ion transporters.
High resolution structures of the α subunit nucleotide binding domain (15, 16) and of its fifth transmembrane helix (17), have been determined by NMR or x-ray crystallography, and the structure of the Na,K-ATPase complex has been determined at 9.5 Å resolution by cryo-electron microscopy analysis of two-dimensional crystals of native kidney membranes (18). In this structure, the electron density map clearly identifies the arrangement of the ten transmembrane helices of the α subunit, as well as distinct regions assigned to the transmembrane domains of the β and FXYD subunits. The map indicates that the FXYD protein associates with the α subunit in a groove, which was assigned to transmembrane helices M2, M4, M6, and M9 by homology modeling based on the 2.6 Å resolution crystal structure of the Ca-ATPase (19). This interaction has been confirmed by cross-linking and mutational studies, showing that the FXYD transmembrane domains associate in close proximity to transmembrane helices M2 and M9 of the α subunit (20, 21).
Recently, we reported that the helical structures of FXYD1, FXYD3, and FXYD4 in micelles mirror the intron-exon structures of their corresponding genes (22). Although the FXYD proteins are relatively small, ranging from about 60 to 160 amino acids, they are all encoded by genes with six to nine small exons, which is highly suggestive of modular gene assembly. The coincidence of helical regions, and connecting segments, with the positions of intron-exon junctions in the genes, indicates that the proteins are assembled from discrete structured domains, which may serve to confer different functional properties in various physiological settings.
Here we present the three-dimensional structure of FXYD1 determined in micelles by NMR spectroscopy. Structure determination was made possible by the measurement of residual dipolar couplings (RDCs) in weakly oriented samples of FXYD1 in micelles, to obtain the relative orientations of the helical segments of the protein. The orientation-dependent information, derived from RDCs, provides very high-resolution restraints for structure determination and refinement of both globular and membrane proteins, as well as detailed information about protein dynamics (23–26), and in our previous FXYD study, the RDCs measured from backbone sites in FXYD1, FXYD3, and FXYD4, were instrumental for the identification of helix breaks that map with the genetic structures (22). Typically, the selection of one correct orientation among the set of four symmetric degenerate solutions consistent with the RDC data, requires the measurement of RDCs in two different alignment media. In this study, however, the RDC analysis was facilitated by considering the protein structure in the frame of the lipid bilayer membrane, and by implementing a micelle depth restraint during structure refinement. The inclusion of this additional membrane-protein-specific restraint may be generally useful for structure determination with RDCs. The structure of FXYD1 provides some insight for the association of FXYD1 with the Na,K-ATPase α subunit, and paves the way for structure determination within the enzyme complex.
MATERIALS AND METHODS
Sample preparation
FXYD1 was expressed, purified, and characterized as described previously (27). SDS-PAGE was performed with the Tris-Tricine system (28), and gels were stained with Coomassie Blue. Solution NMR samples were prepared by dissolving pure lyophilized FXYD1 in 300 μL of buffer (20 mM sodium citrate pH 5, 10 mM DTT, 10% D2O, and 500 mM SDS). To measure RDCs, the samples were weakly aligned in 7% polyacrylamide gels by means of either vertical compression or expansion, as described previously (22). For the spin-label-induced paramagnetic relaxation enhancement (PRE) experiments, one of the cysteines in the sequence of FXYD1 was changed to serine (C40S), using the QuickChange mutagenesis kit (Strategene). The remaining single cysteine (Cys42) was modified by reaction with the paramagnetic spin label 1-oxy-2,2,5,5-tetramethyl-d-3-pyrroline-3-methyl (MTSL; Toronto Research Chemicals), or its acetylated diamagnetic analog 1-acetoxy-2,2,5,5-tetramethyl-d-3-pyrroline-3-methyl. Both reactions were performed in parallel, with two equal portions of purified FXYD1(C40S), as described by Tamm and coworkers (29). For the Mn-induced PRE experiments, the resonance intensities in the 1H/15N HSQC spectrum of FXYD1 were measured after the addition of MnCl2 to the micelle sample, to final concentrations of 0.5, 0.8, or 1.6 mM.
NMR experiments
The 1H/13C/15N NMR experiments, used to assign resonances, and measure chemical shifts, 1H/15N RDCs, hydrogen exchange profiles, and 1H-15N heteronuclear NOEs, for the FXYD family proteins, were described previously (22). NOEs were obtained using the three-dimensional 15N-NOESY-HSQC experiment (30) with a NOESY mixing time of 150 ms. NOE intensities were measured and calibrated using known inter-proton distances in regions of regular secondary structure, and converted into three distance restraint categories with ranges of 1.8–2.7 Å, 1.8–3.5 Å, and 1.8–5.0 Å. All NMR experiments were performed at 40 °C. The chemical shifts were referenced to the 1H2O resonance, set to its expected position of 4.5999 ppm at 40°C (31). NMR experiments were performed on a Bruker AVANCE 600 MHz spectrometer. The NMR data were processed using NMRPipe (32), and the spectra were assigned and analyzed using Sparky (33).
Analysis of MTSL-PRE data
The MTSL PRE data were analyzed as described by Battiste and Wagner (34). The global correlation time of the protein-detergent micelle (10 nsec) was taken from the values, measured by Girvin and coworkers, for micelle samples of membrane proteins with size and aggregation state similar to FXYD1 (35). The PRE distance restraints were divided in three categories with ranges of <15 Å, 15–21 Å, and >21 Å. In the working PRE range of 15–21 Å, a 50% uncertainty in the correlation time corresponds to an uncertainty of ±1 Å in the calculated distance. To account for uncertainty in the correlation time and in the conformation of the MTSL label, we used upper and lower limits of ±1.5 Å in the structure calculations.
Structure calculations
Structure calculations were performed with the program XPLOR-NIH (36). Order tensor analysis of the RDC data, and analysis of molecular fragment orientations, were performed with the program REDCAT (37). R factors for the RDC data were calculated as described by Clore and Garrett (38). Dihedral angles were derived from the analysis of chemical shifts with the program TALOS (39). Coordinate rotations and translations were performed using MacPyMol (DeLano Scientific LLC) or Fortran scripts. Structures were rendered in MacPymol. The protein surface electrostatic potential was calculated with the program GRASP (40), with inner and outer dielectric constants of 4 and 80, respectively. Structures were validated with PROCHECK (41). All calculations were performed on a 2.16 GHz Intel Core Duo MacBook Pro running OS X 10.4.8.
A starting structure was generated from extended random coil coordinates using a high-temperature simulated annealing protocol (42) with XPLOR-NIH internal dynamics (43). Dihedral angle restraints were imposed by a quadratic harmonic potential with a force constant of 400 kcal mol−1 rad−2. Hydrogen bonds for backbone sites in the transmembrane helix (residues 18–38) were derived from hydrogen exchange profiles (22) and imposed by restraining the O-HN distances to 2 ± 0.2 Å, and the O-N distances to 3 ± 0.2 Å, with a force constant of 50 kcal mol−1 Å−2. Distance restraints derived from NOEs were imposed by a flat-well harmonic potential with a force constant of 50 kcal mol−1 Å−2. The torsion angle database Rama potential (44), was used with a force constant of 0.2 to select preferred side-chain conformations relative to the backbone dihedral angles. Other force constants were as described by Nilges and coworkers (42).
A second simulated annealing step was performed at low-temperature (45), to impose RDC restraints with the XPLOR-NIH SANI potential (46). In this step, we used only the RDCs measured for residues in helical segments of the protein, with similar degree of order and values of the heteronuclear 1H/15N NOE greater than 0. During simulated annealing, the temperature was cooled from 300 to 20 K in steps of 10 K, with 7 ps of internal dynamics at each temperature, the RDC restraint force constant was ramped from 0.01 to 5.0 kcal Hz−2, the NOE and hydrogen bond distance restraint force constants were ramped from 2 to 20 kcal mol−1 Å−2, the dihedral angle restraint force constant was held at 300 kcal mol−1 rad−2, and the Rama potential force constant was held at 0.2. The other force constants were as described by Bax and coworkers (45).
A third simulated annealing step was performed with semi rigid-body internal dynamics, to link the molecular fragments in their correct relative positions. In this step, the coordinates of the backbone atoms (N, O, C, CA, HA, HN) of residues 13 to 43 were fixed, while those of residues 60 to 69 were grouped as a rigid body which was allowed to undergo translational, but not rotational, motion. The atoms in the side-chains, and in the termini and connecting loop, were allowed both rotational and translational degrees of freedom. Spin label PREs were imposed by restraining the OS1-HN distances between the paramagnetic oxygen atom of MTSL and the backbone amide proton, with a force constant of 40 kcal mol−1 Å−2. The MTSL parameter and topology files developed by Battiste and Wagner (34), and available with the XPLOR-NIH package, were used. Dihedral angle and NOE restraints were imposed with force constants of 400 kcal mol−1 rad−2 and 40 kcal mol−1 Å−2, respectively. Mn PRE restraints were implemented by applying harmonic coordinate plane restraints with a force constant of −300 kcal mole−1 Åe, to the backbone HN atoms of residues 60 and 68 to restrict their translational motion within an xz plane parallel to the membrane surface.
RESULTS AND DISCUSSION
Oligomerization state
FXYD1 has been shown to induce ionic conductance when it is expressed in Xenopus oocytes or when it is incorporated in planar lipid bilayers (13, 14), and similar observations have been made for its homologs FXYD2-4 (1–4), raising the possibility that channel formation could be another mechanism by which the proteins regulate ion transport across the plasma membrane. Recent studies, however, showed that FXYD3 has no effect on chloride conductance across the membranes of T3M4 pancreatic cancer cells where it is overexpressed (47), and that FXYD4 modulates ionic currents through its interaction with the KCNQ1 depolarization activated channels (48). Furthermore, ion channel formation would entail protein homo-oligomerization, however, there is no evidence of FXYD1 or other FXYD proteins associating as homo-multimeric assemblies, free from the Na,K-ATPase. In contrast, phospholamban, the Ca-ATPase regulatory protein to which channel activity is also ascribed, can adopt a pentameric structure in SDS and other detergents (49–51).
On SDS-PAGE, in the presence of reducing agent, FXYD1 migrates with an estimated molecular mass corresponding to monomeric protein (8.4 kD; Figure 1C), while in the absence of reducing agent, bands corresponding to dimers and higher order aggregates, up to hexamers (50.4 kD; Figure 1B), are also seen. We have observed similar results for the other family members, FXYD2 to FXYD5, indicating that they are also monomeric in SDS, and that oligomerization is through the formation of disulfide bonds between the free Cys side-chains. Indeed, we have found that a mutant of FXYD3, where all four cysteines in the sequence are changed to serine, always migrates as a single band corresponding to monomer, in both SDS and PFO, a detergent that has been shown to maintain the quaternary oligomeric structures of membrane proteins (52). In all FXYD proteins the cysteines are exposed to the reducing environment of the cytoplasm or the plasma membrane, and do not participate in disulfide bonds. Two independent electrophoresis studies found that 20-residue peptides corresponding to the transmembrane domains of FXYD1 (53) or FXYD2 (54), where no cysteines are present, migrate as monomers in SDS, but form oligomers in PFO. While the results from the FXYD1 peptide were suggested to reflect channel formation, the propensity for self-association in the FXYD2 peptide was interpreted as a reflection of its ability to associate with the transmembrane domain of the Na,K-ATPase α subunit, rather than intrinsic channel activity.
Figure 1.
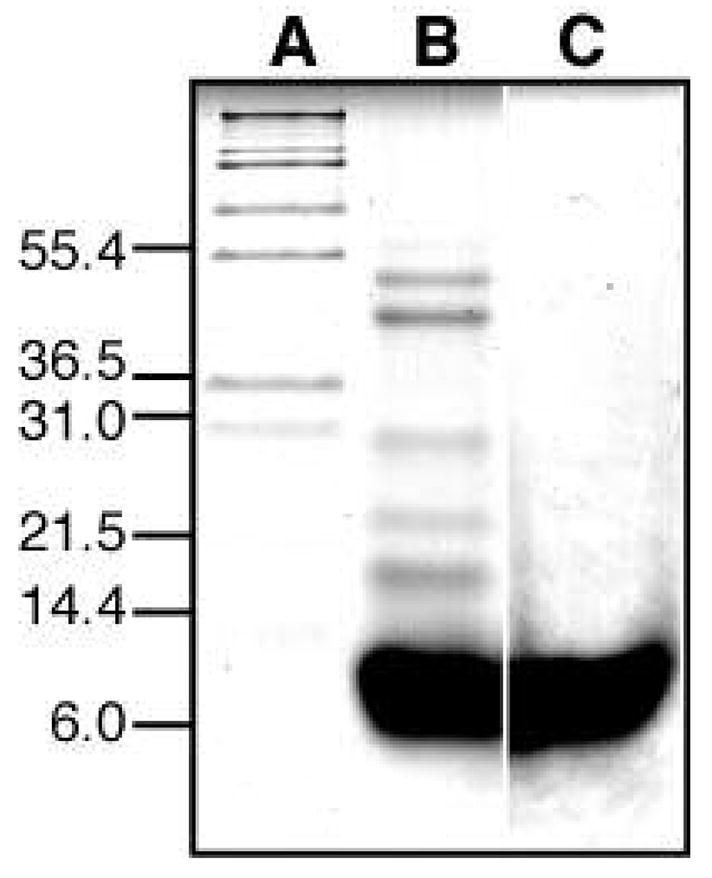
SDS-PAGE of purified FXYD1 obtained with or without the reducing agent β-mercaptoethanol. (A) molecular weight markers. (B) FXYD1 without β-mercaptoethanol. (C) FXYD1 with β-mercaptoethanol.
In NMR studies of membrane proteins, very high detergent concentrations are typically used to ensure the presence of no more than one protomer per micelle (one protein per micelle for monomers, or one protein complex per micelle for oligomers). In our NMR samples, where the critical micelle concentration is estimated to be 4 mM and the aggregation number 90 (55), the probability of finding more than one protein per micelle is 0.93 at 25 mM SDS, but decreases to 0.08 at 200 mM SDS, and plateaus to 0.02 at 400 mM SDS.
Association with the micelle
We examined the 1H/15N HSQC spectra of FXYD1 to FXYD4 in several well-characterized micelle-forming detergents, and obtained the best results in SDS micelles (56). Although SDS is widely assumed to be a universal protein-denaturing detergent because of its common use in protein electrophoresis, many integral membrane proteins retain their structures in SDS micelles (57). Since the majority of functional and structural studies of the Na,K-ATPase have been done on enzyme purified in the presence of SDS (18), and the non-covalent associations of α, β and FXYD subunits are maintained through the SDS purification process, we reasoned that this detergent would also be a good choice for FXYD structural studies.
Previously we showed that the secondary structure of FXYD1 mirrors its genetic organization, with helix breaks occurring at the sites of the intron-exon junctions of the gene (22). The overall structural features are summarized in Figure 2B. The transmembrane helix (H2) is preceded by a short helical segment (H1) of approximately five residues, beginning at Asp12 of the FXYD signature sequence, and is followed by a third short helix (H3), spanning the conserved basic RRCRCK sequence of the protein. The 1H/15N heteronuclear NOEs (Figure 2C) and the HSQC peak intensities (Figure 2D; black bars) indicate that H1, H2, and H3 have similar backbone dynamics. A fourth helix (H4), containing the S63 and S68 consensus sites for phosphorylation by PKA and PKC, is separated from H3 by a flexible linker with significantly smaller values of the 1H/15N NOE, and significantly greater peak intensities.
Figure 2.
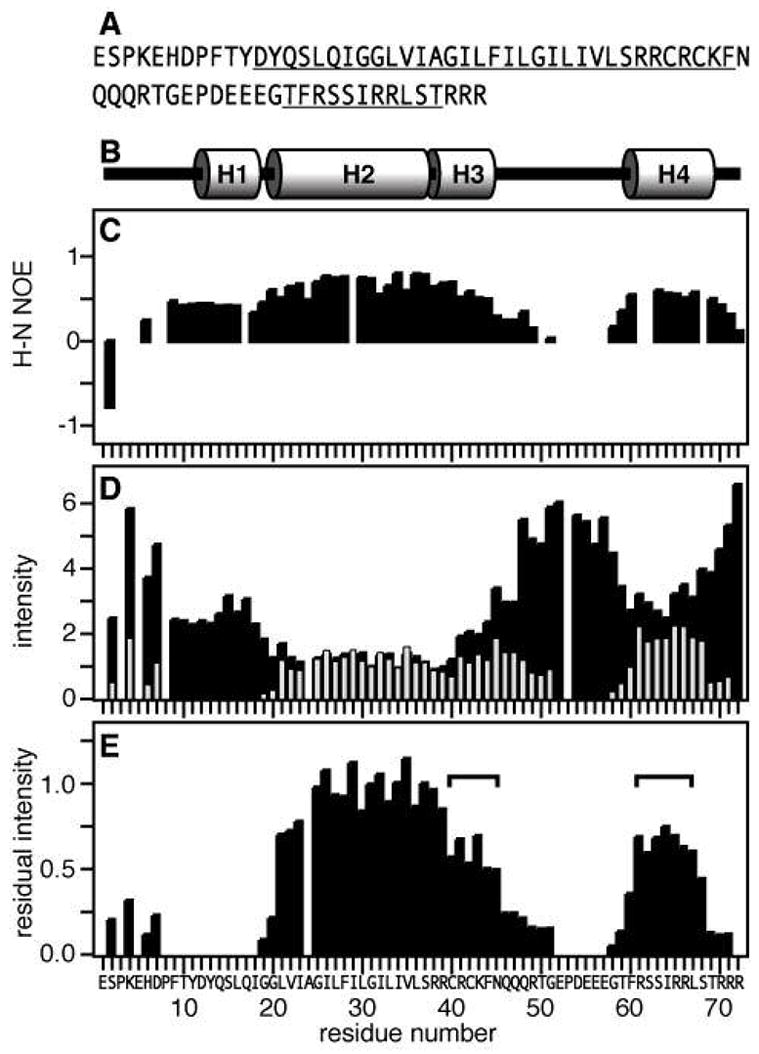
Effect of Mn-induced PRE on the resonance intensities. (A) Amino acid sequence of human FXYD1 with helical regions underlined. Residue numbering begins at 1 after the signal sequence (NCBI protein accession: NP_068702). (B) Protein secondary structure. (C) 1H/15N heteronuclear NOEs. (D) Normalized 1H/15N HSQC peak intensities obtained without (I−Mn, black bars) or with (I+Mn, gray bars) 1.6 mM MnCl2. Positions that are left blank correspond to prolines (P3, P8, P53) or overlapped resonances (E5, A24). (E) Residual normalized peak intensity (I−Mn/I+Mn). The horizontal square brackets mark residues in H3 and H4 with similar protection from aqueous Mn.
To probe the association of FXYD1 with the micelle, we examined the effect of MnCl2 on the 1H/15N HSQC spectrum. The paramagnetic electrons in the Mn ion induce distance-dependent broadening of peaks from protein sites that are solvent-exposed, while residues in the hydrophobic interior of the micelle, such as those in transmembrane helices, are mainly unaffected. The addition of MnCl2 to FXYD1 resulted in substantial line broadening and disappearance of peaks from amino acids in the N- and C-terminal regions of the protein, in H1, and in the flexible linker (Figures 2D and 2E). In contrast, peaks from H2 and H3 retained significant or full intensity, indicative of association with the micelle. Notably, the peaks from residues in H4 also retained much of their intensity, indicating that this region of the protein also associates strongly with the hydrophobic micelle.
The Mn PRE profile in Figure 2E displays the residual intensity measured for each peak after the addition of MnCl2, and reflects the depth of insertion of the respective protein sites in the micelle. The profile indicates that residues 60 to 68 in H4 are protected from interaction with the aqueous Mn ions to a similar extent as residues 39 to 45 in H3, indicating that both segments are buried at a similar depth within the micelle. Residues 22 to 38, in the transmembrane helix (H2), are buried deeper in the micelle, since their amide HN resonances retain nearly 100% of their intensity after addition of MnCl2. In contrast, peaks from H1, and from sites in the flexible loop and in the termini, experience a total or substantial loss of resonance intensity in the presence of Mn, reflecting the exposure of these sites to the aqueous environment. Since the Mn PRE profile effectively serves as a ruler for the depth of insertion of FXYD1 in the micelle, we implemented it as a restraint to define the relative positions of the helices in the structure calculations.
Structure determination
Structure determination relied primarily on restraints from RDCs, dihedral angles, H/D exchange measurements, Mn PREs, and MTSL PREs. No long-range NOEs could be identified connecting helix H4 to the rest of the protein across the 10-residue flexible linker. In a first stage of simulated annealing, a starting structure was generated using dihedral angles, hydrogen bonds, and short- or medium-range NOEs, but no RDCs. Order tensor analysis of the RDC data yielded similar values of the alignment tensor’s magnitude (Da = 12.7), rhombicity (R = 0.4), and generalized degree of order (GDO = 0.00074), for residues in the helical regions of the protein, indicating that they experience similar ordering in the aligned detergent micelles. Since H4 is connected to the rest of the protein by a long flexible segment, the observation of a common order tensor reflects the independent association of the transmembrane and cytoplasmic helical regions with the micelle. In contrast, the flexible loop and termini have a substantially reduced and completely different degree of order. A second stage of simulated annealing, performed at low temperature with the RDC restraints, yielded a refined structure with excellent agreement (RMSD = 0.31; R factor = 0.07) between experimental and calculated RDCs across all four helical regions of the protein (Figure 3).
Figure 3.
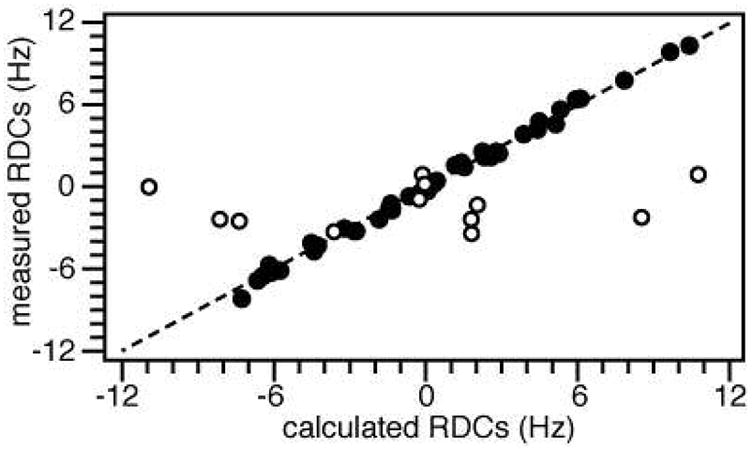
Correlation between HN RDCs measured experimentally and HN RDCs back-calculated from the refined structure of FXYD1. The RDCs from the flexible linker and termini (white circles) correlate poorly with those from the helical region of the protein (black circles; RMSD = 0.31; R factor = 0.07).
The relative orientations of helices H1 to H3 are well defined by the dihedral angles and the RDCs. However, since H4 is loosely connected to the rest of the protein, this initial refined structure represents only one of four possible orientations of H4 that are consistent with the RDC data. To determine the orientation of helix H4, two molecular fragments were generated by dividing the coordinates of the refined initial structure into two sets, for independent order tensor analysis with the experimental RDCs.
Fragment A (residues 1 to 50) includes helices H1 to H3, and fragment B (residues 51 to 72) includes the flexible linker and helix H4. Rotation of each molecular fragment into the principal axis frame of its order tensor, followed by 180° rotations around the principal Sxx, Syy, or Szz axes to generate four degenerate orientations, yielded four possible combinations of fragment A with fragment B, each consistent with the experimental RDCs (Figure 4). The selection of a single fragment combination among these four degenerate solutions was made possible by further rotating the fragments in the frame of the membrane, and considering the Mn PRE profile, as well as the amphiphilic character of H4 which dictates the sidedness of its association with the membrane-water or micelle-water interface.
Figure 4.
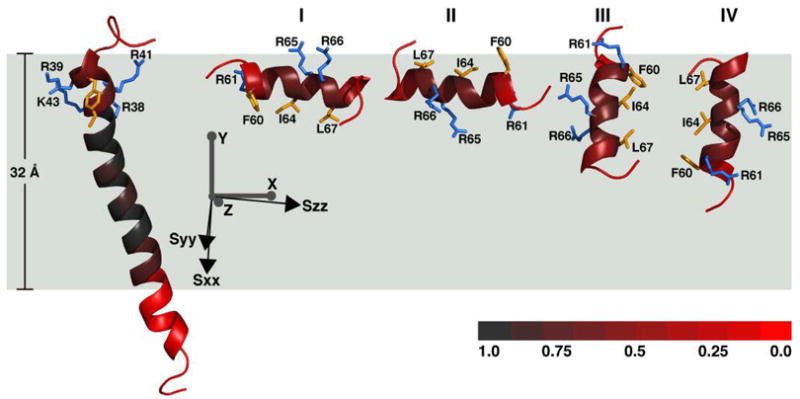
Molecular fragments of FXYD1 oriented in the frame of the alignment tensor (Sxx, Syy, Szz; black axes) and of the membrane (X, Y, Z; gray axes). Fragment A (shown at left) can be linked with one of four possible orientations of fragment B (I to IV). The red color gradation reflects the degree of protection from Mn-induced PRE, with full protection in the center of the membrane (gray), and no protection outside the membrane (red). The total Mn protection profile corresponds to a length of 32 Å along the membrane Y axis. Side-chains from basic residues in helix H3 (R37, R39, R41, K43), and helix H4 (R61, R65, R66), are shown in blue. Side-chains from apolar residues in H3 (C40, C42, F44), and H4 (F60, I64, L67) are shown in yellow.
The absolute orientation of Fragment A in the membrane is not determined by the solution NMR data obtained in micelles, however, we found that by viewing the structure in a membrane frame, there is only one solution for fragment B (Figure 4, solution I) that can simultaneously satisfy both the Mn PRE data and the amphiphilic polarity of amino acids in helix H4, with apolar residues (Phe60, Ile64, Leu67) facing the membrane interior, and charged residues (Arg61, Arg65, Arg66) facing the aqueous exterior. The Arg hydrocarbon side-chains are sufficiently long to reach the membrane surface, while allowing H4 to penetrate the membrane below the water-lipid interface. Solution I is also consistent with the solid-state NMR spectra of FXYD1 in lipid bilayers, which indicate the presence of a helical segment associated with the membrane surface (56). Solution II, on the other hand, has an opposite amphiphilic polarity to the membrane, with apolar side-chains facing out and charged side-chains facing in.
The membrane Y and Z axes were selected to allow the transmembrane helix to cross the membrane at an angle of 15°, in agreement with the solid-state NMR data for FXYD1 and other FXYD proteins in oriented lipids bilayers (56). Rotation around the membrane X axis satisfies both the 15° transmembrane tilt and the amino acid amphiphilic polarity of H4. The resulting orientation of fragment A also satisfies the amphiphilic character of H3, allowing the charged Arg and Lys side-chains (Arg38, Arg39, Arg41, Lys43) to point away from the membrane interior and into the aqueous phase, which, in vivo, corresponds to the cytoplasm.
In the membrane frame of reference, it becomes apparent that solutions III and IV of fragment B both send H4 plunging into the hydrophobic interior of the membrane, in violation of the Mn PRE data. For example, to satisfy the Mn PRE profile of Arg61, whose amide peak retains 60–70% of its intensity in the presence of Mn, solution III would have to place Thr69, which is nearly fully exposed to Mn, at the center of the membrane hydrophobic core, at a similar depth as residues in the transmembrane helix that are fully protected. A similar but opposite situation is encountered with solution IV, which would have to place Thr59 (~90% exposed) in the hydrophobic interior of the micelle to satisfy the Mn PRE profile of Leu67 (~70% protected).
The molecular fragments were linked in their correct relative orientations and positions by a third and final simulated annealing calculation, where the backbone coordinates of residues 13 to 44 (H1 to H3) were fixed, while those of residues 60 to 69 (H4) were allowed to undergo only translational motions as a rigid group, and atoms in the termini, flexible loop, and side-chains were free to undergo both rotational and translational motions. In this step, the dynamics were restrained by the PRE-derived distances between the MTSL label at Cys42 and residues in H2 and H4, by dihedral angle restraints, and by short- to medium-range NOE distance restraints.
In addition, the Mn PRE micelle depths were implemented as harmonic coordinate plane restraints for the backbone HN atoms of residues 60 and 68, to confine residues in H4 within the same XZ plane, parallel to the membrane surface, as that of residue 42, which experiences a similar Mn PRE effect. The Mn PRE profile effectively places residues in H4 at a similar depth as residues 39 to 45 in H3. The resulting structure of FXYD1 in micelles is represented in Figures 5 and 6.
Figure 5.
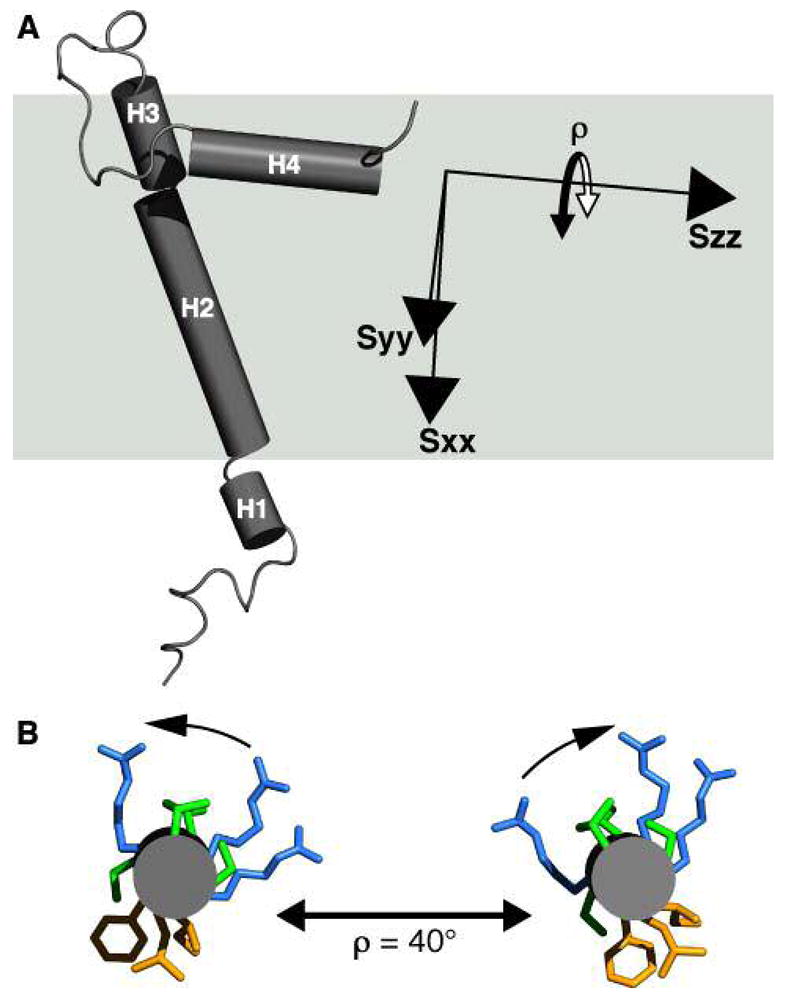
Tube representation of the FXYD1 structure in the frame of the membrane (gray box), and of the alignment tensor (Sxx, Syy, Szz). (A) Helix H4 is nearly parallel to Szz. This is consistent with helix reorientation around Szz, resulting both in a GDO value similar to that of helices H1–H3, as well as greater dynamics observed from the heteronuclear NOEs and resonance intensities. (B) Helix H4 viewed from the C-terminus. A maximum helix reorientation of ρ≈ 40°, around Szz, maintains the amphiphilic polarity of hydrophobic and hydrophilic side-chains at the membrane surface.
Figure 6.
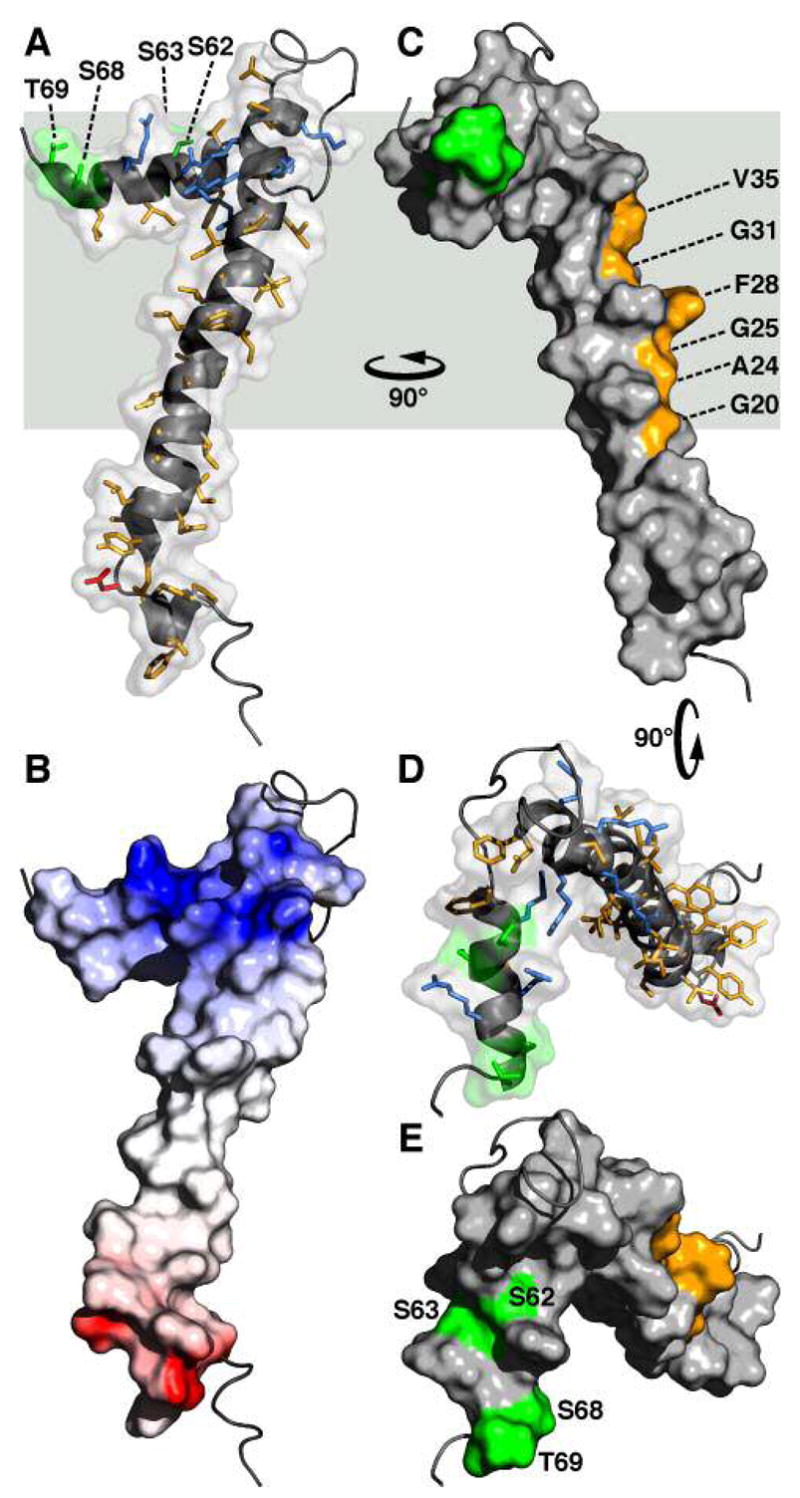
Molecular backbone and surface representations of FXYD1. (A, D) In the helical regions, basic side-chains are shown in blue, acidic side-chains are red, and apolar side-chains are yellow. The three Ser residues (S62, S63, S68) and Thr69 in the cytoplasmic helix are in green. (B) The surface is color coded with regions of electrostatic potential <−8kBT in red, and regions of electrostatic potential >+8kBT in blue, where kB is the Boltzmann constant and T is the temperature. (C) The structure is viewed 90° around the membrane Y axis from (A, B). Residues in the transmembrane helix (G20, A24, G25, F28, G31, V35), predicted to interact with the Na,K-ATPase α subunit are shown in yellow. (D, E) The structures are viewed down the membrane surface from the cytoplasm, 90° around the membrane X axis from (C). The structure has been deposited in the databank (PDB accession code: 2JOL).
Implications for protein dynamics
RDCs contain information about molecular orientation and dynamics, and are sensitive to motions spanning a wide range of time scales. For FXYD1, while the RDC data indicate that helix H4 experiences a similar degree of order in the micelle as the transmembrane helix, the heteronuclear NOEs and resonance intensities indicate increased dynamics in this region. The presence of different degrees of internal motions between two domains in a protein can affect the parameters describing the degree of order and mean orientation of each domain relative to the magnetic field, and Tolman and coworkers have discussed these effects in detail (26).
The effects of dynamics on the generalized degree of order depend on the direction of the axis for internal dynamic reorientation relative to the alignment frame; coincidence of the reorientation axis with the Szz principal axis of the alignment tensor has the minimum effect on degree of order, while coincidence with the Sxx alignment axis produces the maximum effect. As seen in Figures 4 and 5, the long axis of helix H4 is nearly parallel to the Szz axis of the FXYD1 alignment tensor, therefore, a reorientation of H4 around its length, on the membrane surface, would be consistent with both a minimum effect on the degree of order, and with the observation of greater dynamics in this region of the protein. While the extent of helix H4 reorientation cannot be gauged by the data, inspection of the structure suggests that rotational excursions as large as 40° (ρ = 40°; Figure 5B) would be consistent with maintaining the helix amphiphilic polarity with respect to the micelle surface, with apolar residues facing the membrane interior and polar residues facing out.
Structure of FXYD1
Different representations of the resulting structure for FXYD1 in micelles are shown in Figure 6. The structure coordinates have been deposited in the Protein Data Bank (PDB accession code: 2JOL). The FXYD motif forms a disordered segment, albeit with some propensity for helical structure, preceding the short helix H1 (Figure 6A). The function of the FXYD sequence is still unclear, although one study has shown that it is required for the stable interaction of FXYD2 or FXYD4 with the α subunit (58). Alternatively the FXYD sequence could constitute a sorting or localization signal.
In the transmembrane helix of FXYD1, the Phe28 aromatic ring forms a significant protrusion out of a groove lined by small residues (Gly20, Ala24, Gly25, Gly31, Val35) along one side of the helix length (Figure 6C). The Gly residues at positions 20 and 31 are fully conserved in the sequences of all FXYD proteins across species (Figure 7), and positions 24, 25, and 35 typically take small side-chains (Ala, Gly, Val or Leu). Position 28, on the other hand, displays more variability in the type and size of amino acid that it can accommodate, and is occupied by Phe in FXYD1, FXYD2 and FXYD5-7, or by the smaller residues Cys and Ala in FXYD3 and FXYD4 (human sequences). A recent study showed that a Cys residue placed at position 28 (FXYD1 numbering) in the transmembrane helices of FXYD1, FXYD2, and FXYD4, allows cross linking to the α subunit M2 transmembrane helix (20), as first suggested by the cryo-electron microscopy structure (18).
Figure 7.
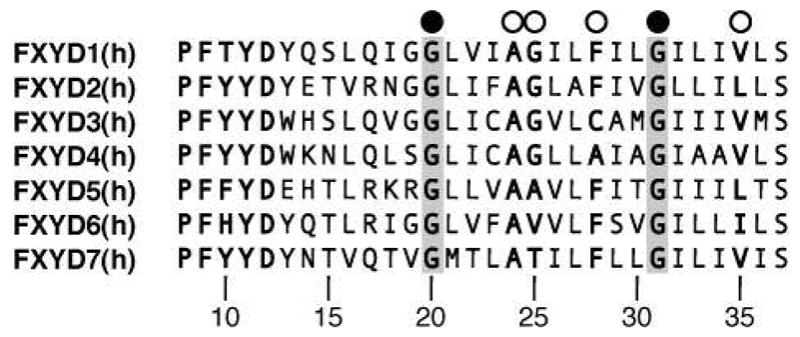
Amino acid sequences of the transmembrane domains from the human FXYD proteins. The fully conserved Gly residues, at positions 20 and 31, are highlighted in gray, and marked by black circles. Other key residues, at positions 24, 25, 28 and 35, are marked by white circles. Residue numbering is according to FXYD1.
The very high conservation of amino acids in the transmembrane helices of all FXYD proteins suggests their involvement in specific intramembrane helix-helix interactions, with the Na,K-ATPase, and possibly other partners, and it is tempting to speculate that the specific activities of the FXYD proteins are related, in part, to subtle differences in their transmembrane sequences. For example, while FXYD2 (Phe at position 28) reduces the Na affinity of the Na,K-ATPase, FXYD4 (Ala at position 28) increases it, and their opposing activities and unique expression patterns in distinct segments of the kidney help define the physiological differences in Na,K-ATPase activity among the kidney nephron segments (1–4). Furthermore, in kidney membranes the transmembrane helix of FXYD2 associates with the transmembrane domain of the α subunit, while the mutation G41R (corresponding to position 31 in FXYD1) causes misrouting of the protein by inhibiting its association with the Na,K-ATPase, and is linked with renal or intestinal familial hypomagnesemia (59). The data, therefore, indicate that the conserved Gly residue at this position plays a role in mediating helix-helix interactions between the FXYD transmembrane helix and the Na,K-ATPase. The structure of FXYD1 shows that Gly31 and the other conserved transmembrane residues line one strip along helix H2 that is unencumbered by the cytoplasmic helix H4, and free to interact with the α subunit.
Helix H4 associates with the micelle surface, and is buried in the micelle to a similar extent as residues 39 to 45 at the end of the transmembrane segment. The Arg and Lys side-chains from residues 38, 39, 41, 43 in H3, and residues 61, 65, 66 in H4, are sufficiently long to reach the membrane surface with their positively charged groups, thus anchoring these segments to the cytoplasmic lipid-water interface. Phosphorylation of FXYD1 by PKA and PKC has been shown to affect both its Na,K-ATPase regulatory activity, and possibly, its mode of association with the α subunit (9–12). In the structure, the cytoplasmic Ser residues that are phosphorylated by PKA (Ser68) or PKC (S63, S68), point to opposite directions of H4 (Figures 6D, 6E), and are exposed to the surface and available for interaction with the kinases. The other two potential phosphorylation sites, S62 and T69, are also surface exposed.
The role of FXYD1 may be analogous to that of phospholamban, which regulates the sarcoplasmic reticulum Ca-ATPase. However, unlike FXYD1, phospholamban can exist either as a monomer or as a homo-pentamer, which may represent a channel active form of the protein. Monomeric phospholamban forms an L-shaped helical structure, where a short N-terminal intracellular helix associates with the membrane surface, and is linked to the transmembrane helix by a short segment which contains the Ser phosphorylation site (60). Phosphorylation increases the dynamics of the linker (61), and could predispose the protein for a conformational rearrangement of the helices. On the other hand, in the pentameric form the N-terminal helix is not membrane associated, but extends out from the membrane to form a funnel-like channel structure (49). The observation of two types of helix orientations in phospholamban indicates a propensity for conformational rearrangement.
The cytoplasmic helix of FXYD1 is highly basic, which helps explain its propensity to associate with the negatively charged micelle surface, or the cellular plasma membrane. However, it is connected to the rest of the protein by a relatively long flexible linker, and therefore would be capable of undergoing reorientation to interact with the cytoplasmic domain of the α subunit. Helix reorientation from the membrane surface could be triggered by phosphorylation, which introduces repulsive negative charges at S63 and S68 in H4. Indeed, a recent study, measuring fluorescence energy transfer between fluorescent-protein-linked FXYD1 and the α subunit, suggests that phosphorylation of FXYD1 by either PKA or PKC may cause a conformational change in their inter-molecular association (12).
Interestingly, the binding of both PKC and its substrate polypeptides to acidic phospholipids is important for phosphorylation (62), and it has been suggested that this lipid binding event may serve not only to co-localize the enzyme and the substrate, but also to stabilize a specific substrate conformation that can be recognized by PKC (63). Thus, the membrane surface association of H4 could be important for FXYD1 substrate recognition by PKC, while the addition of negatively charged phosphate groups could trigger helix reorientation, thereby providing a mechanism for modulating the interaction of FXYD1 with the α subunit.
CONCLUSIONS
The structure of FXYD1 is the first to be determined for the FXYD family of Na,K-ATPase regulatory subunits, and highlights some key features that suggest its mode of association with the α subunit of the enzyme. A long groove formed by highly conserved amino acids runs parallel to the length of the transmembrane helix, delineating the likely binding interface of FXYD1 with the α subunit. The groove is interrupted by the aromatic ring of Phe28, which protrudes from the transmembrane helix near the middle of the hydrophobic lipid core. Cross-linking experiments indicate that Phe28 of FXYD1 is located near Cys145 in the M2 transmembrane helix of the α subunit (20). Interestingly, the proximity of Phe28 to this site also places it near Phe146 in M2, raising the possibility that aromatic ring stacking could stabilize the FXYD-α interaction within the membrane. Preliminary docking studies with the structure of FXYD1 and a model of the Na,K-ATPase generated by homology with the crystal structure of the Ca-ATPase (18, 64) indicate that this is possible.
FXYD1 differs from FXYD2, FXYD3, and FXYD4 in the presence of a well defined cytoplasmic helix loosely connected to the rest of the protein by a long flexible linker. The emerging structures of FXYD2 and FXYD3 show considerably less structural order in this region, while the cytoplasmic helix of FXYD4 is more rigidly connected to the transmembrane domain. This may reflect the ability of FXYD1 to undergo helix reorientation as a means to modulate its interaction with the α subunit. Initial NMR experiments with FXYD1 phosphorylated at Ser68 by PKA indicate that phosphorylation increases the dynamics around helix H4, and it will be interesting to see whether this is accompanied by a conformational change and detachment from the lipid surface.
Table 1.
NMR and structure refinement statistics.
| Experimental restraints used in structure calculation and refinement | |
| Distance restraints | |
| NOE restraints | 447 |
| hydrogen bond restraints | 17 |
| MTSL PRE restraints | 18 |
| Orientation restraints | |
| HN RDC restraints | 55 |
| Dihedral angle restraints | |
| fromTALOS | 41 |
| Micelle depth restraints | |
| Mn PRE restraints | 2 |
| aStructure statistics parameters (RMSD) | |
| Violations from restraints | |
| Distance restraints (Å) | 0.082 |
| Dihedral angle restraints (°) | 2.029 |
| HN RDC restraints (Hz) | 0.310 |
| b HN RDC R factor (Hz) | 0.070 |
| Deviation from idealized geometry | |
| bonds (Å) | 0.005 |
| angles (°) | 0.804 |
| impropers (°) | 0.764 |
| c φ/ψ in most favored region (%) | 88.5 |
| dAverage pairwise RMSD | |
| backbone atoms (Å) | 0.128 |
| heavy atoms (Å) | 7.515 |
| dAverage RMSD from mean structure | |
| backbone atoms (Å) | 0.882 |
| heavy atoms (Å) | 5.257 |
Calculated for 20 lowest energy structures out of a total 40 calculated structures.
Calculated according to Clore and Garrett (38).
Calculated with PROCHECK.
Calculated for the structured regions of the protein from residues 13–44 and 60–69.
Acknowledgments
We thank Homayoun Valafar for his assistance with REDCAT and Charles Schwieters for his assistance with XPLOR-NIH. We also thank Jinghua Yu for her assistance with NMR experiments, and Denise Li and Khang Thai for their assistance with sample preparation.
ABBREVIATIONS
- Da
magnitude of the alignment tensor
- DTT
dithiothreitol
- FXYD1
phospholemman, plm
- GDO
generalized degree of order
- HSQC
heteronuclear single quantum correlation
- MTSL
1-oxy-2,2,5,5-tetramethyl-d-3-pyrroline-3-methyl
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser effect spectroscopy
- PAGE
polyacrylamide electrophoresis
- PKA
protein kinase A
- PKC
protein kinase C
- PRE
paramagnetic relaxation enhancement
- R
rhombicity of the alignment tensor
- RDC
residual dipolar coupling
- RMSD
root mean square deviation
- SDS
sodium dodecyl sulfate
Footnotes
This research was supported by a grant from the National Institutes of Health (CA082864). The NMR studies utilized the Burnham Institute NMR Facility, supported by a grant from the National Institutes of Health (CA030199).
References
- 1.Sweadner KJ, Rael E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- 2.Crambert G, Geering K. FXYD proteins: new tissue-specific regulators of the ubiquitous Na, K-ATPase. Sci STKE 2003. 2003:RE1. doi: 10.1126/stke.2003.166.re1. [DOI] [PubMed] [Google Scholar]
- 3.Cornelius F, Mahmmoud YA. Functional modulation of the sodium pump: the regulatory proteins “Fixit”. News Physiol Sci. 2003;18:119–124. doi: 10.1152/nips.01434.2003. [DOI] [PubMed] [Google Scholar]
- 4.Garty H, Karlish SJ. Role of fxyd proteins in ion transport. Annu Rev Physiol. 2006;68:431–459. doi: 10.1146/annurev.physiol.68.040104.131852. [DOI] [PubMed] [Google Scholar]
- 5.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Phospholemman (FXYD1) associates with Na, K-ATPase and regulates its transport properties. Proc Natl Acad Sci U S A. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer CJ, Scott BT, Jones LR. Purification and complete sequence determination of the major plasma membrane substrate for cAMP-dependent protein kinase and protein kinase C in myocardium. J Biol Chem. 1991;266:11126–11130. [PubMed] [Google Scholar]
- 7.Mounsey JP, John JE, 3rd, Helmke SM, Bush EW, Gilbert J, Roses AD, Perryman MB, Jones LR, Moorman JR. Phospholemman is a substrate for myotonic dystrophy protein kinase. J Biol Chem. 2000;275:23362–23367. doi: 10.1074/jbc.M000899200. [DOI] [PubMed] [Google Scholar]
- 8.Walaas SI, Czernik AJ, Olstad OK, Sletten K, Walaas O. Protein kinase C and cyclic AMP-dependent protein kinase phosphorylate phospholemman, an insulin and adrenaline-regulated membrane phosphoprotein, at specific sites in the carboxy terminal domain. Biochem J. 1994;304(Pt 2):635–640. doi: 10.1042/bj3040635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silverman BZ, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF, Shattock MJ. Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Fuller W, Eaton P, Bell JR, Shattock MJ. Ischemia-induced phosphorylation of phospholemman directly activates rat cardiac Na/K-ATPase. Faseb J. 2004;18:197–199. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- 11.Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, Tucker AL, Bers DM. Phospholemman-phosphorylation mediates the beta-adrenergic effects on Na/K pump function in cardiac myocytes. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- 12.Bossuyt J, Despa S, Martin JL, Bers DM. Phospholemman phosphorylation alters its fluorescence resonance energy transfer with the Na/K-ATPase pump. J Biol Chem. 2006;281:32765–32773. doi: 10.1074/jbc.M606254200. [DOI] [PubMed] [Google Scholar]
- 13.Moorman JR, Ackerman SJ, Kowdley GC, Griffin MP, Mounsey JP, Chen Z, Cala SE, O’Brian JJ, Szabo G, Jones LR. Unitary anion currents through phospholemman channel molecules. Nature. 1995;377:737–740. doi: 10.1038/377737a0. [DOI] [PubMed] [Google Scholar]
- 14.Moorman JR, Palmer CJ, John JE, 3rd, Durieux ME, Jones LR. Phospholemman expression induces a hyperpolarization-activated chloride current in Xenopus oocytes. J Biol Chem. 1992;267:14551–14554. [PubMed] [Google Scholar]
- 15.Hakansson KO. The crystallographic structure of Na, K-ATPase N-domain at 2.6A resolution. J Mol Biol. 2003;332:1175–1182. doi: 10.1016/j.jmb.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Hilge M, Siegal G, Vuister GW, Guntert P, Gloor SM, Abrahams JP. ATP-induced conformational changes of the nucleotide-binding domain of Na, K-ATPase. Nat Struct Biol. 2003;10:468–474. doi: 10.1038/nsb924. [DOI] [PubMed] [Google Scholar]
- 17.Underhaug J, Jakobsen LO, Esmann M, Malmendal A, Nielsen NC. NMR studies of the fifth transmembrane segment of Na+, K+-ATPase reveals a non-helical ion-binding region. FEBS Lett. 2006;580:4777–4783. doi: 10.1016/j.febslet.2006.07.063. [DOI] [PubMed] [Google Scholar]
- 18.Hebert H, Purhonen P, Vorum H, Thomsen K, Maunsbach AB. Three-dimensional structure of renal Na, K-ATPase from cryo-electron microscopy of two-dimensional crystals. J Mol Biol. 2001;314:479–494. doi: 10.1006/jmbi.2001.5137. [DOI] [PubMed] [Google Scholar]
- 19.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 20.Lindzen M, Gottschalk KE, Fuzesi M, Garty H, Karlish SJ. Structural interactions between FXYD proteins and Na+, K+-ATPase: alpha/beta/FXYD subunit stoichiometry and cross-linking. J Biol Chem. 2006;281:5947–5955. doi: 10.1074/jbc.M512063200. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Grosdidier A, Crambert G, Horisberger JD, Michielin O, Geering K. Structural and functional interaction sites between Na, K-ATPase and FXYD proteins. J Biol Chem. 2004;279:38895–38902. doi: 10.1074/jbc.M406697200. [DOI] [PubMed] [Google Scholar]
- 22.Franzin CM, Yu J, Thai K, Choi J, Marassi FM. Correlation of Gene and Protein Structures in the FXYD Family Proteins. J Mol Biol. 2005;354:743–750. doi: 10.1016/j.jmb.2005.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 24.Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. Chem Rev. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- 25.Hus JC, Marion D, Blackledge MJ. De novo Determination of Protein Structure by NMR using Orientational and Long-range Order Restraints. J Mol Biol. 2000;298:927–936. doi: 10.1006/jmbi.2000.3714. [DOI] [PubMed] [Google Scholar]
- 26.Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH. Structural and dynamic analysis of residual dipolar coupling data for proteins. J Am Chem Soc. 2001;123:1416–1424. doi: 10.1021/ja002500y. [DOI] [PubMed] [Google Scholar]
- 27.Thai K, Choi J, Franzin CM, Marassi FM. Bcl-XL as a fusion protein for the high-level expression of membrane-associated proteins. Protein Sci. 2005;14:948–955. doi: 10.1110/ps.041244305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 29.Liang B, Bushweller JH, Tamm LK. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J Am Chem Soc. 2006;128:4389–4397. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bax A, Grzesiek S. Methodological advances in protein NMR. Accounts of Chemical Research. 1993;26:131–138. [Google Scholar]
- 31.Cavanagh J. Protein NMR spectroscopy: principles and practice. Academic Press; San Diego: 1996. [Google Scholar]
- 32.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 33.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 2004. [Google Scholar]
- 34.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 35.Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, Rivera-Torres IO, Cahill SM, Hicks DB, Grinius L, Krulwich TA, Girvin ME. An evaluation of detergents for NMR structural studies of membrane proteins. J Biomol NMR. 2004;28:43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- 36.Schwieters CD, Kuszewski JJ, Tjandra N, Marius Clore G. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 37.Valafar H, Prestegard JH. REDCAT: a residual dipolar coupling analysis tool. J Magn Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Clore GM, Garrett DS. R-factor, Free R, and Complete Cross-Validation for Dipolar Coupling Refinement of NMR Structures. J Am Chem Soc. 1999;121:9008–9012. [Google Scholar]
- 39.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 40.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283. [Google Scholar]
- 42.Nilges M, Clore GM, Gronenborn AM. Determination of three-dimensional structures of proteins from interproton distance data by dynamical simulated annealing from a random array of atoms. Circumventing problems associated with folding. FEBS Lett. 1988;239:129–136. doi: 10.1016/0014-5793(88)80559-3. [DOI] [PubMed] [Google Scholar]
- 43.Schwieters CD, Clore GM. Internal coordinates for molecular dynamics and minimization in structure determination and refinement. J Magn Reson. 2001;152:288–302. doi: 10.1006/jmre.2001.2413. [DOI] [PubMed] [Google Scholar]
- 44.Kuszewski J, Gronenborn AM, Clore GM. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J Magn Reson. 1997;125:171–177. doi: 10.1006/jmre.1997.1116. [DOI] [PubMed] [Google Scholar]
- 45.Chou JJ, Li S, Bax A. Study of conformational rearrangement and refinement of structural homology models by the use of heteronuclear dipolar couplings. J Biomol NMR. 2000;18:217–227. doi: 10.1023/a:1026563923774. [DOI] [PubMed] [Google Scholar]
- 46.Clore GM, Gronenborn AM, Tjandra N. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J Magn Reson. 1998;131:159–162. doi: 10.1006/jmre.1997.1345. [DOI] [PubMed] [Google Scholar]
- 47.Kayed H, Kleeff J, Kolb A, Ketterer K, Keleg S, Felix K, Giese T, Penzel R, Zentgraf H, Buchler MW, Korc M, Friess H. FXYD3 is overexpressed in pancreatic ductal adenocarcinoma and influences pancreatic cancer cell growth. Int J Cancer. 2005;118:43–54. doi: 10.1002/ijc.21257. [DOI] [PubMed] [Google Scholar]
- 48.Jespersen T, Grunnet M, Rasmussen HB, Jorgensen NB, Jensen HS, Angelo K, Olesen SP, Klaerke DA. The corticosteroid hormone induced factor: a new modulator of KCNQ1 channels? Biochem Biophys Res Commun. 2006;341:979–988. doi: 10.1016/j.bbrc.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 49.Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci U S A. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karim CB, Paterlini MG, Reddy LG, Hunter GW, Barany G, Thomas DD. Role of cysteine residues in structural stability and function of a transmembrane helix bundle. J Biol Chem. 2001;276:38814–38819. doi: 10.1074/jbc.M104006200. [DOI] [PubMed] [Google Scholar]
- 51.Simmerman HK, Kobayashi YM, Autry JM, Jones LR. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J Biol Chem. 1996;271:5941–5946. doi: 10.1074/jbc.271.10.5941. [DOI] [PubMed] [Google Scholar]
- 52.Ramjeesingh M, Huan LJ, Garami E, Bear CE. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J. 1999;342:119–123. [PMC free article] [PubMed] [Google Scholar]
- 53.Therien AG, Deber CM. Oligomerization of a peptide derived from the transmembrane region of the sodium pump gamma subunit: effect of the pathological mutation G41R. J Mol Biol. 2002;322:583–550. doi: 10.1016/s0022-2836(02)00781-7. [DOI] [PubMed] [Google Scholar]
- 54.Beevers AJ, Kukol A. Secondary structure, orientation, and oligomerization of phospholemman, a cardiac transmembrane protein. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bales BL. A Definition of the Degree of Ionization of a Micelle Based on Its Aggregation Number. J Phys Chem B. 2001;105:6798–6804. [Google Scholar]
- 56.Franzin CM, Gong XM, Thai K, Yu J, Marassi FM. NMR of membrane proteins in micelles and bilayers: The FXYD family proteins. Methods. 2007;41:398–408. doi: 10.1016/j.ymeth.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanford C, Reynolds JA. Characterization of membrane proteins in detergent solutions. Biochim Biophys Acta. 1976;457:133–170. doi: 10.1016/0304-4157(76)90009-5. [DOI] [PubMed] [Google Scholar]
- 58.Beguin P, Crambert G, Guennoun S, Garty H, Horisberger JD, Geering K. CHIF, a member of the FXYD protein family, is a regulator of Na, K-ATPase distinct from the gamma-subunit. Embo J. 2001;20:3993–4002. doi: 10.1093/emboj/20.15.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+), K(+)-ATPase gamma-subunit. Nat Genet. 2000;26:265–266. doi: 10.1038/81543. [DOI] [PubMed] [Google Scholar]
- 60.Zamoon J, Mascioni A, Thomas DD, Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys J. 2003;85:2589–2598. doi: 10.1016/s0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metcalfe EE, Traaseth NJ, Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry. 2005;44:4386–4396. doi: 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- 62.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinton BB, Wertz SL, Jacob J, Steere J, Grisham CM, Cafiso DS, Sando JJ. Influence of lipid on the structure and phosphorylation of protein kinase C alpha substrate peptides. Biochem J. 1998;330(Pt 3):1433–1442. doi: 10.1042/bj3301433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sweadner KJ, Donnet C. Structural similarities of Na, K-ATPase and SERCA, the Ca(2+)-ATPase of the sarcoplasmic reticulum. Biochem J. 2001;356:685–704. doi: 10.1042/0264-6021:3560685. [DOI] [PMC free article] [PubMed] [Google Scholar]