

Abstract
A strategy is outlined for construction of the fungal immunosuppressant FR901483 (1). It was possible to convert 1,4-cyclohexanedione monoethylene ketal in five simple steps to iodoacetamide ketone 10, which was cyclized in good yield to the key bridged keto lactam 11 containing the A/B 2-azabicyclo[3.3.1]nonane ring system of the natural product. This intermediate could be transformed to N-Boc lactam 16, whose derived enolate underwent stereoselective hydroxylation with the Davis oxaziridine to produce alcohol 17 having the desired C-2 configuration. Compound 17 was then converted in three steps to alkoxy carbamate 20. The N-acyliminium ion derived from intermediate 20 could be alkylated in good overall yield with p-methoxybenzylmagnesium chloride to afford a 5:4 mixure of the desired PMB product 21 and the epimer 23. In an attempt to improve the stereoselectivity in this alkylation, the inverted C-4 protected alcohol N-Boc lactam 33 was prepared and its enolate was hydroxylated. Inexplicably, the product of this reaction was the undesired equatorial alcohol 34. Some model systems were investigated towards annulation of the C-ring of the natural product. It was found that homoallylic amine 40 could be cyclized with PhSCl in the presence of silica gel to generate the desired 5-endo tetracyclic product 42 in moderate yield. This cyclization protocol was also successfully applied to the actual FR901483 system 22, leading to the requisite tricycle 43.
Introduction and Background
The fungal metabolite FR901483 (1), which was isolated in 1996 from a Cladobotryum species, has a unique constitution unprecedented in natural products chemistry.1 The structure and relative stereochemistry of 1 were originally determined by X-ray crystallography, and its absolute configuration was later established by total synthesis.2 The crystal structure of FR901483 shows some interesting conformational features. For example, the B-ring of the molecule exists in a boat, perhaps due to the large C-4 phosphate group (which would be axial in the A/B chair-chair form). It is not clear from published data, however, if the molecule assumes the same conformation in solution. Furthermore, the pyrrolidine nitrogen has an equatorial lone pair, giving a cis fusion of the [6,5]-A/C-rings, but at the same time creating a 1,3-diaxial interaction between C-11 and the C-2 hydroxyl group. FR901483 shows immunosuppressive activity both in vitro and in vivo. Interestingly, the dephosphorylated compound is completely inactive. It was suggested that the compound acts by inhibiting purine nucleotide biosynthesis.1

Several successful total syntheses of FR901483 have appeared in the past few years.2 The first synthesis of this metabolite, reported by Snider in 1999, was biogenetically inspired starting from (S)-tyrosine, and established the absolute stereochemistry of 1, but was not highly stereoselective.2a Subsequent enantioselective total syntheses described by Sorensen2b and Ciufolini2c,e are also closely patterned after the putative biogenesis of FR901483 originating from two molecules of tyrosine. In 2001 Funk reported an elegant synthesis of racemic 1,2d and more recently syntheses have been described by the Fukuyama2f and Brummond2g groups. This continues to be an active area of research, and a number of other strategies leading toward this natural product have appeared in the literature.3
Retrosynthetic Plan
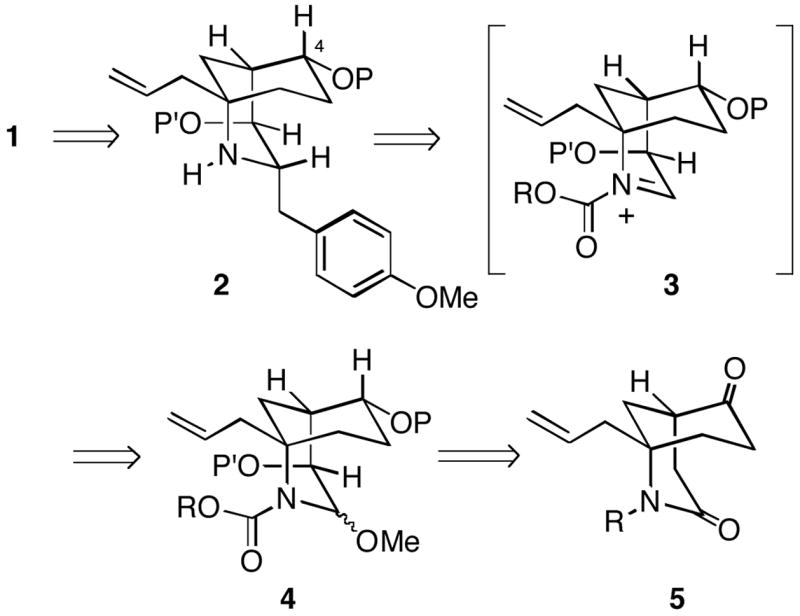
We have been investigating a new strategy for the construction of FR901483 and in this paper we report our progress to date.4 Retrosynthetically, our strategy for synthesis of 1 involved forming the third and final C-ring from a bridged bicyclic intermediate 2 containing the azabicyclo[3.3.1]nonane A/B-ring system (Scheme 1). This strategy is different from the previous total syntheses, where an azaspirocyclic core containing the [6,5]-B/Crings is generally formed first, and subsequently the A-ring is generated via an intramolecular aldol reaction. This latter process in general seems to produce either mixtures of stereoisomeric products or the incorrect isomer at C-2. We envisioned that the C-1 (FR901483 numbering used throughout) p-methoxybenzyl group of bicycle 2 could be installed via attack of a PMB organometallic reagent from the least hindered face of an N-acyliminium ion 3 generated from the α-alkoxycarbamate 4. We also anticipated that the C-4 hydroxyl group stereochemistry, initally set as shown in 4, would have to be inverted at some point (vide infra). The plan was to prepare intermediate 4 from the simple 2-azabicyclo[3.3.1]nonanone 5.
Scheme 1.
Results and Discussion
Construction of the A/B Ring System
The first challenge in the synthesis of FR901483 was to efficiently prepare the key A/B bicyclic ketolactam 11 (Scheme 2). The route began by condensing commercially available 1,4-cyclohexanedione monoethylene ketal with benzylamine to give imine 6, which without purification was treated with allylmagnesium bromide to form homoallylic amine 7. N-Acylation of amine 7 with chloroacetyl chloride then afforded chloroacetamide 8. α-Iodo amidoketone 10 was then formed from α-chloroamide 8 following ketal hydrolysis to 9 and Finkelstein halogen exchange.
Scheme 2.
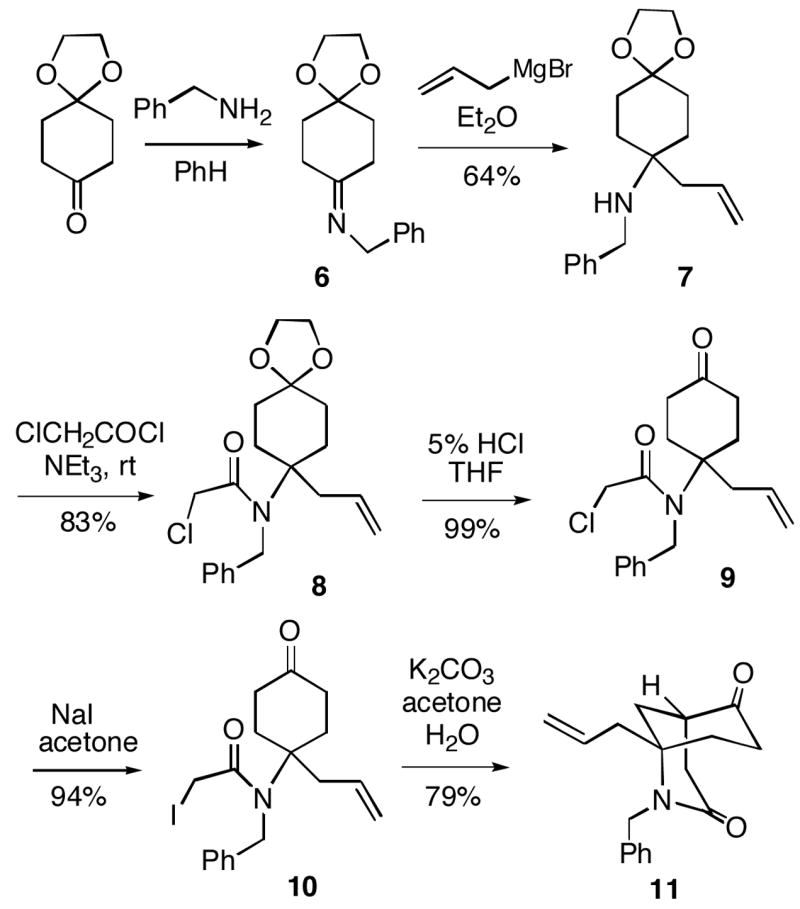
A few examples have been reported of intramolecular cyclizations involving alkylation of an enolate of an α-/α′-unsubstituted cyclohexanone and a tethered 4-alkyl electrophile to form bicyclo[3.3.1]nonane ring systems, but these annulations often occur in poor to mediocre yields.5,6 Indeed, we found initially that cyclization could be achieved if iodoketone 10 is treated with KOt-Bu in t-BuOH/THF (1:1) at room temperature, furnishing the desired bicyclo[3.3.1]nonanone 11, but only in moderate 46–58% yields, depending upon the reaction scale. However, we were pleased to find that the cyclization to 11 could be effected in a much improved 79% yield from ketone 10 if K2CO3 (2 equiv.) is used as the base in acetone/H2O at room temperature.7
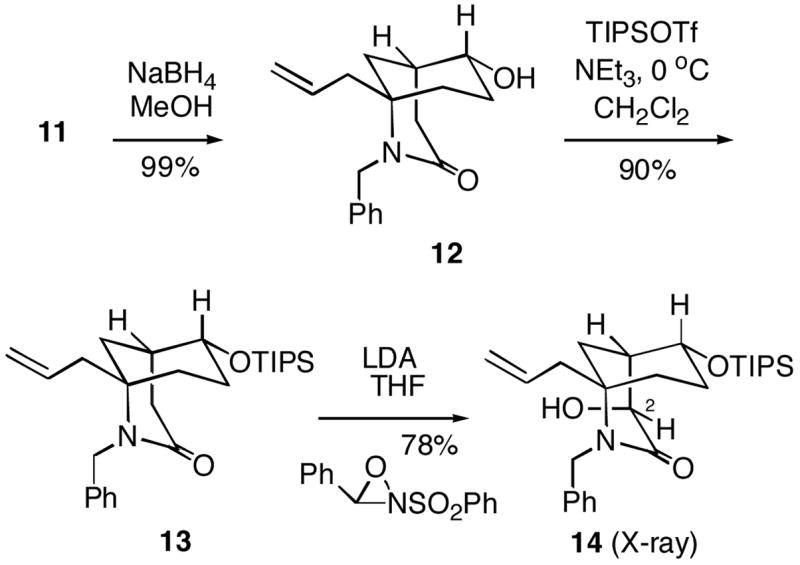
Reduction of ketone 11 with sodium borohydride was completely stereoselective, cleanly affording the equatorial alcohol 12 which was then protected as the triisopropylsilyl ether 13 (Scheme 3). We next investigated the introduction of oxygen at C-2. Thus, bicyclic lactam 13 was first deprotonated with LDA, followed by treatment of the resulting enolate with the Davis oxaziridine reagent8 led to a single α-hydroxylation product 14 in good yield. X-ray analysis of this compound indicated that it did indeed have the correct C-2 stereochemistry for FR901483. However, since dissolving metal reductive removal of the N-benzyl substituent of 14 (and O-protected derivatives) led to loss of the C-2 oxygen, an alternative sequence of reactions was explored.
Scheme 3.
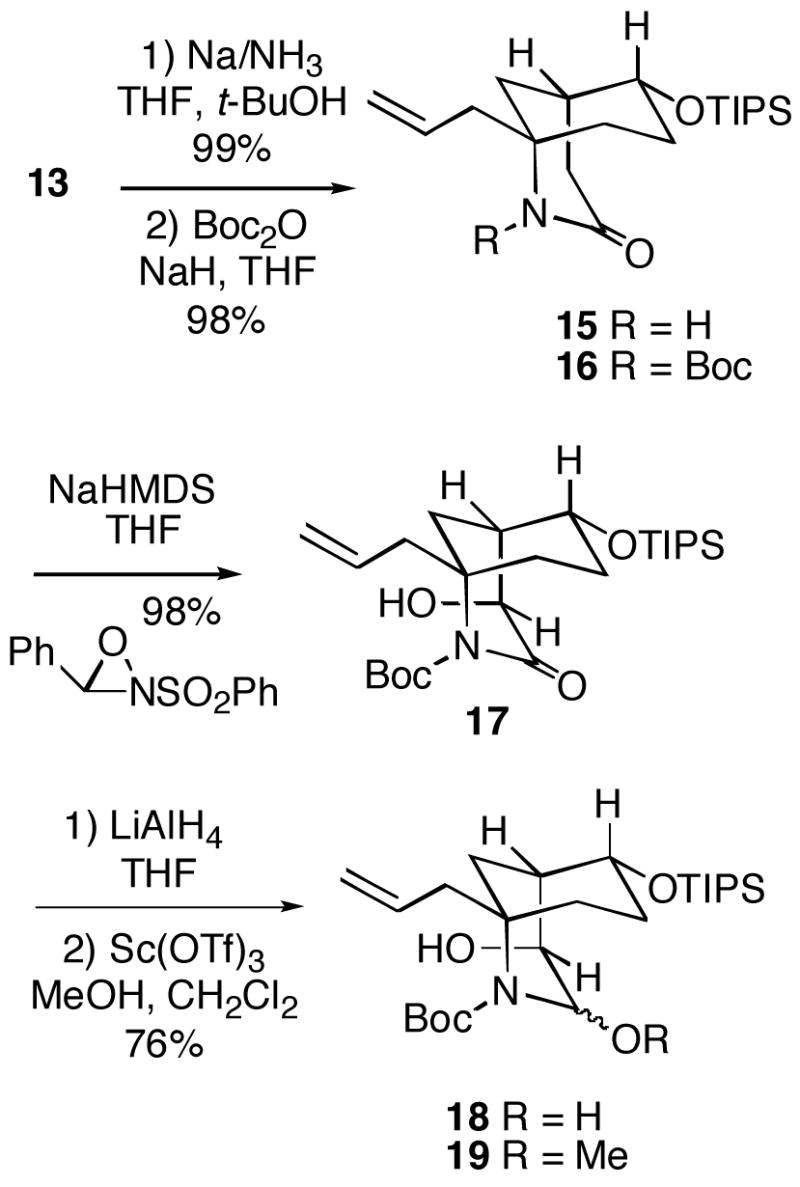
It was possible to first remove the N-benzyl group of 13 using sodium in liquid ammonia to generate lactam 15 (Scheme 4). A Boc group could then be installed on 15 to produce the N-acyl lactam 16. This intermediate was converted to the corresponding enolate with sodium hexamethyldisilazide, followed by exposure to the Davis oxaziridine reagent to produce hydroxy lactam 17. The stereochemistry of this alcohol was established by analysis of a later intermediate (vide infra). Subsequent chemoselective partial reduction of N-Boc lactam 17 with lithium aluminum hydride afforded α-hydroxycarbamate 18, which without purification was converted to methoxy compound 19 with scandium triflate in methanol.9 We had hoped that the hydroxyl group in 19 would promote alkylation at C-1 from the desired direction via chelation-controlled delivery of an organometallic reagent. Unfortunately, all attempts to directly couple alcohol 19 with excess p-methoxybenzylmagnesium chloride in the presence of a variety of Lewis acids were unsuccessful.10
Scheme 4.
Since we were concerned that deprotonation of the alcohol moiety in 19 by the Grignard reagent might possibly be interfering with the coupling step, a protected compound, benzyl ether 20, was prepared (Scheme 5). Addition of p-methoxybenzylmagnesium chloride (3.5 equiv.) and TiCl4 to the O-protected system 20 did in fact produce coupled product in good total yield, but as a mixture of epimers 21 and 23 which was not easily separated. However, removal of the Boc protecting group led to a chromatographically separable 5:4 mixture of the desired PMB product 22 along with the undesired epimer 24. The configurations of amines 22 and 24 were established by 1H NMR nOe experiments (key enhancements indicated). Interestingly, these nOe studies indicate that the A-ring in the minor isomer 24 appears to exist in a boat as shown in conformer 24b. This latter conformation avoids the non-bonded interactions between the axial PMB group and the B-ring in conformer 24a. These structural and stereochemical assignments were also supported by the coupling constants between the protons at C-1,2 (i.e. 2.8 Hz for 22, and 9.2 Hz for the diaxial arrangement in 24b).
Scheme 5.

In view of the disappointingly low level of stereoselectivity in the introduction of the PMB group, we turned to the alternative strategy outlined in Scheme 6. The plan was to first invert the stereochemistry at C-4 to produce a synthon like 25, which has the correct FR901483 configuration at this position. Addition of p-methoxybenzylmagnesium chloride to 25 would then afford 26. The expectation was that this intermediate would have the B-ring in a boat conformation with the large OTIPS group equatorial, similar to the case in the natural product (Cf. 1). Compound 26 would subsequently be converted to the desired PMB product 28 via hydride reduction which we believed should occur from the less hindered axial face of intermediate acyliminium ion 27.
Scheme 6.
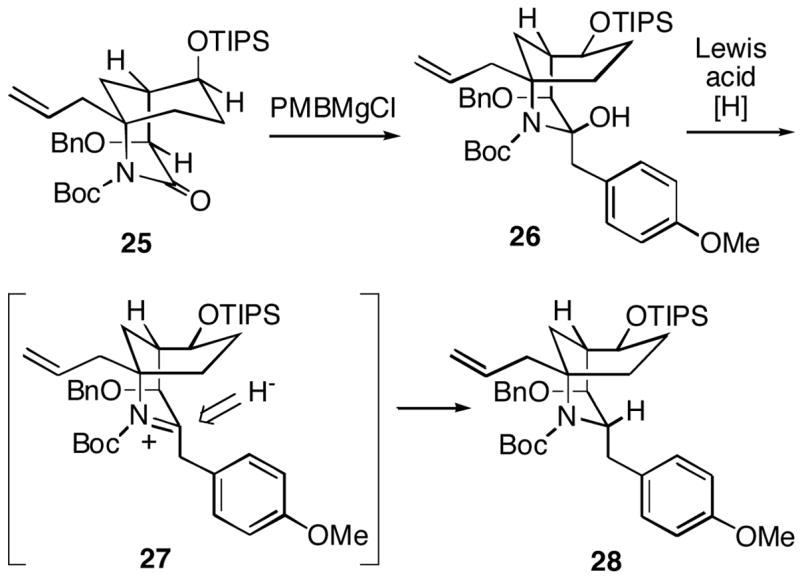
In order to test this approach, equatorial alcohol 12 was first inverted via a Mitsunobu procedure to afford the C-4 epimeric axial alcohol 29 (Scheme 7).11 The alcohol functionality was then protected as the TIPS ether 30. This lactam could be hydroxylated stereoselectively in 60% yield using LDA, followed by the Davis oxaziridine, to produce the axial C-2 alcohol 31. The configuration of this product was confirmed by its conversion to diol 32, whose structure was established by X-ray crystallography. Interestingly, the B-ring of this compound in the crystal was observed to be a chair, with the C-4 hydroxyl group axial. As we had experienced earlier, attempted removal of the N-benzyl group of 31 with sodium/liquid ammonia led to only a low yield (~20%) of the desired NH lactam.
Scheme 7.
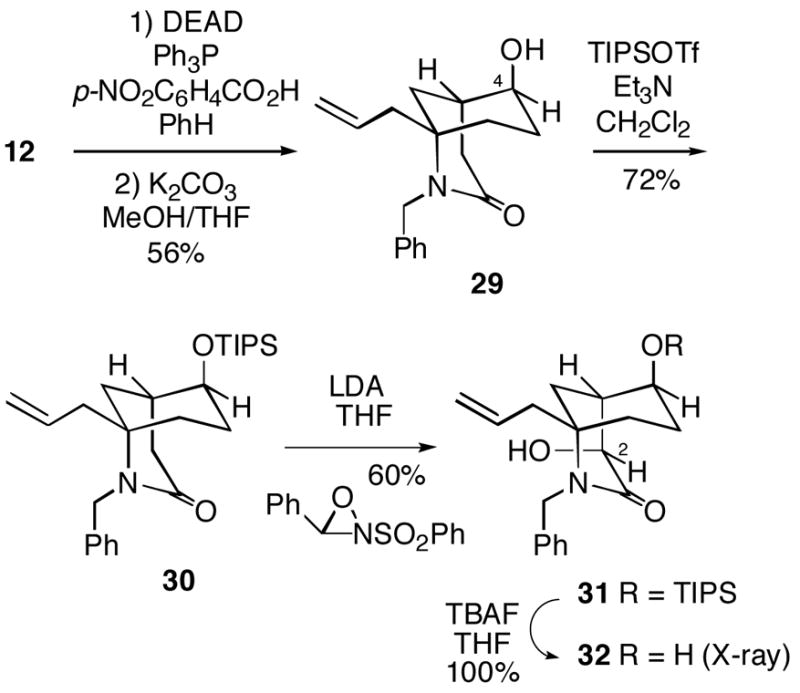
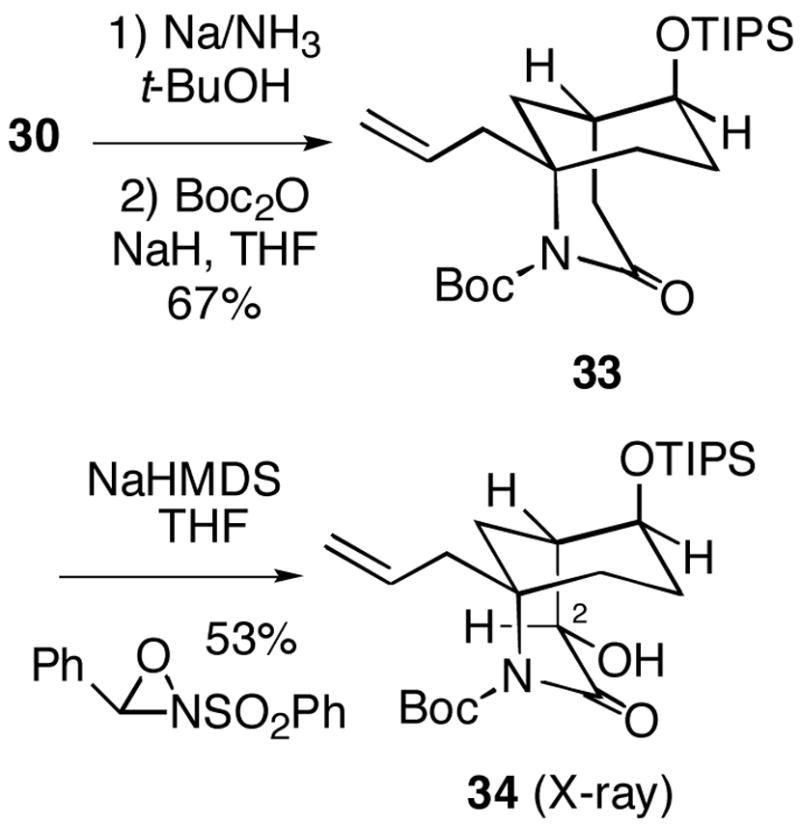
It was therefore necessary to again revise the sequence of steps here, which in this case led to some unexpected results. Removal of the N-benzyl group from lactam 30 proceeded uneventfully, and the resulting product was converted to N-Boc compound 33 (Scheme 8). However, we were surprised to find that hydroxylation of 33 as previously done with N-Boc lactam 16 (Cf. Scheme 4) led exclusively to the incorrect equatorial C-2 alcohol 34, whose structure was unambiguously determined by X-ray crystallography.12 Moreover, the crystal structure shows that like alcohol 32, hydroxylation product 34 exists with the B-ring in a chair, putting the large OTIPS group axial. At this point we are unable to offer a compelling rationale as to why the enolate of N-benzyllactam 30 leads to the desired axial hydroxylation product 31, whereas the corresponding N-Boc system undergoes hydroxylation to produce the undesired equatorial product 34.
Scheme 8.
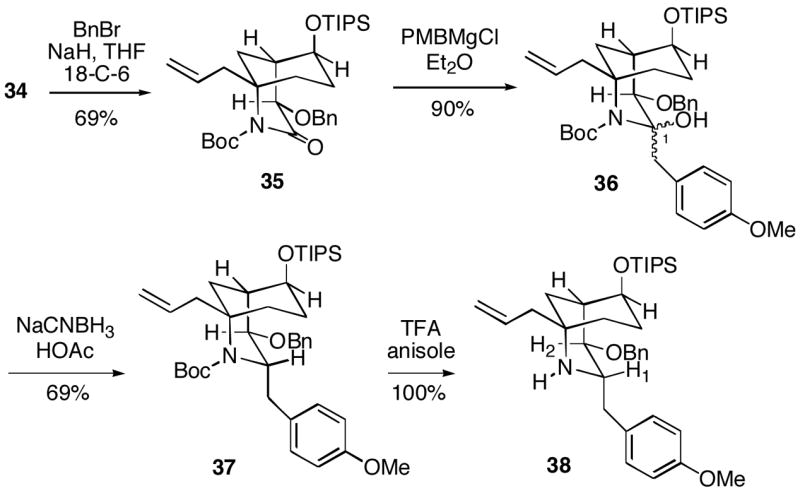
Prior to becoming aware that alcohol 34 in fact had the incorrect C-2 stereochemistry, we continued to explore the strategy outlined in Scheme 6 for introduction of the PMB group. After alcohol protection, benzyl ether 35 underwent clean addition of p-methoxybenzylmagnesium chloride to afford adduct 36 which appears to be one stereoisomer, although its configuration was not established (Scheme 9). This intermediate could be reduced with sodium cyanoborohydride in acetic acid to give a single stereoisomeric product 37 which was deacylated to amine 38. Based on the large observed coupling constant for H1/H2 of 9.0 Hz, we have tentatively assigned the stereochemistry to 38 as shown with an equatorial PMB group.
Scheme 9.
Studies on Annulation of the C-ring
The plan for construction of the C-ring of FR901483 was to effect a 5-endo trig cyclization of a suitable homoallylic amine to produce a properly C-10 functionalized pyrrolidine system. Initial experiments on formation of the C-ring were conducted with simple model bicyclic amino alkenes 39 and 40 (Scheme 10). There are a number of examples in the literature of this type of cyclization process starting from both homoallyic amines (as well as N-acylated amine derivatives) and homoallylic alcohols.13–15 Table 1 indicates some of the results of the cyclizations which were tested. Promising results were obtained using the selenium-induced cyclization methodology of Paulmier et al. (entries 3 and 4).13 Thus, tricyclic selenide 41 (X = SePh) could be prepared as a mixture of stereoisomers from homoallylic amine 39 in moderate yields. The cyclization of 39 using phenylsulfenyl chloride,15 however, proceeded in an improved 52% yield to afford tricyclic sulfide 41 (X = SPh, entry 5). With TIPS-protected substrate 40, tricycle 42 (X = SPh, entry 9) could be formed in 61% yield as a 3:2 mixture of stereoisomers. In this latter case, silica gel proved to be an effective additive.14 In none of these cases did we observe any azetidine formation via a 4-exo trig cyclization mode.13
Scheme 10.
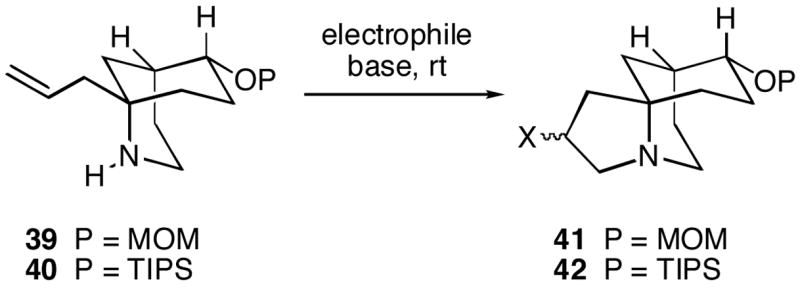
Table 1.
5-Endo Trig Cyclizations of Homoallylic Amines 39 and 40.
| entry | amine | electrophile (eq) | base (eq) | additive (eq) | product (%) |
|---|---|---|---|---|---|
| 1 | 39 | I2 (3) | NaHCO3 (3) | - | complex mixture |
| 2 | 39 | Hg(CO2CF3)2 (2) | NaHCO3 (10) | NaCl (10) | complex mixture |
| 3 | 39 | PhSeBr (1.5), MeCN | Na2CO3 (1.9) | - | 41 (41) X = SePh |
| 4 | 39 | PhSeBr (3), MeCN | Na2CO3 (1.9) | - | 41 (37) X = SePh |
| 5 | 39a | PhSCl (1.4), CH2Cl2 | K2CO3 (4.5) | NaI (4) | 41 (52) X = SPh |
| 6 | 40 | I2 (3) | NaHCO3 (3) | - | complex mixture |
| 7 | 40 | NIS (1.1) | - | - | complex mixture |
| 8 | 40 | PhSeBr (1.5), MeCN | NaHCO3 (1.9) | - | no reaction |
| 9 | 40a | PhSCl (1.4), CH2Cl2 | - | SiO2 | 42 (61) X = SPh |
pyridinium p-toluenesulfonate was added first to protect the amine
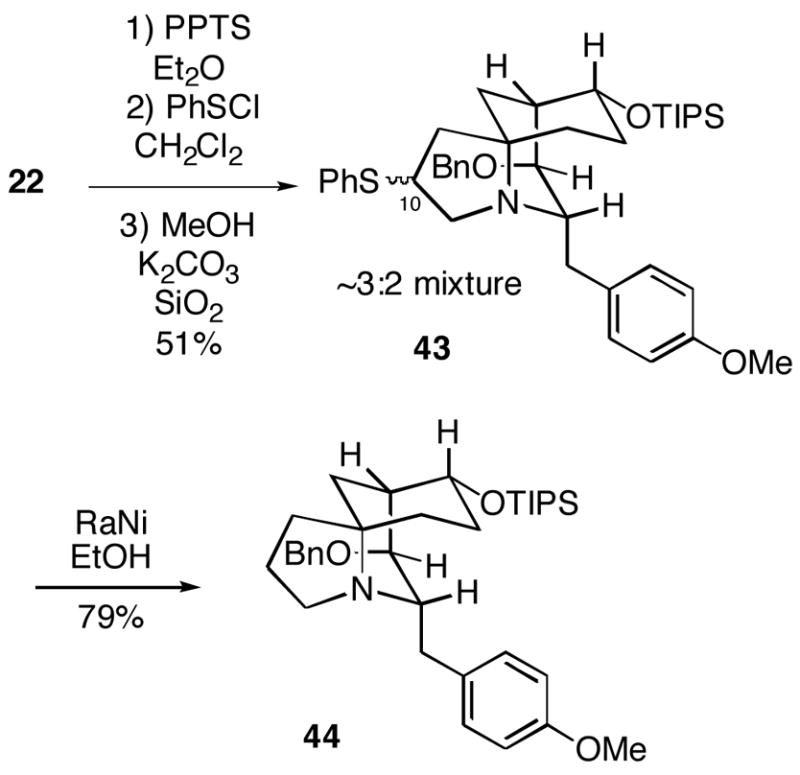
Using the best reaction conditions found for the simple homoallylic amine 40, we have examined cyclization of the actual FR901483 substrate 22 (Scheme 11). The cyclization conditions involved initial addition of phenylsulfenyl chloride to amine 22 which was premixed with the mild acid pyridinium trifluoromethanesulfonate (PPTS). The cyclized product 43 then was produced upon addition of K2CO3 and silica gel. Tricyclic sulfide 43 was formed in 51% yield as a chromatographically separable 3:2 mixture of C-10 stereoisomers whose configurations were not established. In order to confirm that this process had led to products of 5-endo trig cyclization, the mixture of sulfides was desulfurized with Raney nickel to afford tricycle 44 in good yield. If the cyclization had in fact produced the 4-exo trig products, an NMR-detectable methyl group would have been produced in this reduction step. The sulfide functionality in tricycles 43 should provide a convenient handle for eventual introduction of the methylamino group of the natural product.
Scheme 11.
Conclusion
In this report we have described a new approach to the synthesis of the tricyclic framework of FR901483 (1). We have shown that bicyclic keto lactam 11 can be efficiently prepared in five steps from 1,4-cyclohexanedione monoethylene ketal. This key A/B-ring synthon can then be elaborated to the advanced intermediate 22. A major obstacle, which will be investigated further in our future work, is the low stereoselectivity in the introduction of the C-1 p-methoxybenzyl group via addition to an N-acyliminium ion. In addition, we hope to probe the unexpected stereochemical differences in the hydroxylations of bridged lactam 30 vs N-Boc imide 33. We have also found that the tricyclic sulfide 43 bearing the C-ring of the natural product can be prepared by cyclization of homoallylic amine 22 mediated by phenylsulfenyl chloride. The expectation is that compound 43 can be converted to FR901483 by a straightforward route. The results of these further studies will be reported in due course.
Experimental Section
(8-Allyl-1,4-dioxaspiro[4.5]dec-8-yl)benzylamine (7)
To a solution of 97% 1,4-cyclohexanedione monoethylene ketal (25.0 g, 0.16 mmol) in benzene (400 mL) in a round bottomed flask fitted with a Dean-Stark trap at rt was added benzylamine (17 mL, 0.16 mol). The mixture was refluxed for 5 h, concentrated in vacuo and the residual imine 6 was dissolved in Et2O (400 mL). The solution was cooled to 0 °C and allylmagnesium bromide (200 mL, 1.0 M in Et2O) was added dropwise. The mixture was warmed to rt, stirred for an additional 2 h, then cooled to 0 °C and quenched by the slow addition of saturated aqueous NH4Cl followed by water. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (4:1, hexanes/EtOAc) to give amine 7 (29.4 g, 64%) as an orange oil: 1H NMR (300 MHz, CDCl3) δ 7.40-7.21 (m, 5H), 5.92-5.81 (m, 1H), 5.19-5.10 (m, 2H), 3.97 (s, 4H), 3.67 (s, 2H), 2.29 (d, J = 7.5 Hz, 2H), 2.03-1.92 (m, 2H), 1.75-1.55 (m, 6H); 13C NMR (90 MHz, CDCl3) δ 141.2, 134.1, 128.2, 126.7, 117.8, 109.1, 64.1, 64.0, 53.0, 45.6, 42.0, 32.7, 30.2; IR (neat) 3063, 3029, 1637, 1372, 1112 cm−1; HRMS (MALDI) calcd for C18H26NO2 (MH+) 288.1963, found 288.1968.
2-Chloro-N-(8-allyl-1,4-dioxaspiro[4.5]dec-8-yl)-N-benzylacetamide (8)
To a solution of amine 7 (6.8 g, 23.5 mmol) in CH2Cl2 (58 mL) at 0 °C was added triethylamine (3.6 mL, 25.8 mmol). A solution of chloroacetyl chloride (2.3 mL, 28.9 mmol) in CH2Cl2 (19 mL) was then added dropwise via cannula and the mixture was stirred at 0 °C for 1 h before warming to rt. After stirring at rt for 2 h, the solution was cooled to 0°C and water was added. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (4:1, hexanes/EtOAc) to give amide 8 (7.1 g, 83%) as a white solid: mp 123–124 °C; 1H NMR (300 MHz, CDCl3) δ 7.42-7.17 (m, 5H), 5.78 (dddd, J = 17.8, 10.3, 7.5, 7.5 Hz, 1H), 5.16-5.05 (m, 2H), 4.63 (s, 2H), 3.94-3.80 (m, 6H), 2.85 (d, J = 7.4 Hz, 2H), 2.44 (br d, J = 13.0, 2H), 2.04-1.86 (m, 2H), 1.74-1.54 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 168.5, 138.5, 133.5, 129.0, 127.3, 125.2, 118.8, 107.4, 64.2 (2C), 63.2, 48.9, 44.2, 35.4, 31.3, 30.6; IR (Nujol mull) 3060, 1653, 1393 cm−1; HRMS (APCI) calcd for C20H27ClNO3 (MH+) 364.1679, found 364.1695.
N-(1-Allyl-4-oxocyclohexyl)-N-benzyl-2-chloroacetamide (9)
To a solution of the amide 8 (29.5 g, 81 mmol) in THF (170 mL) at rt was added 5% HCl (170 mL). After stirring the mixture overnight, another portion of 5% HCl (85 mL) was added and the mixture was stirred for an additional 2 d. The mixture was then cooled to 0 °C and neutralized with saturated aqueous NaHCO3. The aqueous layer was extracted with Et2O. The combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (3:2, hexanes/EtOAc) to give ketone 9 (25.8 g, 99%) as an orange gum: 1H NMR (360 MHz, CDCl3) δ 7.44 (t, J = 7.7 Hz, 2H), 7.33 (t, J = 7.4 Hz, 1H), 7.25 (d, J = 7.4 Hz, 2H), 5.90 (dddd, J = 17.7, 10.3, 7.4, 7.4 Hz, 1H), 5.28-5.19 (m, 2H), 4.74 (s, 2H), 4.07 (s, 2H), 3.01 (d, J = 7.4 Hz, 2H), 2.63 (ddd, J = 17.5, 11.2, 4.8 Hz, 2H), 2.45-2.17 (m, 6H); 13C NMR (90 MHz, CDCl3) δ 209.6, 168.4, 137.7, 132.6, 129.0, 127.5, 125.0, 119.3, 62.3, 49.0, 43.9, 36.9, 36.4, 32.2; IR (neat) 3065, 3031, 1713, 1651, 1396 cm−1; HRMS (APCI) calcd for C18H23ClNO2 (MH+) 320.1417, found 320.1427.
N-(1-Allyl-4-oxocyclohexyl)-N-benzyl-2-iodoacetamide (10)
To a solution of the chloride 9 (25.8 g, 80.7 mmol) in acetone (91 mL) at rt was added sodium iodide (36 g, 240 mmol). After stirring for 3 h, the mixture was concentrated in vacuo and the residue was dissolved in water and EtOAc. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with saturated aqueous Na2S2O3 and brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (7:3, hexanes/EtOAc) to give iodide 10 (31.0 g, 94%) as an orange solid: 1H NMR (360 MHz, CDCl3) δ 7.35 (t, J = 7.2 Hz, 2H), 7.25 (t, J = 7.3 Hz, 1H), 7.15 (d, J = 7.2 Hz, 2H), 5.84 (dddd, J = 17.7, 10.3, 7.5, 7.5 Hz, 1H), 5.18-5.09 (m, 2H), 4.59 (s, 2H), 3.62 (s, 2H), 2.93 (d, J = 7.4 Hz, 2H), 2.54 (ddd, J = 17.9, 11.5, 5.1 Hz, 2H), 2.37-2.09 (m, 6H); 13C NMR (90 MHz, CDCl3) δ 209.9, 169.9, 137.7, 132.6, 129.1, 127.6, 125.0, 119.6, 62.1, 50.6, 37.0, 36.7, 32.4; IR (neat) 3063, 3030, 1714, 1650, 1387 cm−1; HRMS (APCI) calcd for C18H23INO2 (MH+) 412.0774, found 412.0787.
1-Allyl-2-benzyl-2-azabicyclo[3.3.1]nonane-3,6-dione (11)
Method 1
To a solution of the iodide 10 (500 mg, 1.2 mmol) in t-BuOH/THF (1:1, 5.2 mL) at rt was added KOt-Bu (175 mg, 0.64 mmol) in two equal portions 5 h apart. After stirring for 2 h, the mixture was concentrated in vacuo and the residue was diluted with water. The solution was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (1:1, hexanes/EtOAc) to give ketone 11 (158 mg, 46%) as a white solid.
Method 2
To a solution of the iodide 10 (5.00 g, 12.2 mmol) in acetone (100 mL) at rt was added a solution of K2CO3 (3.25 g, 23.5 mmol) in water (42.5 mL). After stirring for 72 h, the mixture was concentrated in vacuo and the residue was diluted with water. The solution was extracted with EtOAc. The combined organic layers were washed with water, and brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (3:2, EtOAc/hexanes) to give ketone 11 (2.73 g, 79%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.35-7.16 (m, 5H), 5.54 (dddd, J = 16.6, 10.2, 8.1, 6.2 Hz, 1H), 5.12 (d, J = 10.4 Hz, 1H), 5.08 (dd, J = 17.1, 1.4 Hz, 1H), 4.90, 4.70 (ABq, J = 15.5 Hz, ΔνAB = 64.9 Hz, 2H), 2.83 (dd, J = 18.1, 7.1 Hz, 1H), 2.79 (m, 1H), 2.65-2.56 (m, 2H), 2.34-2.07 (m, 4H), 1.96 (ddd, J = 13.7, 3.0, 3.0 Hz, 1H), 1.85-1.77 (m, 1H), 1.66 (ddd, J = 13.5, 13.5, 5.3 Hz, 1H); 13C NMR (90 MHz, CDCl3) δ 211.1, 170.3, 138.7, 131.7, 128.5, 127.6, 127.2, 120.0, 58.1, 44.7, 43.2, 42.6, 37.2, 35.5, 34.8, 34.5; IR (neat) 2939, 1713, 1637, 984 cm−1; HRMS (APCI) calcd for C18H22NO2 (MH+) 284.1651, found 284.1661.
1-Allyl-6-hydroxy-2-benzyl-2-azabicyclo[3.3.1]nonan-3-one (12)
To a solution of the ketone 11 (16.4 g, 57.8 mmol) in methanol (332 mL) at 0 °C was added sodium borohydride (2.70 g, 71.4 mmol). The mixture was warmed to rt and was stirred for an additional 1 h. The mixture was then concentrated in vacuo, diluted with Et2O, washed with water and the aqueous layer was further extracted with Et2O. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (95:5, EtOAc/MeOH) to give alcohol 12 (16.3 g, 99%) as a white foam: 1H NMR (300 MHz, CDCl3) δ 7.30-7.15 (m, 5H), 5.57 (dddd, J = 16.8, 10.3, 8.0, 6.4 Hz, 1H), 5.07-4.98 (m, 2H), 4.68 (ABq, J = 15.6 Hz, ΔνAB = 19.1 Hz, 2H), 3.74 (ddd, J = 10.8, 4.6, 4.6 Hz, 1H), 2.88 (dd, J = 18.6, 1.8 Hz, 1H), 2.56-2.31 (m, 3H), 2.26-2.12 (m, 2H), 1.95 (ddd, J = 13.6, 3.4, 3.4 Hz, 1H), 1.71-1.43 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 173.3, 139.1, 132.4, 128.2, 127.3, 126.6, 118.9, 70.6, 58.0, 44.5, 43.6, 35.7, 32.7, 32.5, 31.0, 26.2; IR (film) 3378, 2936, 1615, 916 cm−1; EIMS m/z (relative intensity) 285 (MH+, 7), 91 (100); HRMS calcd for C18H23NO2 285.1727, found 285.1729.
1-Allyl-2-benzyl-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonan-3-one (13)
To a solution of alcohol 12 (792 mg, 2.77 mmol) in CH2Cl2 (11 mL) at 0 °C was added triethylamine (1.0 mL, 7.2 mmol) and triisopropylsilyl trifluoromethanesulfonate (1.0 mL, 3.7 mmol). After 2.5 h at 0 °C, the mixture was diluted with water, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (9:1, hexanes/EtOAc) to give silyl ether 13 (1.1 g, 90%) as a white solid: mp 96–97 °C; 1H NMR (300 MHz, CDCl3) δ 7.31-7.17 (m, 5H), 5.47 (ddt, J = 16.8, 9.1, 7.7 Hz, 1H), 4.98 (d, J = 10.2 Hz, 1H), 4.93 (dd, J = 17.0, 1.3 Hz, 1H), 4.67 (ABq, J = 12.4 Hz, ΔνAB = 34.0 Hz, 2H), 3.79-3.74 (m, 1H), 2.97 (d, J = 18.5 Hz, 1H), 2.47 (dd, J = 18.5, 7.2 Hz, 1H), 2.40 (dd, J = 14.4, 6.3 Hz, 1H), 2.20-2.16 (m, 2H), 1.92 (ddd, J = 10.4, 3.2, 3.2 Hz, 1H), 1.60-1.28 (m, 5H), 1.12-0.95 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 173.0, 139.4, 132.6, 128.2, 127.4, 126.6, 118.9, 71.7, 57.8, 44.4, 43.8, 35.9, 33.5, 32.8, 31.0, 27.2, 17.9, 12.1; IR (neat) 3065, 3028, 1634, 1399 cm−1; HRMS (MALDI) calcd for C27H44NO2Si (MH+) 442.3141, found 442.3155.
1-Allyl-2-benzyl-4-hydroxy-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonan-3-one (14)
To a solution of N, N-diisopropylamine (0.19 mL, 1.3 mmol) in THF (45 mL) at −78 °C was added n-BuLi (0.70 mL, 1.81 M in hexanes). The mixture was warmed to −30 °C and stirred for 10 min. The mixture was then cooled to −78 °C and a solution of lactam 13 (470 mg, 1.1 mmol) in THF (5 mL) was added via cannula. The mixture was slowly warmed to 0 °C and stirred for 45 min. The mixture was recooled to −78 °C and 2-(phenylsulfonyl)-3-phenyloxaziridine8b (420 mg, 1.6 mmol) was then added. The resulting mixture was slowly warmed to rt while stirring for 18 h. The mixture was then carefully poured into saturated aqueous NaHCO3. The organic layer was washed with water and brine. The combined aqueous layers were extracted with CH2Cl2, and the extract was dried with MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (9:1, hexanes/EtOAc) to give hydroxy lactam 14 (380 mg, 78%) as a white solid: mp 110–111 °C; 1H NMR (300 MHz, CDCl3) δ 7.32-7.18 (m, 5H), 5.60 (dddd, J = 16.9, 10.2, 7.7, 7.7 Hz, 1H), 5.11-4.98 (m, 2H), 4.68 (ABq, J = 15.6 Hz, ΔνAB = 26.1 Hz, 2H), 4.43 (s, 1H), 3.77 (ddd, J = 9.5, 4.7, 4.7 Hz, 1H), 2.97 (s, 1H), 2.42 (dd, J = 14.3, 6.6 Hz, 1H), 2.31-2.26 (m, 1H), 2.24-2.07 (m, 2H), 1.70-1.60 (m, 1H), 1.48-1.37 (m, 2H), 1.39-1.15 (m, 2H), 1.10-0.91 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 174.8, 138.7, 132.4, 128.5, 127.4, 127.0, 119.5, 70.4, 66.9, 58.9, 44.9, 43.8, 40.4, 33.1, 31.9, 28.2, 18.0, 12.3; HRMS (MALDI) calcd for C27H44NO3Si (MH+) 458.3090, found 458.3087.
1-Allyl-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonan-3-one (15)
In a 1 L three-neck round bottomed flask fitted with a cold finger condenser at −78 °C was condensed ammonia (380 mL). Sodium (4.0 g, 172 mmol) followed by t-butanol (8.2 mL, 86 mmol) were carefully added to the ammonia, resulting in a blue mixture. After 45 min, a solution of benzyl lactam 13 (7.5 g, 17 mmol) in Et2O (55 mL) was added to the mixture via a gastight syringe. The mixture was stirred at reflux for 75 min and then solid ammonium chloride (10 g) was carefully added until the blue color dissipated. The mixture was slowly warmed to rt while occasionally replacing the evaporating ammonia with THF. The resulting mixture was filtered and the filtrate was concentrated in vacuo. The crude residue was purified by flash chromatography (1:1, hexanes/EtOAc) to give lactam 15 (6.0 g, 99%) as a colorless oil sufficiently pure for use in the next step: 1H NMR (360 MHz, CDCl3) δ 6.39 (s, 1H), 5.74 (dddd, J = 18.8, 10.2, 8.5, 6.5 Hz, 1H), 5.15-5.08 (m, 2H), 3.80-3.71 (m, 1H) 2.80-2.71 (m, 1H), 2.26-2.13 (m, 4H), 1.75-1.64 (m, 2H), 1.55-1.39 (m, 4H), 1.06-0.91 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 173.7, 131.7, 119.9, 71.9, 52.7, 45.6, 35.8, 35.2, 34.4, 29.5, 27.5, 18.0, 12.2; IR (neat) 3192, 3076, 1651, 1401 cm−1; HRMS (APCI) calcd for C20H38NO2Si (MH+) 352.2672, found 352.2688.
1-Allyl-3-oxo-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (16)
To a solution of lactam 15 (430 mg, 1.2 mmol) in THF (60 mL) at 0 °C was added 60% NaH in mineral oil (98 mg, 4.1 mmol), and the mixture was heated at reflux for 1 h. The mixture was then cooled to rt and di-t-butyl dicarbonate (1.4 mL, 6.1 mmol) was added, reheated to reflux and stirred for 18 h. The mixture was cooled to rt and saturated aqueous NH4Cl (20 mL) was carefully added. The aqueous layer was extracted with Et2O. The combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (20:1, hexanes/EtOAc) to give carboxy imide 16 (540 mg, 98%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 5.78 (dddd, J = 16.6, 10.2, 8.3, 6.2 Hz, 1H), 5.12-4.99 (m, 2H), 3.77 (ddd, J = 9.4, 4.6, 4.6 Hz, 1H), 2.82 (dd, J = 18.1, 1.1 Hz, 1H), 2.52 (dd, J = 17.4, 9.1 Hz, 1H), 2.34-2.18 (m, 2H) 2.17-2.04 (m, 2H), 1.94 (dd, J = 13.6, 3.5 Hz, 1H), 1.80-1.65 (m, 1H), 1.53-1.44 (m, 11H), 1.43-1.25 (m, 1H), 1.04-0.90 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 172.2, 153.5, 132.3, 119.1, 83.5, 71.7, 58.7, 43.0, 35.0, 34.5, 33.8, 30.9, 27.5, 17.9, 12.1; IR (neat) 2942, 1737, 1670, 1385 cm−1; HRMS (APCI) calcd for C20H45NO2Si (MH+-C5H9O2) 352.2672, found 352.2662.
1-Allyl-4-hydroxy-3-oxo-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (17)
To a solution of carboxyimide 16 (540 mg, 1.2 mmol) in THF (90 mL) at −78 °C was added sodium bis(trimethylsilyl)amide (1.8 mL, 1.0 M in THF). After the mixture was stirred for 1 h, 2-(phenylsulfonyl)-3-phenyloxaziridine8b (630 mg, 2.4 mmol) was then added. After 30 min, saturated aqueous NH4Cl was added, and the aqueous layer was extracted with Et2O. The combined organic layers were washed with 28% NH4OH, water and brine, dried with MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (toluene) to give α-hydroxyimide 17 (550 mg, 98%) as a colorless oil: 1H NMR (360 MHz, CDCl3) δ 5.78 (dddd, J = 14.8, 7.9, 6.5, 6.5 Hz, 1H), 5.17-5.02 (m, 2H), 4.39-4.32 (m, 1H), 3.84-3.69 (m, 1H), 3.11 (d, J = 1.5 Hz, 1H), 2.71 (dd, J = 14.2, 6.4 Hz, 1H), 2.30–2.08 (m, 4H), 1.89-1.73 (m, 1H), 1.51 (s, 9H), 1.44-1.30 (m, 3H), 1.11-0.90 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 174.7, 152.2, 132.2, 119.4, 83.9, 70.3, 67.7, 60.4, 43.1, 41.4, 32.9, 32.7, 28.6, 27.7, 18.0, 12.2; IR (Nujol mull) 3423, 3078, 1742, 1659, 1462 cm−1; HRMS (APCI) calcd for C20H37NO3Si (MH+ -C5H9O2) 368.2621, found 368.2596.
1-Allyl-4-hydroxy-3-methoxy-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (19)
To a solution of α-hydroxyimide 17 (66 mg, 0.14 mmol) in THF (4.7 mL) at 0 °C was added lithium aluminum hydride (21 mg, 0.56 mmol). After stirring for 30 min at 0 °C, the mixture was carefully quenched with H2O (0.5 mL), 15% aqueous NaOH (0.5 mL) and water (0.5 mL). This mixture was dried with Na2SO4 and concentrated in vacuo. To the crude residue was added CH2Cl2/MeOH (1:1, 0.3 mL) and scandium(III) trifluoromethanesulfonate (1.0 mg) at rt. After 4 h, saturated aqueous NaHCO3 was added. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (15:1 –9:1, hexanes/EtOAc gradient) to give α-methoxyimide 19 (52 mg, 76%) as a white solid: mp 97–98 °C; 1H NMR (300 MHz, CDCl3) δ 5.77 (dddd, J = 17.5, 8.7, 6.3, 6.3 Hz, 1H), 5.67-5.24 (m, 1H), 5.09-4.99 (m, 2H), 3.93 (dd, J = 9.7, 6.0 Hz, 1H), 3.75-3.59 (m, 1H), 3.56 (s, 3H), 3.03-2.50 (m, 3H), 2.15 (dd, J = 13.7, 5.9 Hz, 1H), 2.07-1.91 (m, 1H), 1.71-1.59 (m, 1H), 1.51 (s, 9H), 1.32-1.18 (m, 1H), 1.12-0.90 (m, 23H); HRMS (APCI) calcd for C25H46NO4Si (MH+-MeOH) 452.3196, found 452.3166.
1-Allyl-4-benzyloxy-3-methoxy-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (20)
To a solution of alcohol 19 (509 mg, 1.1 mmol) and 18-crown-6 (14 mg, 0.05 mmol) in THF (3 mL) at rt was added 60% NaH in mineral oil (84 mg, 3.5 mmol). After 30 min, the mixture was cooled to 0 °C and benzyl bromide (250 μL, 2.1 mmol) was added. The mixture was warmed to rt and stirred for 18 h. Water was then carefully added to the mixture, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (30:1, hexanes/EtOAc) to give benzyl ether 20 (580 mg, 96%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.40-7.20 (m, 5H), 5.90-5.22 (m, 2H, rotamer), 5.10-4.92 (m, 2H, rotamer), 4.72-4.48 (m, 2H, rotamer), 3.82-3.62 (m, 2H), 3.46-3.22 (m, 3H, rotamer), 2.99-2.16 (m, 5H, rotamer), 1.73-1.62 (m, 1H, rotamer), 1.58-1.20 (m, 11H), 1.18-0.91 (m, 22H); 1H NMR (300 MHz, toluene-d8, 70 °C) δ 7.36-7.30 (m, 2H), 7.17-6.95 (m, 3H), 5.82 (dddd, J = 16.9, 10.2, 8.1, 6.7 Hz, 1H), 5.70 (br s, 1H), 5.09-4.98 (m, 2H), 4.59-4.47 (m, 2H), 3.76 (d, J = 5.0 Hz, 1H), 3.68 (ddd, J = 10.9, 5.0, 5.0 Hz, 1H), 3.30 (s, 3H), 2.97-2.81 (m, 2H), 2.63-2.50 (m, 1H), 2.30-2.18 (m, 2H), 1.70-1.57 (m, 1H), 1.49-1.32 (m, 10H), 1.11-0.88 (m, 23H); 13C NMR (75 MHz, toluene-d8, 70 °C) δ 139.9, 135.3, 128.3, 127.9 (2C), 127.4, 117.2, 84.0, 79.9, 75.1, 72.4, 71.1, 65.8, 56.8 (2C), 42.6, 34.2, 29.5, 28.6, 18.5, 18.4, 13.0; IR (neat) 3070, 1700, 1367 cm−1; HRMS (APCI) calcd for C32H52NO5Si (MH+-MeOH) 542.3666, found 542.3617.
1-Allyl-4-benzyloxy-3-(4-methoxybenzyl)-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (21 and 23)
To a solution of methoxyimide 20 (210 mg, 0.36 mmol) in Et2O (5 mL) at −30 °C was added TiCl4 (140 μL, 1.3 mmol). After 2 min, p-methoxybenzylmagnesium chloride16 (1.6 mL, 0.80 M17 in THF) was added to the mixture. The mixture was slowly warmed to -10 °C over 90 min. Saturated aqueous NH4Cl was then added to the mixture. The aqueous layer was extracted with Et2O. The combined organics were washed with saturated aqueous NaHCO3 and brine, dried with MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (toluene) to give a mixture of carbamates 21 and 23 (210 mg, 87%) as a colorless oil. Data for the 5:4 mixture of isomers: 1H NMR (300 MHz, CDCl3) δ 7.35-7.11 (m, 7H), 6.84-6.72 (m, 2H), 5.90-5.72 (m, 1H × 0.56), 5.45-5.29 (m, 1H × 0.44), 5.14-5.04 (m, 1H), 4.94-4.85 (m, 1H), 4.68 (d, J = 11.2 Hz, 1H × 0.56); 4.58 (br s, 1H × 0.44), 4.40 (d, J = 11.2 Hz, 1H × 0.56), 4.33 (br s, 1H × 0.44), 3.99 (d, J = 8.4 Hz, 1H × 0.56), 3.95-3.83 (m, 1H × 0.44), 3.82-3.73 (m, 7H), 3.66-3.57 (m, 1H), 3.32-3.18 (m, 1H), 2.84-2.22 (m, 5H), 1.83 (dd, J = 13.4, 8.9 Hz, 1H × 0.56), 1.77-1.22 (m, 13H), 1.18-1.12 (m, 21H), 0.98 (ddd, J = 7.6, 7.6, 4.6 Hz, 1H × 0.44); 13C NMR (75 MHz, CDCl3) δ 157.7 (minor), 157.6 (major), 155.3, 138.8 (minor), 138.7 (major), 134.8 (minor), 134.3 (major), 132.2, 130.4 (minor), 130.2 (major), 128.3 (minor), 128.0 (major), 127.7 (minor), 127.5, 127.4 (major), 127.1 (minor), 127.6 (major), 118.1 (minor), 117.5 (major), 113.5 (minor), 113.1 (major), 79.3, 75.6 (minor), 74.6 (major), 72.0, 71.6 (minor), 71.1 (major), 58.8 (minor), 57.6 (major), 55.3 (minor), 55.2 (major), 44.1, 43.7 (minor), 42.3 (major), 34.8 (minor), 34.2 (major), 33.7 (major), 29.2 (minor), 28.6 (major), 28.4, 18.3 (minor), 18.2, 12.6 (minor), 12.4 (major); IR (neat) 3066, 3031, 1691, 1366 cm−1; HRMS (APCI) calcd for C40H62NO5Si (MH+) 664.4397, found 664.4343.
1-Allyl-4-benzyloxy-3-(4-methoxybenzyl)-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonanes (22 and 24)
To a solution of the mixture of carbamates 21 and 23 (290 mg, 0.43 mmol) and anisole (0.5 mL, 4.3 mmol) in CH2Cl2 (5.7 mL) at rt was added trifluoroacetic acid (0.7 mL, 8.6 mmol). After 18 h, saturated aqueous NaHCO3 was carefully added. The aqueous layer was extracted with CH2Cl2. The combined organics were washed with saturated aqueous NaHCO3 and brine, dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (19:1 – 4:1, hexanes/EtOAc gradient) to give amines 22 (125 mg, 51%) and 24 (96 mg, 39%) as colorless oils.
22
1H NMR (400 MHz, CDCl3) δ 7.40-7.25 (m, 5H), 7.11 (d, J = 8.6 Hz, 2H), 6.79 (d, J = 8.6 Hz, 2H), 5.89-5.75 (m, 1H), 5.12-5.04 (m, 2H), 4.47 (d, J = 11.6 Hz, 1H), 4.21-4.08 (m, 2H), 3.78 (s, 3H), 3.76-3.69 (m, 1H), 3.50 (t, J = 2.8 Hz, 1H), 2.80-2.69 (m, 2H), 2.51-2.40 (m, 1H), 2.11-1.86 (m, 5H), 1.75-1.66 (m, 1H), 1.54 (ddd, J = 7.9, 13.5, 13.5 Hz, 1H), 1.15 (dd, J = 12.8, 2.6 Hz, 1H), 1.19-0.90 (m, 21H); 13C NMR (100 MHz, CDCl3) δ 157.7, 139.2, 133.8, 131.6, 130.0, 128.2, 127.6, 127.3, 118.4, 113.6, 72.5, 71.0, 70.8, 57.0, 55.2, 50.3, 48.4, 38.4, 38.3, 35.5, 32.8, 32.4, 18.1, 18.0, 12.3; HRMS (ESI+) calcd for C35H54NO3Si (MH+) 564.3873, found 564.3861.
24
1H NMR (300 MHz, CDCl3) δ 7.37-7.24 (m, 5H), 7.19-7.13 (m, 2H), 6.85-6.78 (m, 2H), 5.62 (dddd, J = 17.8, 10.2, 7.7, 7.7 Hz, 1H), 4.87 (dd, J = 10.1, 2.2 Hz, 1H), 4.78 (d, J = 11.5 Hz, 1H), 4.71-4.62 (m, 1H), 4.42 (d, J = 11.5 Hz, 1H), 3.81-3.66 (m, 4H), 3.51-3.45 (m, 1H), 3.20 (dd, J = 13.4, 2.7 Hz, 1H), 3.06 (ddd, J = 2.9, 9.2, 9.2 Hz, 1H), 2.41 (dd, J = 13.4, 9.1 Hz, 1H), 2.28-2.20 (m, 1H), 2.10 (ddd, J = 13.3, 3.6, 3.6 Hz, 1H) 1.96-1.82 (m, 2H), 1.81-1.54 (m, 2H), 1.24-0.95 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 157.9, 139.2, 133.7, 131.5, 130.2, 128.3, 127.7, 127.3, 117.9, 113.6, 78.6, 72.2, 70.7, 55.2, 54.5, 50.1, 47.5, 43.3, 39.3, 37.8, 31.5, 28.8, 18.4, 18.2, 12.7; IR (neat) 3067, 3030 cm−1; HRMS (ESI+) calcd for C35H54NO3Si (MH+) 564.3873, found 564.3848.
1-Allyl-6-hydroxy-2-benzyl-2-azabicyclo[3.3.1]nonan-3-one (29)
To a solution of alcohol 12 (1.3 g, 4.4 mmol), p-nitrobenzoic acid (3.2 g, 19.3 mmol) and triphenylphosphine (5.7 g, 21.9 mmol) in benzene (83 mL) at rt was added diethyl azodicarboxylate (3.5 mL, 21.9 mmol) dropwise. After 1 h, the mixture was concentrated in vacuo. The crude residue was dissolved with MeOH/THF (5:3, 110 mL) and a solution of K2CO3 (1.2 g, 8.8 mmol) in H2O (11 mL) was added at rt. The mixture was stirred for 18 h, and the aqueous layer was extracted with Et2O. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (1:2, hexanes/EtOAc) to give alcohol 29 (700 mg, 56%) as a white foam: 1H NMR (300 MHz, CDCl3) δ 7.30-7.11 (m, 5H), 5.54 (dddd, J = 16.7, 10.3, 7.9, 6.4 Hz, 1H), 5.04-4.91 (m, 2H), 4.60 (ABq, J = 15.6 Hz, ΔνAB = 64.5 Hz, 2H), 3.76 (d, J = 2.5 Hz, 1H), 2.93 (br s, 1H), 2.70 (dd, J = 18.4, 7.5 Hz, 1H), 2.36 (dd, J = 14.4, 6.3 Hz, 1H), 2.27 (dd, J = 18.4, 1.8 Hz, 1H), 2.22-2.06 (m, 2H), 2.05-1.97 (m, 1H), 1.72-1.38 (m, 4H), 1.32-1.22 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 172.3, 139.1, 132.5, 128.2, 127.3, 126.8, 118.9, 69.0, 58.9, 44.7, 44.4, 35.9, 32.8, 31.1, 28.1, 25.1; IR (film) 3388, 3064, 3027, 1611 cm−1; HRMS (ESI+) calcd for C18H24NO2 (MH+) 286.1807, found 286.1802.
1-Allyl-2-benzyl-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonan-3-one (30)
To a solution of alcohol 29 (860 mg, 3.0 mmol) in CH2Cl2 (12 mL) at 0 °C was added triethylamine (1.1 mL, 7.5 mmol) and triisopropylsilyl trifluoromethanesulfonate (1.1 mL, 3.9 mmol). The mixture warmed to rt, stirred for 2 h and water (10 mL) was then added. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (8:1, hexanes/EtOAc) to give silyl ether 30 (950 mg, 72%) as a colorless oil: 1H NMR (360 MHz, CDCl3) δ 7.32-7.16 (m, 5H), 5.57 (dddd, J = 16.9, 10.2, 7.6, 7.6 Hz, 1H), 5.06-4.96 (m, 2H), 4.66 (ABq, J = 15.5 Hz, ΔνAB = 42.0 Hz, 2H), 3.91-3.81 (m, 1H), 2.75 (dd, J = 18.4, 7.5 Hz, 1H), 2.41 (dd, J = 13.9, 5.8 Hz, 1H), 2.36-2.28 (m, 1H), 2.21 (dd, J = 14.3, 7.9 Hz, 1H), 2.16-2.04 (m, 2H), 1.71 (ddd, J = 4.7, 13.1, 13.1 Hz, 1H), 1.64-1.57 (m, 1H), 1.55-1.39 (m, 2H), 1.30-1.19 (m, 1H), 1.11-0.96 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 171.9, 139.4, 132.6, 128.1, 127.5, 126.6, 118.9, 70.0, 58.7, 44.5, 44.3, 35.8, 33.6, 31.2, 28.4, 25.7, 17.9, 12.1; IR (neat) 3063, 3027, 1623, 1398 cm−1; HRMS (ESI+) calcd for C27H44NO2Si (MH+) 442.3141, found 442.3103.
1-Allyl-2-benzyl-4-hydroxy-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonan-3-one (31)
A solution of carboxyimide 30 (1.00 g, 2.26 mmol) in THF (6.8 mL) was added dropwise via cannula to LDA (7.14 mL, 0.47 M in THF) at −78 °C. The mixture was warmed to 0 °C and stirred for 1 h, recooled to −78 °C and 2-(phenylsulfonyl)-3-phenyloxaziridine8b (886 mg, 3.39 mmol) was added. The mixture was allowed to warm to rt overnight, saturated aqueous NaHCO3 was added and the aqueous layer was extracted with Et2O. The combined organic layers were washed with saturated aqueous NaHCO3, water and brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (4:1, hexanes/EtOAc) to give α-hydroxyimide 31 (628 mg, 60%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.39-7.19 (m, 5H), 5.64 (dddd, J = 16.9, 10.3, 7.5, 6.6 Hz, 1H), 5.12 (d, J = 8.4 Hz, 1H), 5.05 (dd, J = 15.1, 1.6 Hz, 1H), 4.78, 4.57 (ABq, J = 15.5 Hz, ΔνAB = 48.2 Hz, 2H), 4.00-3.88 (m, 1H), 3.85 (s, 1H), 3.40 (s, 1H), 2.41 (dd, J = 14.4, 6.5 Hz, 1H), 2.24 (dd, J = 14.2, 7.7 Hz, 1H), 2.00 (ddd, J = 12.1, 3.1, 1.3 Hz, 1H), 1.88-1.65 (m, 3H), 1.57-1.46 (m, 1H), 1.22-1.14 (m, 2H), 1.10-0.89 (m, 21H).
1-Allyl-2-benzyl-4,6-dihydroxy-2-azabicyclo[3.3.1]nonan-3-one (32)
To a stirred solution of lactam alcohol 31 (200 mg, 0.437 mmol) in anhydrous THF (4.4 mL) was added tetrabutylammonium fluoride (525 μL, 1.0 M in THF, 0.525 mmol). After stirring the mixture at rt for 5 h, a further portion of tetrabutylammonium fluoride (1.0 mL, 1.0 M in THF, 1.0 mmol) was added and the mixture was stirred overnight. The reaction was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (EtOAc) to afford diol 32 (132 mg, 100%) as a white crystalline solid. Crystals suitable for X-ray crystallography were obtained by recrystallization from MeOH: 1H NMR (400 MHz, CDCl3) δ 7.31-7.20 (m, 5H), 5.62 (dddd, J = 16.9, 10.2, 7.6, 6.7 Hz, 1H), 5.08 (d, J = 10.7 Hz, 1H), 5.04 (dd, J = 16.9, 1.3 Hz, 1H), 4.75, 4.61 (ABq, J = 13.4 Hz, ΔνAB = 28.1 Hz, 2H), 3.94 (d, J = 2.5 Hz, 1H), 3.89 (s, 1H), 3.76 (br s, 1H), 2.41 (dd, J = 14.4, 6.5 Hz, 1H), 2.24 (dd J = 13.0, 7.3 Hz, 2H), 1.98-1.87 (m, 3H), 1.69 (ddd, J = 12.8, 12.8, 3.6 Hz, 1H), 1.38-1.21 (m, 2H), 1.58-1.49 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 173.7, 138.4, 132.1, 128.5, 127.5, 127.1, 119.5, 70.2, 66.9, 59.9, 44.9, 44.3, 39.9, 28.1, 27.1, 25.7.
1-Allyl-3-oxo-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (33)
In a 500 mL three-neck round bottomed flask fitted with a cold finger condenser at −78 °C was condensed ammonia (60 mL). Sodium (500 mg, 27.7 mmol) followed by t-butanol (1.0 mL, 10.9 mmol) were carefully added to the ammonia changing the color of the mixture to blue. After 45 min, a solution of N-benzyl lactam 30 (950 mg, 2.2 mmol) in Et2O (17 mL) was added to the mixture dropwise via a gastight syringe. The mixture was stirred at reflux for 75 min, and then solid ammonium chloride (3 g) was carefully added until the blue color dissipated. The mixture was then slowly warmed to rt, while occasionally replacing the evaporating ammonia with THF. The resulting mixture was concentrated in vacuo. The crude residue was dissolved in THF (32 mL) and 60% NaH in mineral oil (170 mg, 7.2 mmol) was added at 0 °C. The mixture was heated at reflux for 1 h, was then cooled to rt and di-t-butyl dicarbonate (2.0 mL, 8.7 mmol) was added. The mixture was again heated at reflux and stirred for 18 h, cooled to rt and saturated aqueous NH4Cl was carefully added. The aqueous layer was extracted with Et2O. The combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (20:1, hexanes/EtOAc) to give carboxyimide 33 (660 mg, 67%) as a colorless oil: 1H NMR (360 MHz, CDCl3) δ 5.84-5.69 (m, 1H), 5.09-4.98 (m, 2H), 3.87-3.71 (m, 1H), 2.55 (ddd, J = 18.1, 7.3, 4.5 Hz, 1H), 2.50-2.41 (m, 1H), 2.16-2.03 (m, 3H), 2.01-1.90 (m, 2H), 1.73-1.49 (m, 4H), 1.48-1.40 (m, 9H), 1.08-0.86 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 171.1, 153.4, 132.2, 118.8, 83.3 (2C), 69.8, 59.7 (2C), 43.4, 35.7, 34.0, 30.1, 29.8, 27.4, 25.8, 17.8 (3C), 12.0; HRMS (APCI) calcd for C25H46NO4Si (MH+) 452.3196, found 452.3193.
1-Allyl-4-hydroxy-3-oxo-6-(triisopropylsilanyloxy)-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (34)
To a solution of carboxyimide 33 (200 mg, 0.45 mmol) in THF (27 mL) at −78 °C was added sodium bis(trimethylsilyl)amide (0.54 mL, 1.0 M in THF). The mixture was warmed to −10 °C over a 1 h period. After the mixture was cooled to −78 °C, 2-(phenylsulfonyl)-3-phenyloxaziridine8b (190 mg, 0.72 mmol) was added and the mixture was stirred for an additional 30 min. Saturated aqueous NH4Cl was added, the mixture was warmed to rt, and the aqueous layer was extracted with Et2O. The combined organic layers were washed with 28% aqueous NH4OH, water and brine, dried with MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (toluene - 25:1, toluene/EtOAc gradient) to give α-hydroxyimide 34 (110 mg, 53%) as a white solid: mp 77–78 °C; 1H NMR (360 MHz, CDCl3) δ 5.78 (dddd, J = 17.0, 10.3, 7.6, 6.8 Hz, 1H), 5.17-5.08 (m, 2H), 4.33 (d, J = 2.7 Hz, 1H), 4.17 (d, J = 6.6 Hz, 1H), 3.47 (s, 1H), 2.64 (dd, J = 14.6, 6.7 Hz, 1H), 2.54-2.48 (m, 1H), 2.21–2.13 (m, 2H), 2.07 (dd, J = 13.7, 4.0 Hz, 1H), 1.85-1.63 (m, 3H), 1.55-1.44 (m, 10H), 1.12-0.98 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 175.9, 153.0, 131.9, 119.4, 83.9, 70.0, 64.5, 62.0, 43.1, 39.7, 30.6, 27.7, 26.2, 18.1 (2C), 12.2; HRMS (ESI+) calcd for C25H45NNaO5Si (MH+ +Na) 490.2965, found 490.2951.
1-Allyl-4-benzyloxy-3-oxo-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (35)
To a solution of alcohol 34 (44 mg, 0.09 mmol) and 18-crown-6 (1 mg, 0.005 mmol) in THF (0.3 mL) at rt was added 60% NaH in mineral oil (7 mg, 0.30 mmol). After 30 min, the mixture was cooled to 0 °C and benzyl bromide (22 μL, 0.18 mmol) was added. The mixture was warmed to rt and stirred for 18 h. Water was then carefully added to the mixture, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (9:1, hexanes/EtOAc) to give benzyl ether 35 (35 mg, 69%) as a colorless oil: 1H NMR (360 MHz, CDCl3) δ 7.39-7.23 (m, 5H), 5.92-5.71 (m, 1H), 5.18-5.06 (m, 3H), 4.79 (d, J = 12.7 Hz, 1H), 4.39 (d, J = 2.7 Hz, 1H), 3.94 (d, J = 6.5 Hz, 1H), 2.54 (dd, J = 14.5, 6.4 Hz, 1H), 2.42-2.36 (m, 1H), 2.25-1.95 (m, 3H), 1.90-1.60 (m, 4H), 1.53 (m, 9H), 1.12-0.93 (m, 21H); 13C NMR (90 MHz, CDCl3) δ 173.7, 153.4, 138.3, 132.9, 128.3, 127.7, 127.6, 119.2, 83.8, 75.8, 73.9, 65.0, 60.7, 43.4, 40.6, 30.9, 30.3, 27.7, 26.3, 18.1 (2C), 12.1 (2C); IR (neat) 3065, 3031, 1739, 1689 cm−1; HRMS (APCI) calcd for C32H52NO5Si (MH+) 558.3615, found 558.3620.
1-Allyl-4-benzyloxy-3-hydroxy-3-(4-methoxybenzyl)-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (36)
To a solution of carboxyimide 35 (17 mg, 0.03 mmol) in Et2O (0.4 mL) at −35 °C was added p-methoxybenzylmagnesium chloride16 (0.09 mL, 1.21 M17 in THF). The mixture was slowly warmed to rt and stirred for 18 h. Saturated aqueous NH4Cl was then added to the mixture, and the aqueous layer was extracted with Et2O. The combined organics were washed with brine, dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (10:1, hexanes/EtOAc) to give adduct 36 (19 mg, 90%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.35-7.24 (m, 5H), 7.16-7.09 (m, 2H), 6.88-6.81 (m, 2H), 5.63 (dddd, J = 17.4, 10.5, 7.3, 7.3 Hz, 1H), 5.10-5.00 (m, 2H), 4.63 (d, J = 2.3 Hz, 1H), 4.48-4.34 (m, 2H), 4.24 (d, J = 10.9 Hz, 1H), 3.90-3.76 (m, 4H), 3.66 (ABq, J = 15.2 Hz, ΔνAB = 22.5 Hz, 2H), 2.71-2.53 (m, 1H), 2.39 (dd, J = 14.2, 7.5 Hz, 1H), 2.22-2.08 (m, 1H), 2.03-1.88 (m, 2H), 1.70-1.48 (m, 4H), 1.45-1.35 (m, 9H), 1.12-0.94 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 209.0, 158.7, 137.9, 133.6, 130.5, 128.2, 127.6, 125.4, 118.2, 114.1, 87.8, 82.4, 72.2, 70.6, 55.2, 53.9, 45.2, 43.5, 33.0, 32.3, 31.3, 29.7, 28.4, 18.4, 18.3, 13.0; IR (neat) 3412, 3044, 3032, 1717, 1512 cm−1; HRMS (ESI+) calcd for C40H62NO6Si (MH+) 680.4346, found 680.4311.
1-Allyl-4-benzyloxy-3-(4-methoxybenzyl)-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane-2-carboxylic Acid t-Butyl Ester (37)
To a solution of alcohol 36 (30 mg, 0.04 mmol) in AcOH (0.5 mL) at rt was added 5 portions of sodium cyanoborohydride (14 mg × 5, 0.22 mmol × 5) every 30 min for 2.5 h. After 48 h at rt, the mixture was poured into H2O, and the aqueous layer was extracted with EtOAc. The combined organics were washed with brine and 10% NaOH, dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (9:1, hexanes/EtOAc) to give carbamate 37 (20 mg, 69%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.38-7.25 (m, 5H), 7.20-7.13 (m, 2H), 6.89-6.83 (m, 2H), 5.82 (dddd, J = 16.7, 9.1, 7.3, 7.3 Hz, 1H), 5.18-5.06 (m, 2H), 4.69 (ABq, J = 11.4 Hz, ΔνAB = 19.9 Hz, 2H), 4.53-4.39 (m, 1H) 4.05-3.95 (m, 2H), 3.88 (ddd, J = 9.8, 9.8, 4.4 Hz, 1H), 3.80 (s, 3H), 2.91-2.78 (m, 1H), 2.77-2.62 (m, 2H), 2.55 (dd, J = 14.5, 7.8 Hz, 1H), 2.19-2.08 (m, 1H), 2.03-1.88 (m, 2H), 1.72-1.50 (m, 2H), 1.45-1.36 (m, 9H), 1.29-1.18 (m, 1H), 1.11-1.00 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 158.7, 156.6, 139.1, 134.5, 131.0, 130.7, 128.8, 128.0, 127.7, 118.7, 114.5, 80.1, 73.6, 73.3, 72.4, 55.7, 54.8, 42.6, 39.2, 37.8, 33.5, 31.9, 30.1, 28.9, 18.9 (2C), 13.7; HRMS (APCI) calcd for C40H62NO5Si (MH+) 664.4397, found 664.4450.
1-Allyl-4-benzyloxy-3-(4-methoxybenzyl)-6-triisopropylsilanyloxy-2-azabicyclo[3.3.1]nonane (38)
To a solution of carbamate 37 (20 mg, 0.03 mmol) and anisole (33 μL, 0.30 mmol) in CH2Cl2 (0.5 mL) at rt was added trifluoroacetic acid (47 μL, 0.60 mmol). After 18 h, saturated aqueous NaHCO3 was carefully added, and the aqueous layer was extracted with CH2Cl2. The combined organics were washed with saturated aqueous NaHCO3 and brine, dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by flash chromatography (9:1, EtOAc/MeOH) to give amine 38 (17 mg, 100%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.37-7.23 (m, 5H), 7.20-7.14 (m, 2H), 6.89-6.81 (m, 2H), 5.96-5.78 (m, 1H), 5.22-5.12 (m, 2H), 4.65 (ABq, J = 11.4 Hz, Δν = 33.4 Hz, 2H), 4.09 (ddd, J = 9.0, 4.7, 4.7 Hz, 1H), 3.89 (br d, J = 3.8 Hz, 1H), 3.85-3.75 (m, 4H), 2.97-2.62 (m, 2H), 2.44-2.17 (m, 2H), 2.05-1.90 (m, 2H), 1.74-1.19 (m, 6H), 1.12-0.99 (m, 21H); IR (neat) 3342, 3064, 3031, 1463 cm−1; APCI m/z (relative intensity) 582.4 (MH++H2O, 100), 408.3 (9).
7-Benzyloxy-6-(4-methoxybenzyl)-3-phenylsulfanyl-9-triisopropylsilanyloxy-5-azatricyclo[6.3.1.01,5]dodecanes (43a/43b)
To a solution of amine 22 (40 mg, 0.07 mmol) in Et2O (1 mL) at rt was added pyridinium p-toluenesulfonate (21 mg, 0.09 mmol). After 30 min, the mixture was concentrated in vacuo and the crude residue was diluted with CH2Cl2 (1 mL). To the mixture was then added PhSCl18 (12 μL, 0.11 mmol) and after 30 min, MeOH (1 mL) was added followed by K2CO3 (98 mg, 0.71 mmol) and silica gel (160 mg). After 18 h, the mixture was filtered and the filtrate was concentrated in vacuo. The crude residue was purified by flash chromatography (9:1 – 1:1, hexanes/EtOAc gradient) to give amines 43a (9 mg, 19%) and 43b (15 mg, 32 %) as colorless oils.
43a
1H NMR (300 MHz, CDCl3) δ 7.32-7.15 (m, 10H), 7.02 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.6 Hz, 2H), 4.32-4.23 (m, 1H), 4.08-3.94 (m, 2H), 3.80-3.59 (m, 6H), 3.41-3.33 (m, 1H), 2.90-2.72 (m, 2H), 2.39-2.26 (m, 1H), 2.21-1.70 (m, 5H), 1.62-1.07 (m, 3H), 0.98-0.73 (m, 21H); HRMS (ESI+) calcd for C41H58NO3SSi (MH+) 672.3907, found 672.3884.
43b
1H NMR (400 MHz, CDCl3) δ 7.39-7.22 (m, 9H), 7.20-7.14 (m, 1H), 7.07 (d, J = 8.6 Hz, 2H), 6.76 (d, J = 8.6 Hz, 2H), 4.46-4.40 (m, 1H), 4.13-4.00 (m, 2H), 3.80-3.62 (m, 5H), 3.49-3.35 (m, 2H), 3.00 (dd, J = 13.1, 10.6 Hz, 1H), 2.89-2.80 (m, 1H), 2.46-2.28 (m, 2H), 2.12-1.49 (m, 6H), 1.34-1.19 (m, 1H), 1.12-0.81 (m, 22H); HRMS (ESI+) calcd for C41H58NO3SSi (MH+) 672.3907, found 672.3884.
7-Benzyloxy-6-(4-methoxybenzyl)-9-triisopropylsilanyloxy-5-azatricyclo[6.3.1.01,5]dodecane (44)
To a solution of amines 43a and 43b (6 mg, 0.009 mmol) in EtOH (1.5 mL) at rt was added Raney-Ni (~50–70 mg) and the mixture was then heated at reflux. After 15 min, the mixture was filtered and the filtrate was concentrated in vacuo. The crude residue was purified by flash chromatography (10:1, hexanes/EtOAc) to give amine 44 (4 mg, 79%) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.38-7.24 (m, 5H), 7.14-7.08 (m, 2H), 6.82-6.75 (m, 2H), 4.41-4.32 (m, 1H), 4.15-3.99 (m, 2H), 3.78 (s, 3H), 3.73-3.61 (m, 1H), 3.51-3.29 (m, 2H), 3.02-2.71 (m, 3H), 2.45-2.32 (m, 1H), 2.18-1.92 (m, 2H), 1.90-1.11 (m, 8H), 1.01-0.79 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 157.8, 139.2, 130.1, 128.2, 127.5, 127.2, 113.6, 72.9, 71.5, 70.6, 61.4, 55.2, 47.8, 38.6, 38.1, 35.9, 32.4, 31.0, 29.7, 19.7, 18.1, 18.0, 12.3; HRMS (ESI+) calcd for C35H54NO3Si (MH+) 564.3873, found 564.3908.
Supplementary Material
Copies of proton and carbon NMR spectra of new compounds, and X-ray data including ORTEP drawings for compounds 14, 32 and 34. This material is available free of charge on the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful to the National Institutes of Health (CA-034303) for financial support of this research. We thank Darren DiPietro for conducting some preliminary experiments on this project. We also thank Dr. Hemant Yennawar (Penn State Small Molecule X-Ray Crystallographic Facility), and Dr. Louis Todaro (Hunter College, CUNY) for crystal structure determinations.
References
- 1.Sakamoto K, Tsujii E, Abe F, Nakanishi T, Yamashita M, Shigematsu N, Izumi S, Okuhara M. J Antibiot. 1996;49:37. doi: 10.7164/antibiotics.49.37. [DOI] [PubMed] [Google Scholar]
- 2.For total syntheses of FR901483 see: Snider BB, Lin H. J Am Chem Soc. 1999;121:7778.Scheffler G, Seike H, Sorensen EJ. Angew Chem Int Ed Engl. 2000;39:4593. doi: 10.1002/1521-3773(20001215)39:24<4593::aid-anie4593>3.0.co;2-x.Ousmer M, Braun NA, Ciufolini MA. Org Lett. 2001;3:765. doi: 10.1021/ol015526i.Maeng JH, Funk RL. Org Lett. 2001;3:1125. doi: 10.1021/ol015506g.Ousmer M, Braun NA, Bavoux C, Perrin M, Ciufolini MA. J Am Chem Soc. 2001;123:7534. doi: 10.1021/ja016030z.Kan T, Fujimoto T, Ieda S, Asoh Y, Kitaoka H, Fukuyama T. Org Lett. 2004;6:2729. doi: 10.1021/ol049074w.For a formal total synthesis see: Brummond KM, Hong S. J Org Chem. 2005;70:907. doi: 10.1021/jo0483567.
- 3.For other synthetic approaches to this metabolite and some analogs see: Snider BB, Lin H, Foxman BM. J Org Chem. 1998;63:6442.Bonjoch J, Diaba F, Puigbo G, Sole D, Segarra V, Santamaria L, Beleta J, Ryder H, Palacios JM. Bioorg Med Chem. 1999;7:2891. doi: 10.1016/s0968-0896(99)00250-3.Suzuki H, Yamazaki N, Kibayashi C. Tetrahedron Lett. 2001;42:3013.Brummond KM, Lu J. Org Lett. 2001;3:1347. doi: 10.1021/ol010029n.Wardrop DJ, Zhang W. Org Lett. 2001;3:2353. doi: 10.1021/ol0161514.Bonjoch J, Diaba F, Puigbo G, Peidro E, Sole D. Tetrahedron Lett. 2003;44:8387.Panchaud P, Ollivier C, Renaud P, Zigmantas S. J Org Chem. 2004;69:2755. doi: 10.1021/jo035843y.
- 4.Taken in part from the Ph.D. thesis of J.E. Kropf, The Pennsylvania State University, 2002.
- 5.See for example: Marvell EN, Sturmer D, Rowell C. Tetrahedron. 1966;22:861.Lednicer D. J Med Chem. 1977;20:171. doi: 10.1021/jm00211a040.Boger DL, Patel M, Mullican MD. Tetrahedron Lett. 1982;23:4559.Quirante J, Escolano C, Merino A, Bonjoch J. J Org Chem. 1998;63:968.
- 6.For a review, see: Zefirov NS, Palyulin VA. Conformational Analysis of Bicyclo[3.3.1]nonanes and Their Hetero Analogs. In: Eliel EL, Wilen SH, editors. Topics in Stereochemistry. Vol. 20. Wiley; New York: 1991. p. 171.
- 7.In model systems, we have investigated the possibility of enantioselectively cyclizing prochiral ketones similar to 10 with chiral amide bases: Kropf JE, Weinreb SM. Chem Commun. 1998:2357.
- 8.(a) Davis FA, Vishwakarma LC, Billmers JM, Finn J. J Org Chem. 1984;49:3241. [Google Scholar]; b Vishwakarma LC, Stringer OD, Davis FA. Org Synth. 1988;66:203. [Google Scholar]
- 9.Okitsu O, Suzuki R, Kobayashi S. J Org Chem. 2001;66:809. doi: 10.1021/jo001297m. [DOI] [PubMed] [Google Scholar]
- 10.CfHarrison JR, O’Brien P. Tetrahedron Lett. 2000;41:6167. [Google Scholar]
- 11.Martin SF, Dodge JA. Tetrahedron Lett. 1991;32:3017. [Google Scholar]; b) Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 12.Exact replication of the hydroxylation conditions previously used with N-benzyl lactam 30 (Cf. Scheme 7), which involved the use of LDA to form the corresponding enolate in place of sodium hexamethyldisilazide, also led to the same incorrect equatorial C-2 alcohol 34, but in low yield.
- 13.(a) Berthe B, Outurquin F, Paulmier C. Tetrahedron Lett. 1997;38:1393. [Google Scholar]; b Pannecoucke X, Outurquin F, Paulmier C. Eur J Org Chem. 2002:995. [Google Scholar]; c Outurquin F, Pannecoucke X, Bénédicte B, Paulmier C. Eur J Org Chem. 2002:1007. [Google Scholar]
- 14.(a) Cooper MA, Francis CL, Holman JW, Kasum B, Taverner T, Tickink ERT, Word DA. Aust J Chem. 2000;53:123. [Google Scholar]; b Toshimitsu A, Terao K, Uemura S. J Org Chem. 1986;51:1724. [Google Scholar]; (c) Clive DLJ, Farina V, Singh A, Wong CK, Kiel WA, Menchen SM. J Org Chem. 1980;45:2120. [Google Scholar]
- 15.Yodo M, Harada H. Chem Pharm Bull. 1989;37:2361. [Google Scholar]
- 16.Iida H, Yamazaki N, Kibayashi C. J Org Chem. 1986;51:1069. [Google Scholar]
- 17.Titrated with salicylaldehyde phenylhydrazone: Love BE, Jones EG. J Org Chem. 1999;64:3755. doi: 10.1021/jo982433e.
- 18.Harpp DN, Friedlander BT, Smith RA. Synthesis. 1979:181. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of proton and carbon NMR spectra of new compounds, and X-ray data including ORTEP drawings for compounds 14, 32 and 34. This material is available free of charge on the Internet at http://pubs.acs.org.